
Multi-Omic Analysis of Two Common P53 Mutations: Proteins Regulated by Mutated P53 as Potential Targets for Immunotherapy

 ,
,  , ,
, ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Cell Adhesion Assays
2.3. Colony Forming Assays
2.4. Proliferation Assays
2.5. Gene Expression Arrays
2.6. Cell Lysis and Homogenization
2.7. Liquid Chromatography with Tandem Mass Spectrometry (LC MS/MS) Analysis (IDA/SWATH)
2.8. SWATH Data Analysis
2.9. Purification of Antibodies and Preparation of Immunoaffinity Columns
2.10. HLA Purification and Peptide Elution
2.11. Analysis of HLA Class I Bound Peptides by LC-MS/MS
2.12. Immunopeptidomic Data Analysis
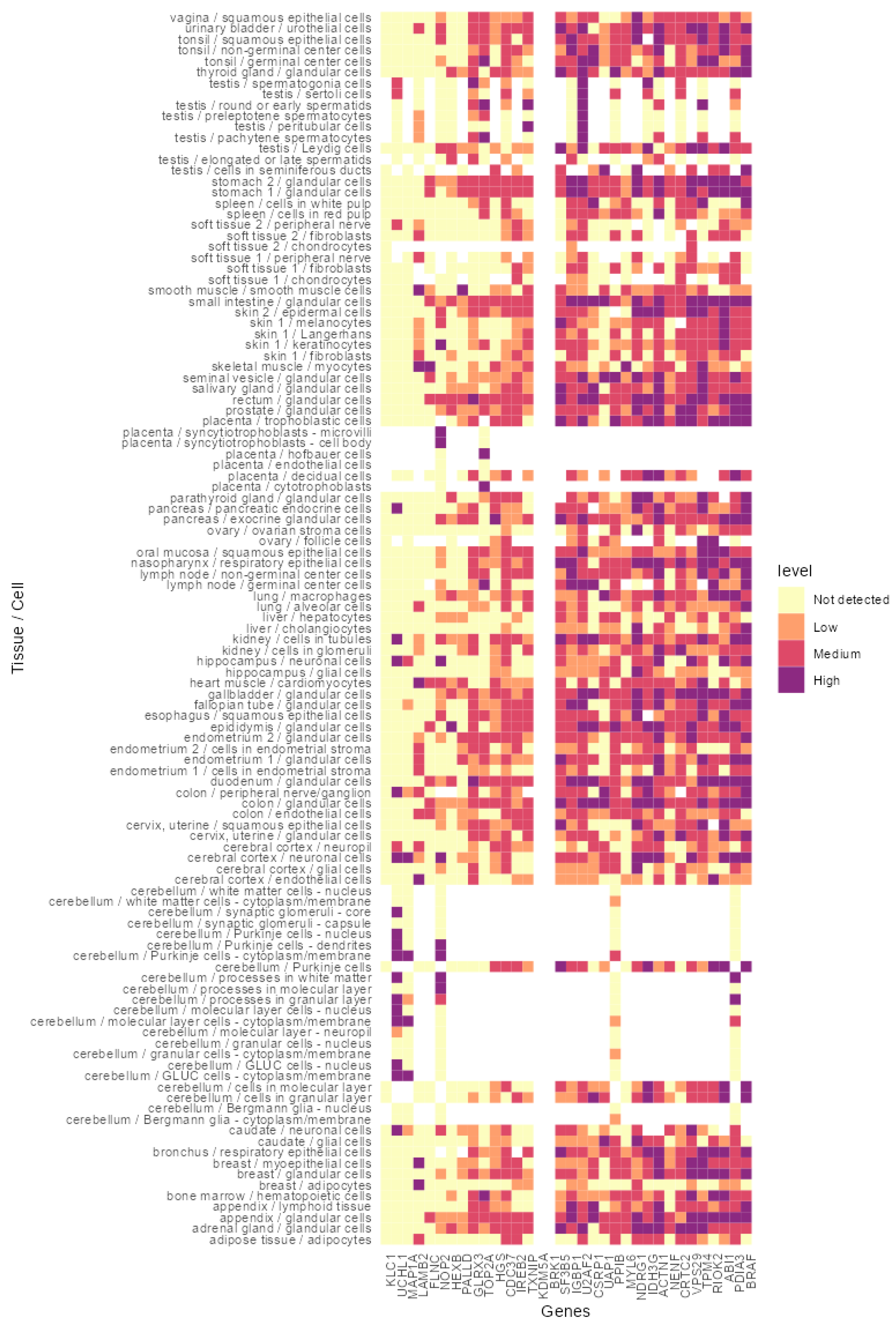
2.13. In Silico Analysis of the Identified Proteins Indicated Their Expression in a Broad Spectrum of Healthy Tissues and Cancer
3. Results
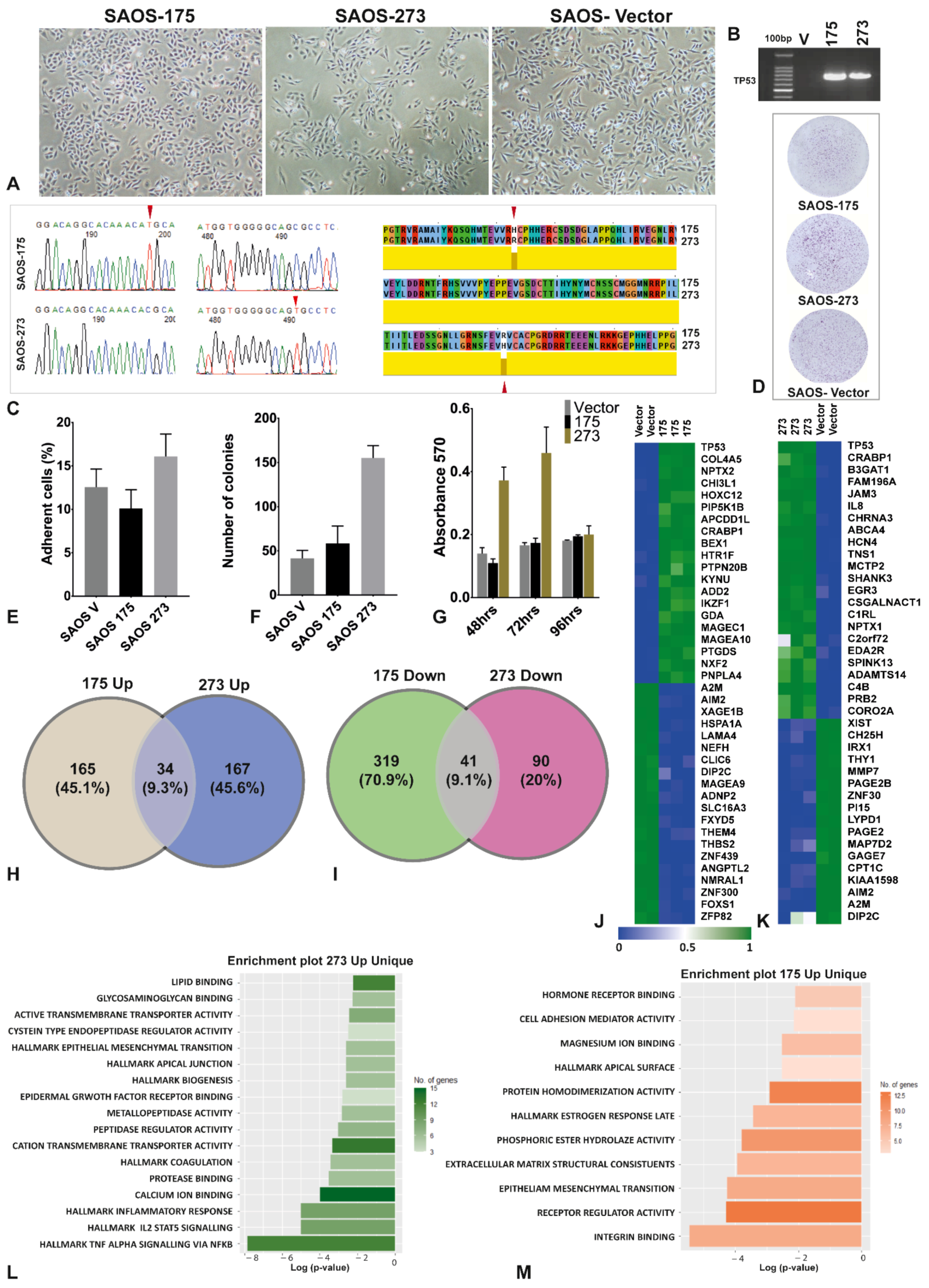
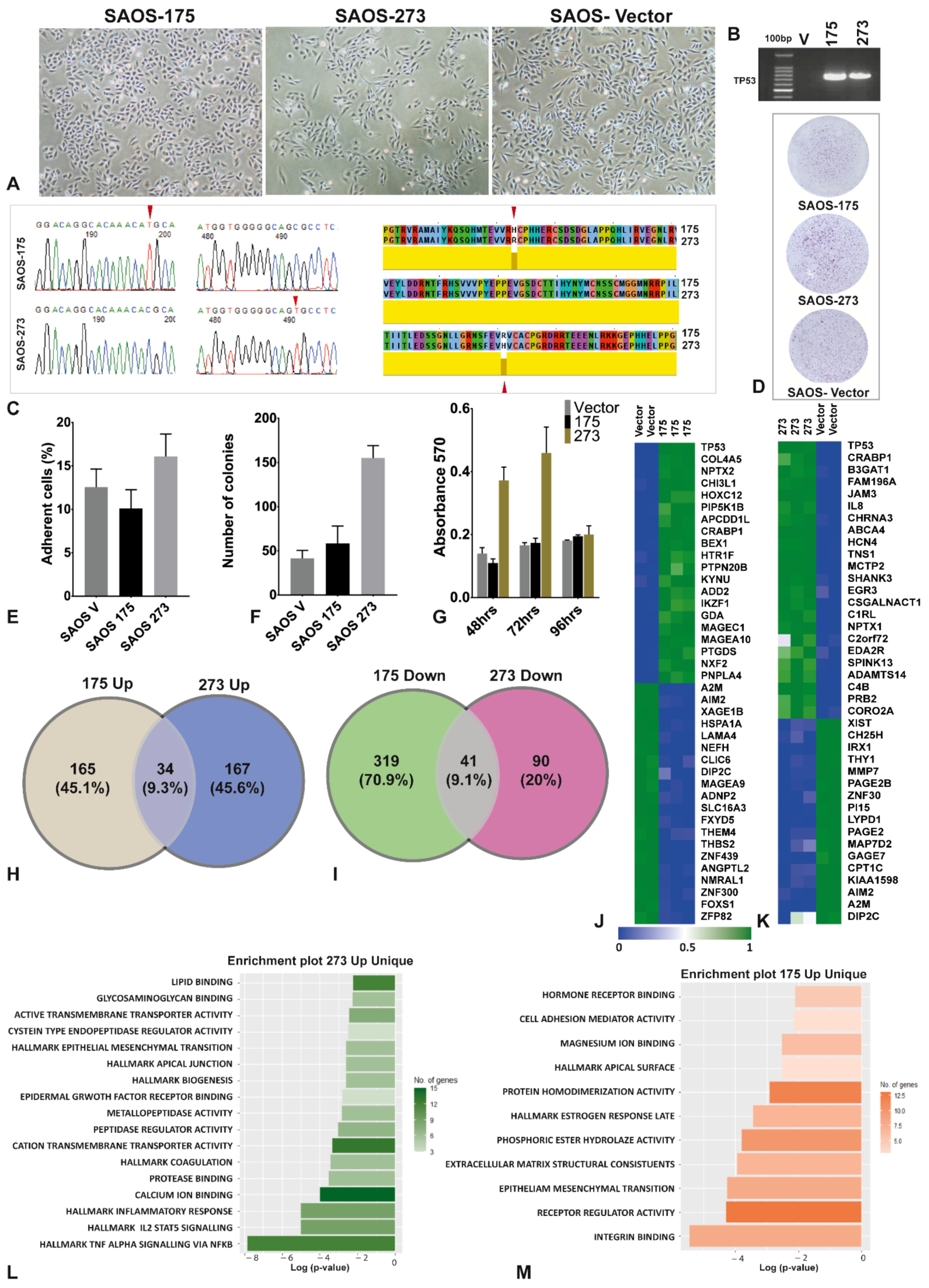
3.1. The Two Mutants of TP53 Had Significantly Different Phenotypical, Functional and Gene Expression
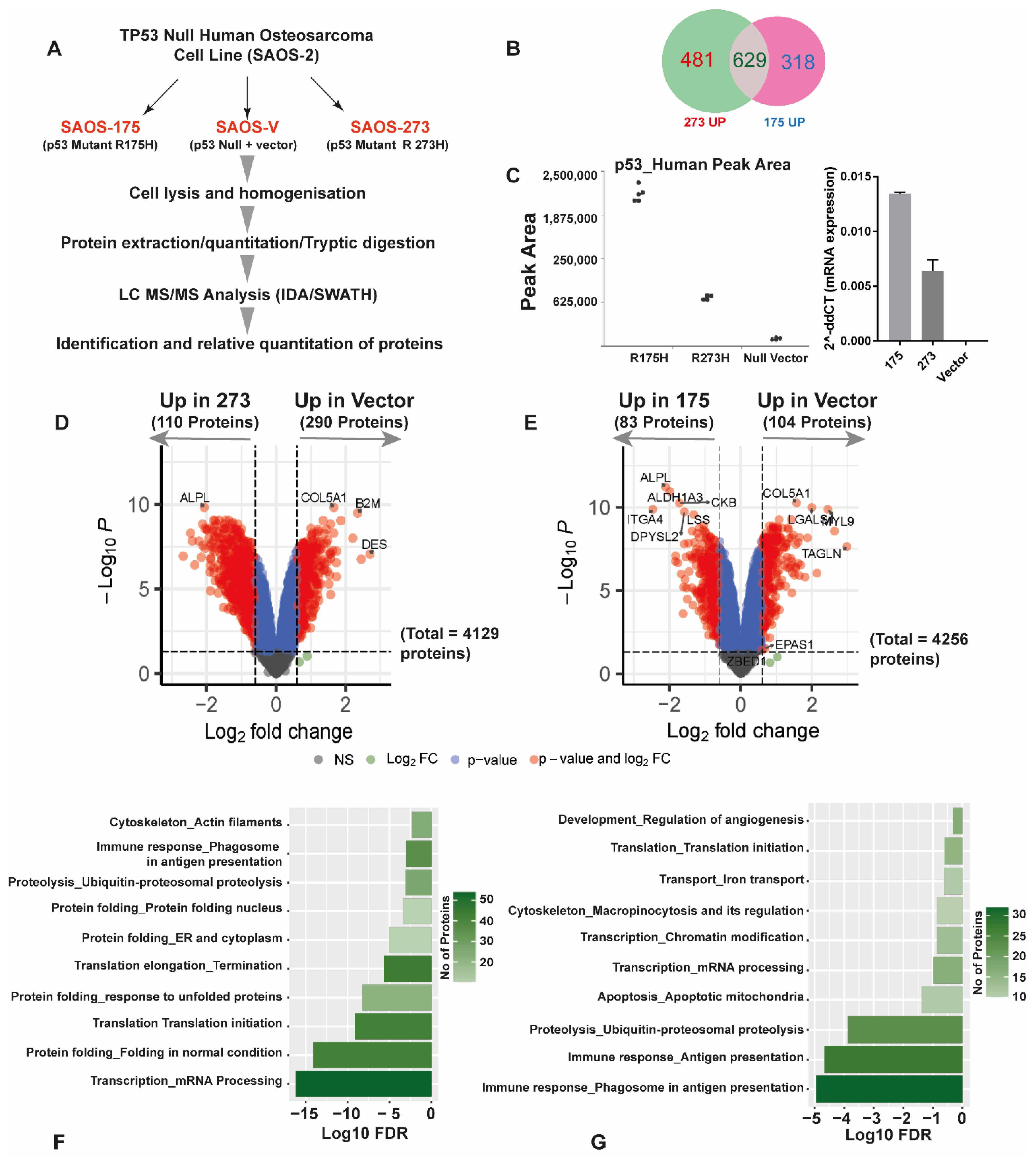
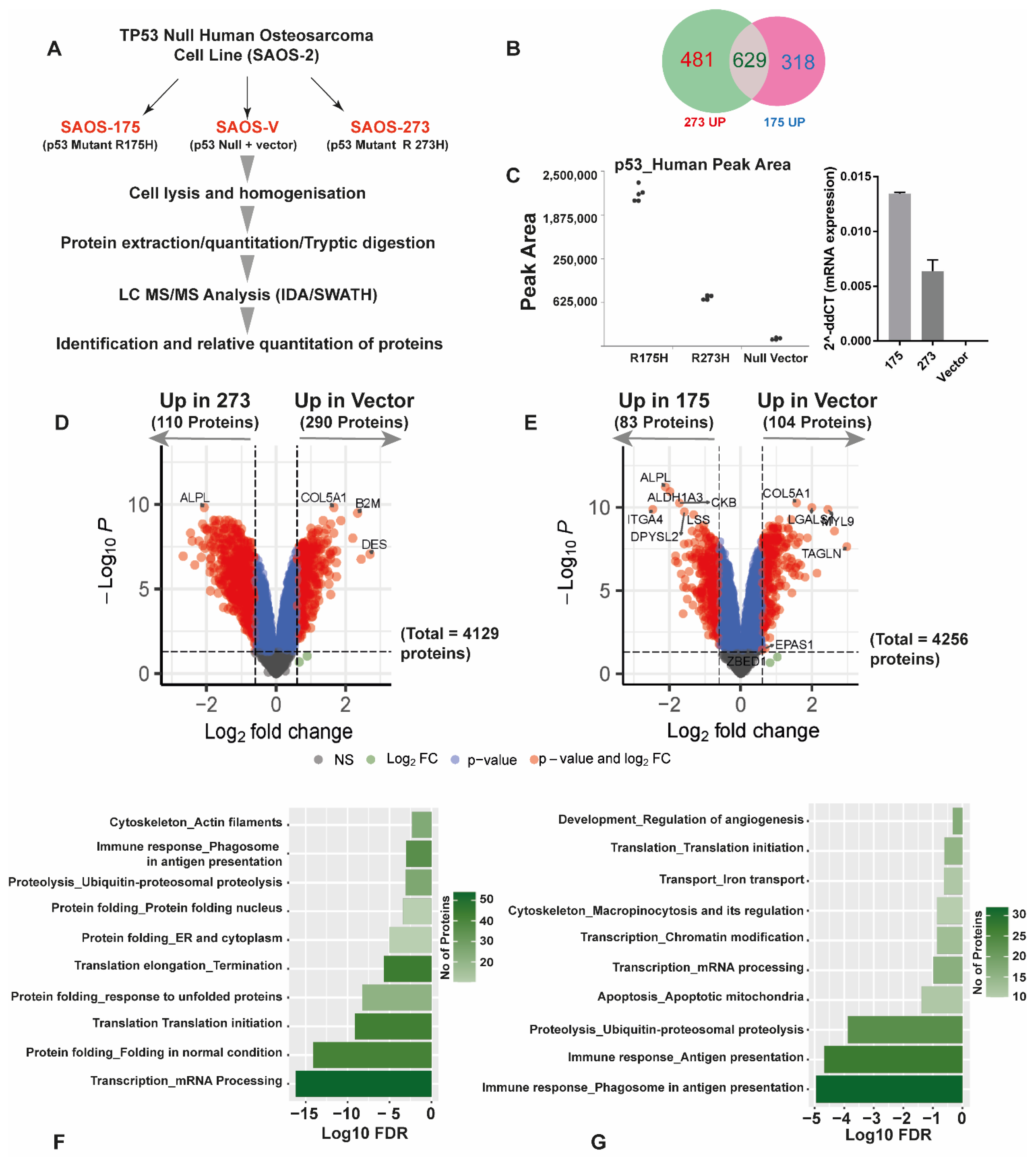
3.2. Quantitative SWATH Proteomics
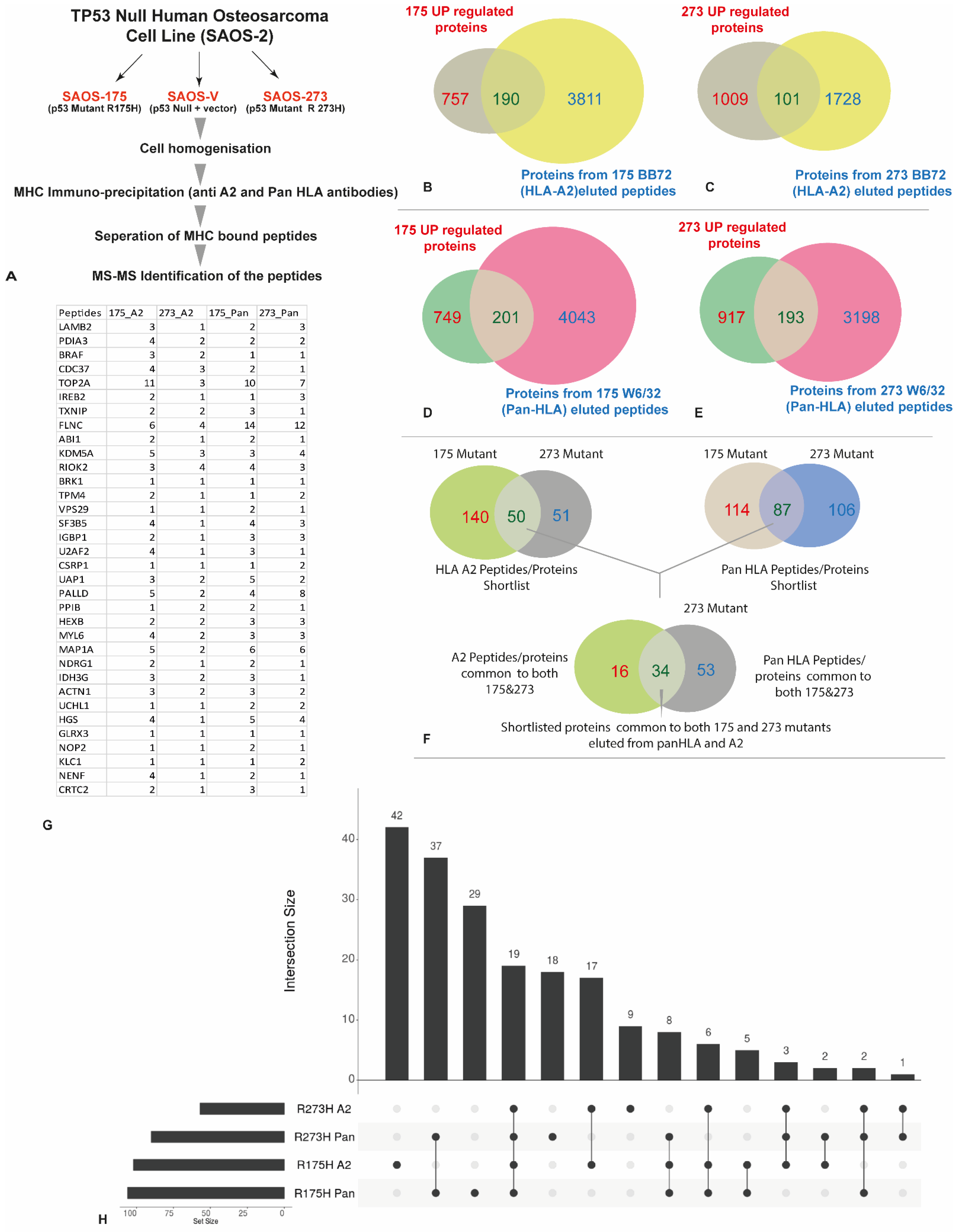
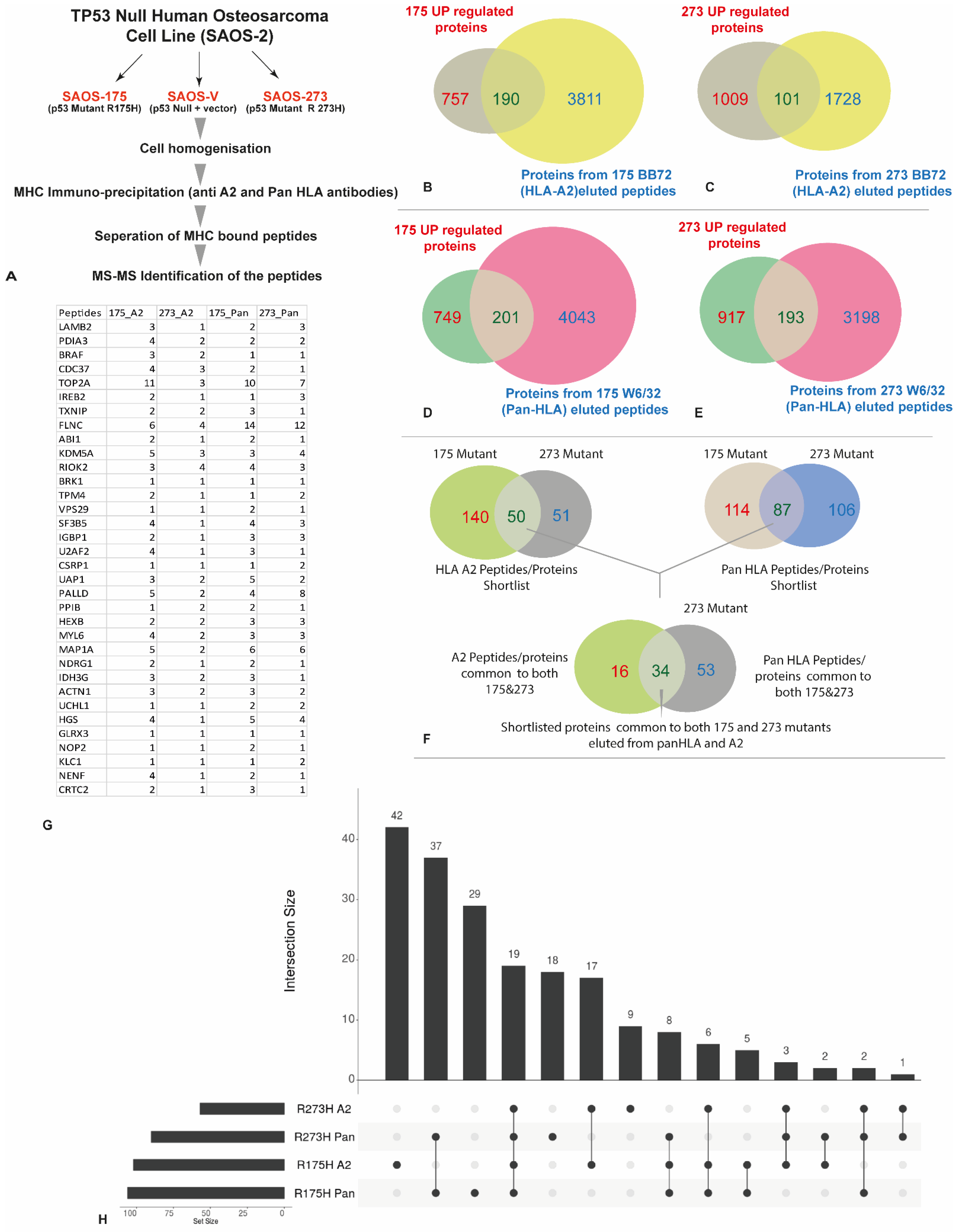
3.3. HLA Immunopeptidome Analysis Identified Unique and Shared Peptides between the SaOS-TP53 Mutants
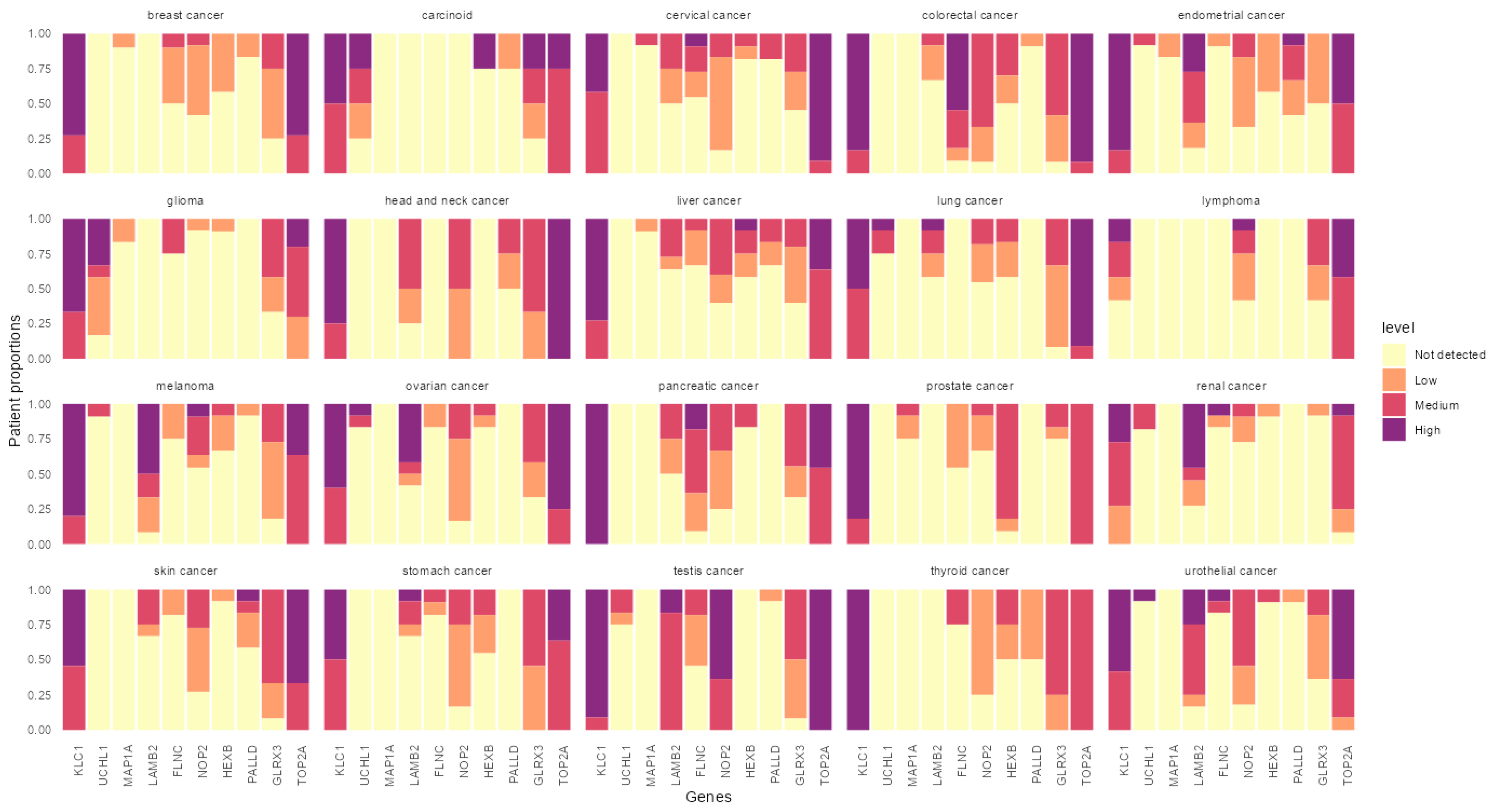
3.4. In Silico Analysis of the Identified Peptides Revealed Targetable Peptides in TP53 Mutants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pitolli, C.; Wang, Y.; Mancini, M.; Shi, Y.; Melino, G.; Amelio, I. Do Mutations Turn p53 into an Oncogene? Int. J. Mol. Sci. 2019, 20, 6241. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, J.; Xu, D.; Zhang, T.; Hu, W.; Feng, Z. Gain-of-function mutant p53 in cancer progression and therapy. J. Mol. Cell Biol. 2020, 12, 674–687. [Google Scholar] [CrossRef]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef]
- Cordani, M.; Pacchiana, R.; Butera, G.; D’Orazi, G.; Scarpa, A.; Donadelli, M. Mutant p53 proteins alter cancer cell secretome and tumour microenvironment: Involvement in cancer invasion and metastasis. Cancer Lett. 2016, 376, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Caswell, P.T.; Doyle, B.; Iwanicki, M.P.; Tan, E.H.; Karim, S.; Lukashchuk, N.; Gillespie, D.A.; Ludwig, R.L.; Gosselin, P.; et al. Mutant p53 drives invasion by promoting integrin recycling. Cell 2009, 139, 1327–1341. [Google Scholar] [CrossRef]
- Redman-Rivera, L.N.; Shaver, T.M.; Jin, H.; Marshall, C.B.; Schafer, J.M.; Sheng, Q.; Hongo, R.A.; Beckermann, K.E.; Wheeler, F.C.; Lehmann, B.D.; et al. Acquisition of aneuploidy drives mutant p53-associated gain-of-function phenotypes. Nat. Commun. 2021, 12, 5184. [Google Scholar] [CrossRef] [PubMed]
- Mello, S.S.; Attardi, L.D. Not all p53 gain-of-function mutants are created equal. Cell Death Differ. 2013, 20, 855–857. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.S.; Tiong, K.H.; Choo, H.L.; Chung, F.F.; Hii, L.W.; Tan, S.H.; Yap, I.K.; Pani, S.; Khor, N.T.; Wong, S.F.; et al. Mutant p53-R273H mediates cancer cell survival and anoikis resistance through AKT-dependent suppression of BCL2-modifying factor (BMF). Cell Death Dis. 2015, 6, e1826. [Google Scholar] [CrossRef]
- Green, D.R.; Kroemer, G. Cytoplasmic functions of the tumour suppressor p53. Nature 2009, 458, 1127–1130. [Google Scholar] [CrossRef]
- Di Minin, G.; Bellazzo, A.; Dal Ferro, M.; Chiaruttini, G.; Nuzzo, S.; Bicciato, S.; Piazza, S.; Rami, D.; Bulla, R.; Sommaggio, R.; et al. Mutant p53 reprograms TNF signaling in cancer cells through interaction with the tumor suppressor DAB2IP. Mol. Cell 2014, 56, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, R.A.; Chan, C.S. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2018, 25, 154–160. [Google Scholar] [CrossRef] [PubMed]
- DeLeo, A.B.; Appella, E. The p53 Saga: Early Steps in the Development of Tumor Immunotherapy. J. Immunol. 2020, 204, 2321–2328. [Google Scholar] [CrossRef] [PubMed]
- Rodan, S.B.; Imai, Y.; Thiede, M.A.; Wesolowski, G.; Thompson, D.; Bar-Shavit, Z.; Shull, S.; Mann, K.; Rodan, G.A. Characterization of a human osteosarcoma cell line (Saos-2) with osteoblastic properties. Cancer Res. 1987, 47, 4961–4966. [Google Scholar]
- McArdle, S.E.; Rees, R.C.; Mulcahy, K.A.; Saba, J.; McIntyre, C.A.; Murray, A.K. Induction of human cytotoxic T lymphocytes that preferentially recognise tumour cells bearing a conformational p53 mutant. Cancer Immunol. Immunother. 2000, 49, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Foulds, G.A.; Vadakekolathu, J.; Abdel-Fatah, T.M.A.; Nagarajan, D.; Reeder, S.; Johnson, C.; Hood, S.; Moseley, P.M.; Chan, S.Y.T.; Pockley, A.G.; et al. Immune-Phenotyping and Transcriptomic Profiling of Peripheral Blood Mononuclear Cells From Patients With Breast Cancer: Identification of a 3 Gene Signature Which Predicts Relapse of Triple Negative Breast Cancer. Front. Immunol. 2018, 9, 2028. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.; Ugun-Klusek, A.; Coveney, C.; Boocock, D.J. Multiomic analysis of stretched osteocytes reveals processes and signalling linked to bone regeneration and cancer. NPJ Regen. Med. 2021, 6, 32. [Google Scholar] [CrossRef]
- Demichev, V.; Messner, C.B.; Vernardis, S.I.; Lilley, K.S.; Ralser, M. DIA-NN: Neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 2020, 17, 41–44. [Google Scholar] [CrossRef]
- Yang, Y.; Cheng, J.; Wang, S.; Yang, H. StatsPro: Systematic integration and evaluation of statistical approaches for detecting differential expression in label-free quantitative proteomics. J. Proteom. 2022, 250, 104386. [Google Scholar] [CrossRef]
- Parham, P.; Brodsky, F.M. Partial purification and some properties of BB7.2. A cytotoxic monoclonal antibody with specificity for HLA-A2 and a variant of HLA-A28. Hum. Immunol. 1981, 3, 277–299. [Google Scholar] [CrossRef]
- Parham, P.; Barnstable, C.J.; Bodmer, W.F. Use of a monoclonal antibody (W6/32) in structural studies of HLA-A,B,C, antigens. J. Immunol. 1979, 123, 342–349. [Google Scholar] [PubMed]
- Purcell, A.W.; Ramarathinam, S.H.; Ternette, N. Mass spectrometry-based identification of MHC-bound peptides for immunopeptidomics. Nat. Protoc. 2019, 14, 1687–1707. [Google Scholar] [CrossRef] [PubMed]
- Pandey, K.; Ramarathinam, S.H.; Purcell, A.W. Isolation of HLA Bound Peptides by Immunoaffinity Capture and Identification by Mass Spectrometry. Curr. Protoc. 2021, 1, e92. [Google Scholar] [CrossRef]
- Escher, C.; Reiter, L.; MacLean, B.; Ossola, R.; Herzog, F.; Chilton, J.; MacCoss, M.J.; Rinner, O. Using iRT, a normalized retention time for more targeted measurement of peptides. Proteomics 2012, 12, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xin, L.; Shan, B.; Chen, W.; Xie, M.; Yuen, D.; Zhang, W.; Zhang, Z.; Lajoie, G.A.; Ma, B. PEAKS DB: De novo sequencing assisted database search for sensitive and accurate peptide identification. Mol. Cell Proteom. 2012, 11, M111.010587. [Google Scholar] [CrossRef] [PubMed]
- Colaert, N.; Helsens, K.; Martens, L.; Vandekerckhove, J.; Gevaert, K. Improved visualization of protein consensus sequences by iceLogo. Nat. Methods 2009, 6, 786–787. [Google Scholar] [CrossRef]
- Jurtz, V.; Paul, S.; Andreatta, M.; Marcatili, P.; Peters, B.; Nielsen, M. NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J. Immunol. 2017, 199, 3360–3368. [Google Scholar] [CrossRef]
- Deutsch, E.W.; Bandeira, N.; Sharma, V.; Perez-Riverol, Y.; Carver, J.J.; Kundu, D.J.; García-Seisdedos, D.; Jarnuczak, A.F.; Hewapathirana, S.; Pullman, B.S.; et al. The ProteomeXchange consortium in 2020: Enabling ‘big data’ approaches in proteomics. Nucleic Acids Res. 2020, 48, D1145–D1152. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef]
- Masuda, H.; Miller, C.; Koeffler, H.P.; Battifora, H.; Cline, M.J. Rearrangement of the p53 gene in human osteogenic sarcomas. Proc. Natl. Acad. Sci. USA 1987, 84, 7716–7719. [Google Scholar] [CrossRef]
- Brazda, V.; Fojta, M. The Rich World of p53 DNA Binding Targets: The Role of DNA Structure. Int. J. Mol. Sci. 2019, 20, 5605. [Google Scholar] [CrossRef] [PubMed]
- Brosh, R.; Rotter, V. When mutants gain new powers: News from the mutant p53 field. Nat. Rev. Cancer 2009, 9, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Synnott, N.C.; O’Grady, S.; Crown, J. Targeting p53 for the treatment of cancer. Semin. Cancer Biol. 2020, 79, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. p53 programmes plough on. Nat. Rev. Drug Discov. 2020, 19, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. The many faces of p53: Something for everyone. J. Mol. Cell Biol. 2019, 11, 524–530. [Google Scholar] [CrossRef]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef]
- Espadinha, M.; Barcherini, V.; Lopes, E.A.; Santos, M.M.M. An Update on MDMX and Dual MDM2/X Inhibitors. Curr. Top. Med. Chem. 2018, 18, 647–660. [Google Scholar] [CrossRef]
- Hsiue, E.H.; Wright, K.M.; Douglass, J.; Hwang, M.S.; Mog, B.J.; Pearlman, A.H.; Paul, S.; DiNapoli, S.R.; Konig, M.F.; Wang, Q.; et al. Targeting a neoantigen derived from a common TP53 mutation. Science 2021, 371, 1–31. [Google Scholar] [CrossRef]
- Vaughan, C.A.; Singh, S.; Windle, B.; Yeudall, W.A.; Frum, R.; Grossman, S.R.; Deb, S.P.; Deb, S. Gain-of-Function Activity of Mutant p53 in Lung Cancer through Up-Regulation of Receptor Protein Tyrosine Kinase Axl. Genes Cancer 2012, 3, 491–502. [Google Scholar] [CrossRef]
- Dong, P.; Karaayvaz, M.; Jia, N.; Kaneuchi, M.; Hamada, J.; Watari, H.; Sudo, S.; Ju, J.; Sakuragi, N. Mutant p53 gain-of-function induces epithelial-mesenchymal transition through modulation of the miR-130b-ZEB1 axis. Oncogene 2013, 32, 3286–3295. [Google Scholar] [CrossRef]
- Putzer, B.M.; Bramson, J.L.; Addison, C.L.; Hitt, M.; Siegel, P.M.; Muller, W.J.; Graham, F.L. Combination therapy with interleukin-2 and wild-type p53 expressed by adenoviral vectors potentiates tumor regression in a murine model of breast cancer. Hum. Gene Ther. 1998, 9, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Aerts, J.L.; Vandenplas, H.; Wang, J.A.; Gorbenko, O.; Chen, J.P.; Giron, P.; Heirman, C.; Goyvaerts, C.; Zacksenhaus, E.; et al. Phosphorylated STAT5 regulates p53 expression via BRCA1/BARD1-NPM1 and MDM2. Cell Death Dis. 2016, 7, e2560. [Google Scholar] [CrossRef] [PubMed]
- Rath, O.; Kozielski, F. Kinesins and cancer. Nat. Rev. Cancer 2012, 12, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.N.; Zheng, L.L.; Wang, D.; Liang, X.X.; Gao, F.; Zhou, X.L. Recent advances in microtubule-stabilizing agents. Eur. J. Med. Chem. 2018, 143, 806–828. [Google Scholar] [CrossRef]
- Togashi, Y.; Soda, M.; Sakata, S.; Sugawara, E.; Hatano, S.; Asaka, R.; Nakajima, T.; Mano, H.; Takeuchi, K. KLC1-ALK: A novel fusion in lung cancer identified using a formalin-fixed paraffin-embedded tissue only. PLoS ONE 2012, 7, e31323. [Google Scholar] [CrossRef]
- Elliott, J.; Bai, Z.; Hsieh, S.C.; Kelly, S.E.; Chen, L.; Skidmore, B.; Yousef, S.; Zheng, C.; Stewart, D.J.; Wells, G.A. ALK inhibitors for non-small cell lung cancer: A systematic review and network meta-analysis. PLoS ONE 2020, 15, e0229179. [Google Scholar] [CrossRef]
- Trahair, T.; Gifford, A.J.; Fordham, A.; Mayoh, C.; Fadia, M.; Lukeis, R.; Wood, A.C.; Valvi, S.; Walker, R.D.; Blackburn, J.; et al. Crizotinib and Surgery for Long-Term Disease Control in Children and Adolescents With ALK-Positive Inflammatory Myofibroblastic Tumors. JCO Precis. Oncol. 2019, 363, 1727–1733. [Google Scholar] [CrossRef]
- Fukano, R.; Mori, T.; Sekimizu, M.; Choi, I.; Kada, A.; Saito, A.M.; Asada, R.; Takeuchi, K.; Terauchi, T.; Tateishi, U.; et al. Alectinib for relapsed or refractory anaplastic lymphoma kinase-positive anaplastic large cell lymphoma: An open-label phase II trial. Cancer Sci. 2020, 111, 4540–4547. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
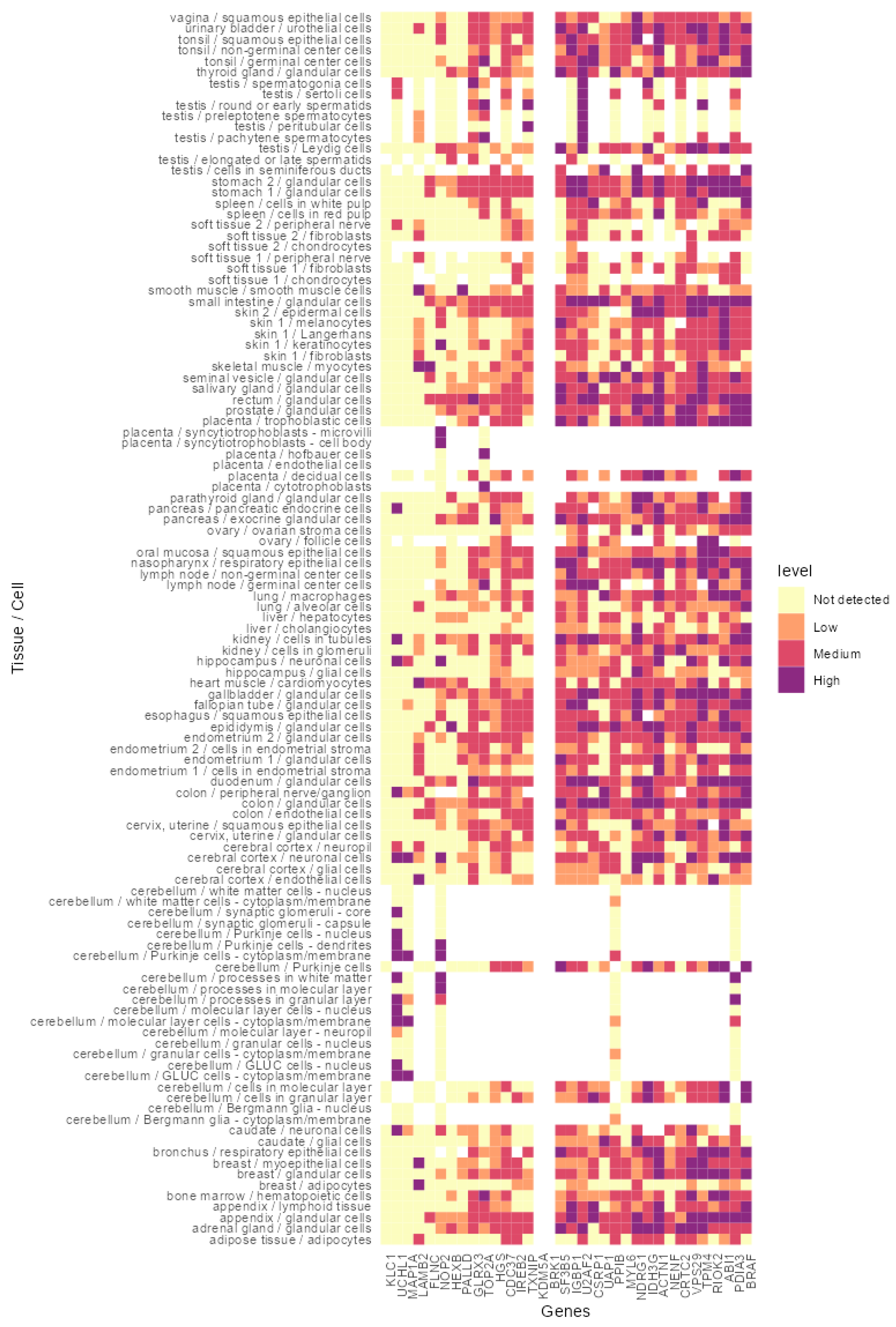{kind=link}
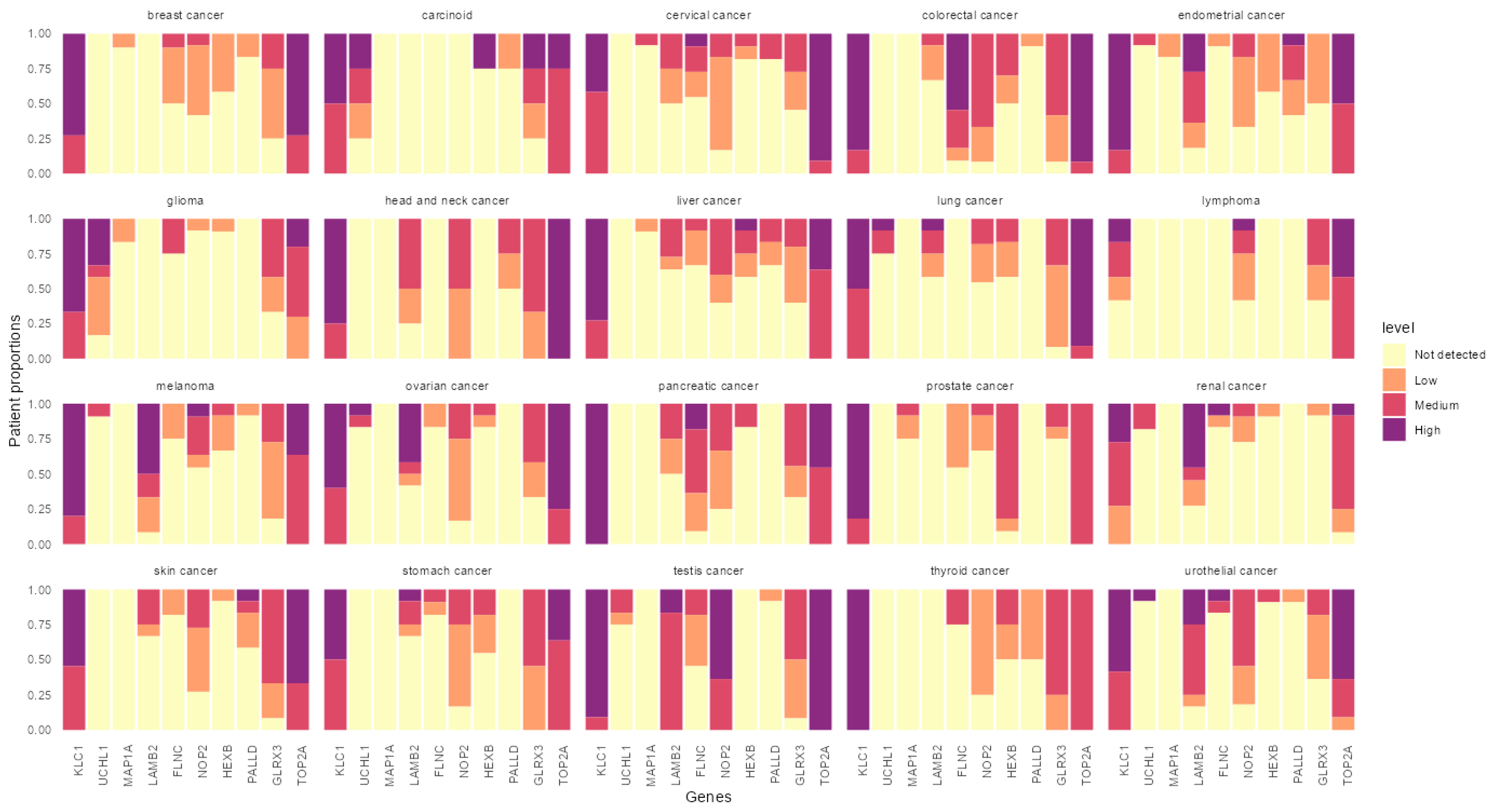{kind=link}
| Condition | HLA-A2 (BB7.2) Peptides | Pan HLA (W632) Peptides | Total |
|---|---|---|---|
| No vector control | 2688 | 8982 | 11,670 |
| R175H | 9127 | 9814 | 18,941 |
| R273H | 2970 | 7145 | 10,115 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vadakekolathu, J.; Boocock, D.J.; Pandey, K.; Guinn, B.-a.; Legrand, A.; Miles, A.K.; Coveney, C.; Ayala, R.; Purcell, A.W.; McArdle, S.E. Multi-Omic Analysis of Two Common P53 Mutations: Proteins Regulated by Mutated P53 as Potential Targets for Immunotherapy. Cancers 2022, 14, 3975. https://doi.org/10.3390/cancers14163975
Vadakekolathu J, Boocock DJ, Pandey K, Guinn B-a, Legrand A, Miles AK, Coveney C, Ayala R, Purcell AW, McArdle SE. Multi-Omic Analysis of Two Common P53 Mutations: Proteins Regulated by Mutated P53 as Potential Targets for Immunotherapy. Cancers. 2022; 14(16):3975. https://doi.org/10.3390/cancers14163975
Chicago/Turabian StyleVadakekolathu, Jayakumar, David J. Boocock, Kirti Pandey, Barbara-ann Guinn, Antoine Legrand, Amanda K. Miles, Clare Coveney, Rochelle Ayala, Anthony W. Purcell, and Stephanie E. McArdle. 2022. "Multi-Omic Analysis of Two Common P53 Mutations: Proteins Regulated by Mutated P53 as Potential Targets for Immunotherapy" Cancers 14, no. 16: 3975. https://doi.org/10.3390/cancers14163975
APA StyleVadakekolathu, J., Boocock, D. J., Pandey, K., Guinn, B.-a., Legrand, A., Miles, A. K., Coveney, C., Ayala, R., Purcell, A. W., & McArdle, S. E. (2022). Multi-Omic Analysis of Two Common P53 Mutations: Proteins Regulated by Mutated P53 as Potential Targets for Immunotherapy. Cancers, 14(16), 3975. https://doi.org/10.3390/cancers14163975