
The Immunomodulatory Role of Hypoxic Tumor-Derived Extracellular Vesicles


Abstract
:Simple Summary
Abstract

1. Introduction
2. Neutrophils
3. Macrophages

{kind=link}
{kind=link}
{kind=link}
| Factor | Effect on Macrophages | Proven to Be EV-Mediated? 1 |
|---|---|---|
| TGF-β1 TGF-β2 TGF-β3 [42] | Induces anti-inflammatory M2 phenotype with expression of TAM-associated genes. | Yes |
| CAIX [49,51] | Involved in extracellular acidification which, in turn, causes a metabolic switch in macrophages, inducing the M2 phenotype | No |
| MIF [42] | Induces anti-inflammatory M2 phenotype with expression of TAM-associated genes. | Yes |
| FTH/FTL [42] | Induces anti-inflammatory M2 phenotype with expression of TAM-associated genes. | Yes |
| MIR-1246 [44,63,64,65] | Induces anti-inflammatory M2 phenotype via NF-κB inhibition. Limits differentiation of monocytes into macrophages via reduced caveolin-1 expression. Increases macrophage-mediated angiogenesis and metastasis formation. | Yes |
| MIR-103a [45] | Induces anti-inflammatory M2 phenotype via reduced PTEN expression. | Yes |
| MIR-301a-3P [46] | Induces anti-inflammatory M2 phenotype via reduced PTEN expression. | Yes |
| MIR-21 [52,53] | Induces anti-inflammatory M2 phenotype via reduced PTEN expression. | No |
| MIR-23 [54] | Induces anti-inflammatory M2 phenotype via reduced PTEN expression. | No |
| MIR-494 [55] | Induces anti-inflammatory M2 phenotype via reduced PTEN expression. | No |
| MIR-135 [56] | Reduces production of pro-inflammatory factors TNF-α and ROS. | No |
| MIR-92a [57] | Enhances production of tumor-supportive IL-6. | No |
| MIR-127 [58,59,60] | Induces anti-inflammatory M2 phenotype via reduced CD64 and Traf1 expression. Induces pro-inflammatory M1 phenotype via reduced BCL-6 expression. | No |
| MIR-210 [62] | Induces necroptosis via reduced DECR1 expression. | No |
| PKM2 [67,68] | Induces anti-inflammatory M2 phenotype via STAT3 phosphorylation. | No |
4. Myeloid-Derived Suppressor Cells
| Factor | Effect on MDSCs | Proven to Be EV-Mediated? 1 |
|---|---|---|
| MIR-10a [73] | Potentiates MDSC function via reduced Rora expression. | Yes |
| MIR-21 [73] | Potentiates MDSC function via reduced Pten expression. | Yes |
| MIR-29a [74] | Increases MDSC proliferation via reduced Hbp1 expression. | Yes |
| MIR-92a [74] | Increases the production of immunosuppressive factors by MDSCs via reduced Prkar1α expression. | Yes |
| MIR-210 [75] | Enhances the immunosuppressive effects of MDSCs via increased ARG activity and NO production. | No |
| MIR-494 [76] | Stimulates MDSCs’ immunosuppressive effects via targeting of PTEN. | No |
| CCL2 [79] | Stimulates immunosuppressive effects of MDSCs. | No |
| IGFBP-3 [80] | Induces a more efficient CD38high MDSC population. | No |
5. Dendritic Cells
6. NK Cells
7. T Cells
7.1. Hypoxic EVs May Limit Differentiation and Regulate Polarization toward Tumor-Supportive Subtypes via Transfer of Factors with Known Immunomodulatory Roles
7.2. Hypoxic EVs May Decrease the Abundance of T Cells (Infiltration and Proliferation) in the Tumor and Limit Antitumor Immunity via Transfer of Factors with Known Immunomodulatory Roles
| Factor | Effect on T Cells | Proven to Be EV-Mediated? 1 |
|---|---|---|
| MIR-23 [117,118,119] | Suppresses T-cell-mediated cytotoxicity by reducing the expression of BLIMP-1. | No |
| MIR-24-3p [103] | Reduces proliferation of CD4+ and CD8+ T cells. Inhibits differentiation towards Th1 and Th17 subtypes. Increases the FOXP3+ Treg cell population. | Yes |
| MIR-125 [104,105,106] | Maintains a naïve T-cell state by decreasing the expression of Ifn-γ, TNF-α, IL-2Rβ, IL-10Rα, BLIMP-1, Stat3, and Il-13. Stabilizes Treg lineage commitment via suppression of STAT3, Il-13, and Ifn-γ expression. | No |
| MIR-210 [110] | Suppresses Th17 differentiation and reduces inflammation via reduced HIF-1α expression. | No |
| Let-7a [128,129] | Reduces T-cell proliferation and infiltration. Reduces IFN-γ secretion via reduced STAT3 expression. | No |
| ADAMTS1 [133,134] | Negatively influences the infiltration of cytotoxic lymphocytes and the expression of antitumor immune gene profiles. | No |
| CAIX [120,121,122] | Induces lymphocyte anergy via extracellular acidification. Hinders T-cell differentiation via extracellular acidification. | No |
| MMP9 [127] | Prevents T-cell proliferation via shedding of IL-2 receptor-α. | No |
| IGFBP-3 [130] | Suppresses immune infiltration into the tumor. | No |
| TSP-1 [131] | Reduces the infiltration of CD8+ lymphocytes. Decreases inflammatory IFN-γ signaling via activation of TGF-β. Induces the differentiation of Treg cells. | No |
| PKM2 [111] | Promotes Th17 differentiation through STAT3 activation. | No |
8. NKT Cells
9. B Cells
10. Immune Stimulation by Hypoxia-Upregulated Factors in EVs
11. Importance of EV Isolation Methodology and Experimental Setup
12. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- WHO. Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 14 July 2022).
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Pang, Y.; Moses, H.L. TGF-beta and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Shields, J.D.; Kourtis, I.C.; Tomei, A.A.; Roberts, J.M.; Swartz, M.A. Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science 2010, 328, 749–752. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Ding, S. The Crosstalk Between Tumor-Associated Macrophages (TAMs) and Tumor Cells and the Corresponding Targeted Therapy. Front. Oncol. 2020, 10, 590941. [Google Scholar] [CrossRef]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef]
- Brown, J.M.; Wilson, W.R. Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer 2004, 4, 437–447. [Google Scholar] [CrossRef]
- Rouschop, K.M.; van den Beucken, T.; Dubois, L.; Niessen, H.; Bussink, J.; Savelkouls, K.; Keulers, T.; Mujcic, H.; Landuyt, W.; Voncken, J.W.; et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Investig. 2010, 120, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Barsoum, I.B.; Koti, M.; Siemens, D.R.; Graham, C.H. Mechanisms of hypoxia-mediated immune escape in cancer. Cancer Res. 2014, 74, 7185–7190. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef]
- Yanez-Mo, M.; Siljander, P.R.; Andreu, Z.; Zavec, A.B.; Borras, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, S.H.; Andrews, A.M.; Paul, D.; Pachter, J.S. Extracellular vesicles: Mediators and biomarkers of pathology along CNS barriers. Fluids Barriers CNS 2018, 15, 19. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.W.; Sceneay, J.; Lima, L.G.; Wong, C.S.; Becker, M.; Krumeich, S.; Lobb, R.J.; Castillo, V.; Wong, K.N.; Ellis, S.; et al. The Biodistribution and Immune Suppressive Effects of Breast Cancer-Derived Exosomes. Cancer Res. 2016, 76, 6816–6827. [Google Scholar] [CrossRef] [PubMed]
- Vanherle, S.; Haidar, M.; Irobi, J.; Jfj, B.; Jja, H. Extracellular vesicle-associated lipids in central nervous system disorders. Adv. Drug Deliv. Rev. 2020. [Google Scholar] [CrossRef]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Meldolesi, J. Exosomes and ectosomes in intercellular communication. Curr. Biol. 2018, 28, R435–R444. [Google Scholar] [CrossRef]
- McKelvey, K.J.; Powell, K.L.; Ashton, A.W.; Morris, J.M.; McCracken, S.A. Exosomes: Mechanisms of uptake. J. Circ. Biomark. 2015, 4, 7. [Google Scholar] [CrossRef]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Zonneveld, M.I.; Keulers, T.G.H.; Rouschop, K.M.A. Extracellular Vesicles as Transmitters of Hypoxia Tolerance in Solid Cancers. Cancers 2019, 11, 154. [Google Scholar] [CrossRef]
- Keulers, T.G.; Libregts, S.F.; Beaumont, J.E.J.; Savelkouls, K.G.; Bussink, J.; Duimel, H.; Dubois, L.; Zonneveld, M.I.; Lopez-Iglesias, C.; Bezstarosti, K.; et al. Secretion of pro-angiogenic extracellular vesicles during hypoxia is dependent on the autophagy-related protein GABARAPL1. J. Extracell. Vesicles 2021, 10, e12166. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Gilkes, D.M.; Takano, N.; Xiang, L.; Luo, W.; Bishop, C.J.; Chaturvedi, P.; Green, J.J.; Semenza, G.L. Hypoxia-inducible factors and RAB22A mediate formation of microvesicles that stimulate breast cancer invasion and metastasis. Proc. Natl. Acad. Sci. USA 2014, 111, E3234–E3242. [Google Scholar] [CrossRef] [PubMed]
- Dorayappan, K.D.P.; Wanner, R.; Wallbillich, J.J.; Saini, U.; Zingarelli, R.; Suarez, A.A.; Cohn, D.E.; Selvendiran, K. Hypoxia-induced exosomes contribute to a more aggressive and chemoresistant ovarian cancer phenotype: A novel mechanism linking STAT3/Rab proteins. Oncogene 2018, 37, 3806–3821. [Google Scholar] [CrossRef]
- Shaul, M.E.; Fridlender, Z.G. Tumour-associated neutrophils in patients with cancer. Nat. Rev. Clin. Oncol. 2019, 16, 601–620. [Google Scholar] [CrossRef]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Kucharzewska, P.; Christianson, H.C.; Welch, J.E.; Svensson, K.J.; Fredlund, E.; Ringnér, M.; Mörgelin, M.; Bourseau-Guilmain, E.; Bengzon, J.; Belting, M. Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc. Natl. Acad. Sci. USA 2013, 110, 7312–7317. [Google Scholar] [CrossRef]
- David, J.M.; Dominguez, C.; Hamilton, D.H.; Palena, C. The IL-8/IL-8R axis: A double agent in tumor immune resistance. Vaccines 2016, 4, 22. [Google Scholar] [CrossRef]
- Baggiolini, M.; Walz, A.; Kunkel, S. Neutrophil-activating peptide-1/interleukin 8, a novel cytokine that activates neutrophils. J. Clin. Investig. 1989, 84, 1045–1049. [Google Scholar] [CrossRef]
- Murata, K.; Yoshitomi, H.; Furu, M.; Ishikawa, M.; Shibuya, H.; Ito, H.; Matsuda, S. MicroRNA-451 down-regulates neutrophil chemotaxis via p38 MAPK. Arthritis Rheumatol. 2014, 66, 549–559. [Google Scholar] [CrossRef]
- Davies, L.C.; Taylor, P.R. Tissue-resident macrophages: Then and now. Immunology 2015, 144, 541–548. [Google Scholar] [CrossRef]
- Perdiguero, E.G.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; De Bruijn, M.F.; Geissmann, F. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Wang, Z.; Fu, L.; Xu, T. Macrophage Polarization in the Development and Progression of Ovarian Cancers: An Overview. Front. Oncol. 2019, 9, 421. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef]
- Galvan-Pena, S.; O’Neill, L.A. Metabolic reprograming in macrophage polarization. Front. Immunol. 2014, 5, 420. [Google Scholar] [CrossRef] [PubMed]
- Viola, A.; Munari, F.; Sanchez-Rodriguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-associated macrophages in tumor immunity. Front. Immunol. 2020, 11, 3151. [Google Scholar] [CrossRef]
- Torroella-Kouri, M.; Silvera, R.; Rodriguez, D.; Caso, R.; Shatry, A.; Opiela, S.; Ilkovitch, D.; Schwendener, R.A.; Iragavarapu-Charyulu, V.; Cardentey, Y. Identification of a subpopulation of macrophages in mammary tumor–bearing mice that are neither M1 nor M2 and are less differentiated. Cancer Res. 2009, 69, 4800–4809. [Google Scholar] [CrossRef]
- Specht, H.; Emmott, E.; Petelski, A.A.; Huffman, R.G.; Perlman, D.H.; Serra, M.; Kharchenko, P.; Koller, A.; Slavov, N. Single-cell proteomic and transcriptomic analysis of macrophage heterogeneity using SCoPE2. Genome Biol. 2021, 22, 1–27. [Google Scholar] [CrossRef]
- Ham, S.; Lima, L.G.; Chai, E.P.Z.; Muller, A.; Lobb, R.J.; Krumeich, S.; Wen, S.W.; Wiegmans, A.P.; Moller, A. Breast Cancer-Derived Exosomes Alter Macrophage Polarization via gp130/STAT3 Signaling. Front. Immunol. 2018, 9, 871. [Google Scholar] [CrossRef]
- Popena, I.; Abols, A.; Saulite, L.; Pleiko, K.; Zandberga, E.; Jekabsons, K.; Endzelins, E.; Llorente, A.; Line, A.; Riekstina, U. Effect of colorectal cancer-derived extracellular vesicles on the immunophenotype and cytokine secretion profile of monocytes and macrophages. Cell Commun. Signal. 2018, 16, 17. [Google Scholar] [CrossRef]
- Park, J.E.; Dutta, B.; Tse, S.W.; Gupta, N.; Tan, C.F.; Low, J.K.; Yeoh, K.W.; Kon, O.L.; Tam, J.P.; Sze, S.K. Hypoxia-induced tumor exosomes promote M2-like macrophage polarization of infiltrating myeloid cells and microRNA-mediated metabolic shift. Oncogene 2019, 38, 5158–5173. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; He, Y.; Peng, F.; Yang, J.; Yuan, C. Endometrial Cancer Cells Promote M2-Like Macrophage Polarization by Delivering Exosomal miRNA-21 under Hypoxia Condition. J. Immunol. Res. 2020, 2020, 9731049. [Google Scholar] [CrossRef]
- Qian, M.; Wang, S.; Guo, X.; Wang, J.; Zhang, Z.; Qiu, W.; Gao, X.; Chen, Z.; Xu, J.; Zhao, R.; et al. Hypoxic glioma-derived exosomes deliver microRNA-1246 to induce M2 macrophage polarization by targeting TERF2IP via the STAT3 and NF-kappaB pathways. Oncogene 2020, 39, 428–442. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.L.; Hung, J.Y.; Chang, W.A.; Jian, S.F.; Lin, Y.S.; Pan, Y.C.; Wu, C.Y.; Kuo, P.L. Hypoxic Lung-Cancer-Derived Extracellular Vesicle MicroRNA-103a Increases the Oncogenic Effects of Macrophages by Targeting PTEN. Mol. Ther. 2018, 26, 568–581. [Google Scholar] [CrossRef]
- Wang, X.; Luo, G.; Zhang, K.; Cao, J.; Huang, C.; Jiang, T.; Liu, B.; Su, L.; Qiu, Z. Hypoxic Tumor-Derived Exosomal miR-301a Mediates M2 Macrophage Polarization via PTEN/PI3Kgamma to Promote Pancreatic Cancer Metastasis. Cancer Res. 2018, 78, 4586–4598. [Google Scholar] [CrossRef]
- Chen, X.; Ying, X.; Wang, X.; Wu, X.; Zhu, Q.; Wang, X. Exosomes derived from hypoxic epithelial ovarian cancer deliver microRNA-940 to induce macrophage M2 polarization. Oncol. Rep. 2017, 38, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhou, J.; Li, X.; Wang, X.; Lin, Y.; Wang, X. Exosomes derived from hypoxic epithelial ovarian cancer cells deliver microRNAs to macrophages and elicit a tumor-promoted phenotype. Cancer Lett. 2018, 435, 80–91. [Google Scholar] [CrossRef]
- Lee, S.H.; McIntyre, D.; Honess, D.; Hulikova, A.; Pacheco-Torres, J.; Cerdan, S.; Swietach, P.; Harris, A.L.; Griffiths, J.R. Carbonic anhydrase IX is a pH-stat that sets an acidic tumour extracellular pH in vivo. Br. J. Cancer 2018, 119, 622–630. [Google Scholar] [CrossRef]
- Erra Diaz, F.; Dantas, E.; Geffner, J. Unravelling the Interplay between Extracellular Acidosis and Immune Cells. Mediat. Inflamm. 2018, 2018, 1218297. [Google Scholar] [CrossRef]
- Jiang, W.; Le, J.; Wang, P.Y.; Cheng, X.; Smelkinson, M.; Dong, W.; Yang, C.; Chu, Y.; Hwang, P.M.; Munford, R.S.; et al. Extracellular Acidity Reprograms Macrophage Metabolism and Innate Responsiveness. J. Immunol. 2021, 206, 3021–3031. [Google Scholar] [CrossRef]
- Das, A.; Ganesh, K.; Khanna, S.; Sen, C.K.; Roy, S. Engulfment of apoptotic cells by macrophages: A role of microRNA-21 in the resolution of wound inflammation. J. Immunol. 2014, 192, 1120–1129. [Google Scholar] [CrossRef] [PubMed]
- Sheedy, F.J.; Palsson-McDermott, E.; Hennessy, E.J.; Martin, C.; O’Leary, J.J.; Ruan, Q.; Johnson, D.S.; Chen, Y.; O’Neill, L.A. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat. Immunol. 2010, 11, 141–147. [Google Scholar] [CrossRef]
- Liu, J.; Fan, L.; Yu, H.; Zhang, J.; He, Y.; Feng, D.; Wang, F.; Li, X.; Liu, Q.; Li, Y.; et al. Endoplasmic Reticulum Stress Causes Liver Cancer Cells to Release Exosomal miR-23a-3p and Up-regulate Programmed Death Ligand 1 Expression in Macrophages. Hepatology 2019, 70, 241–258. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; He, K.; Zhou, W.; Cao, J.; Jin, Z. miR4943p regulates lipopolysaccharideinduced inflammatory responses in RAW264.7 cells by targeting PTEN. Mol. Med. Rep. 2019, 19, 4288–4296. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Fan, J.B.; Gao, Y.; Zhang, M.; Zhang, L.; Yang, N.; Zhao, X. miR-135b-5p inhibits LPS-induced TNFalpha production via silencing AMPK phosphatase Ppm1e. Oncotarget 2016, 7, 77978–77986. [Google Scholar] [CrossRef]
- Casadei, L.; Calore, F.; Creighton, C.J.; Guescini, M.; Batte, K.; Iwenofu, O.H.; Zewdu, A.; Braggio, D.A.; Bill, K.L.; Fadda, P.; et al. Exosome-Derived miR-25-3p and miR-92a-3p Stimulate Liposarcoma Progression. Cancer Res. 2017, 77, 3846–3856. [Google Scholar] [CrossRef]
- Xie, T.; Liang, J.; Liu, N.; Wang, Q.; Li, Y.; Noble, P.W.; Jiang, D. MicroRNA-127 inhibits lung inflammation by targeting IgG Fcgamma receptor I. J. Immunol. 2012, 188, 2437–2444. [Google Scholar] [CrossRef]
- Chen, C.; Lin, S.; Zhou, L.; Wang, J.; Chen, J.; Yu, R.; Luo, H.; Lu, J.; Xue, Z.; Chen, M. MicroRNA-127-5p attenuates severe pneumonia via tumor necrosis factor receptor-associated factor 1. Exp. Med. 2020, 20, 2856–2862. [Google Scholar] [CrossRef]
- Ying, H.; Kang, Y.; Zhang, H.; Zhao, D.; Xia, J.; Lu, Z.; Wang, H.; Xu, F.; Shi, L. MiR-127 modulates macrophage polarization and promotes lung inflammation and injury by activating the JNK pathway. J. Immunol. 2015, 194, 1239–1251. [Google Scholar] [CrossRef]
- Tadokoro, H.; Umezu, T.; Ohyashiki, K.; Hirano, T.; Ohyashiki, J.H. Exosomes derived from hypoxic leukemia cells enhance tube formation in endothelial cells. J. Biol. Chem. 2013, 288, 34343–34351. [Google Scholar] [CrossRef]
- Karshovska, E.; Wei, Y.; Subramanian, P.; Mohibullah, R.; Geissler, C.; Baatsch, I.; Popal, A.; Corbalan Campos, J.; Exner, N.; Schober, A. HIF-1alpha (Hypoxia-Inducible Factor-1alpha) Promotes Macrophage Necroptosis by Regulating miR-210 and miR-383. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Kanlikilicer, P.; Bayraktar, R.; Denizli, M.; Rashed, M.H.; Ivan, C.; Aslan, B.; Mitra, R.; Karagoz, K.; Bayraktar, E.; Zhang, X.; et al. Exosomal miRNA confers chemo resistance via targeting Cav1/p-gp/M2-type macrophage axis in ovarian cancer. EBioMedicine 2018, 38, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Moore, X.L.; Lee, M.K.; Fernandez-Rojo, M.A.; Parat, M.O.; Parton, R.G.; Meikle, P.J.; Sviridov, D.; Chin-Dusting, J.P. Caveolin-1 plays a critical role in the differentiation of monocytes into macrophages. Arterioscler. Thromb. Vasc. Biol. 2012, 32, e117–e125. [Google Scholar] [CrossRef] [PubMed]
- Celus, W.; Di Conza, G.; Oliveira, A.I.; Ehling, M.; Costa, B.M.; Wenes, M.; Mazzone, M. Loss of Caveolin-1 in Metastasis-Associated Macrophages Drives Lung Metastatic Growth through Increased Angiogenesis. Cell Rep. 2017, 21, 2842–2854. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Deng, X.; Liu, Y.; Liu, Y.; Sun, L.; Chen, F. PKM2, function and expression and regulation. Cell Biosci. 2019, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.-P.; Luo, L.-J.; Chen, H.-Z.; Chen, Q.-T.; Bian, X.-L.; Wu, S.-F.; Zhou, J.-X.; Zhao, W.-X.; Liu, J.-M.; Wang, X.-M. Ectosomal PKM2 promotes HCC by inducing macrophage differentiation and remodeling the tumor microenvironment. Mol. Cell 2020, 78, 1192–1206.e1110. [Google Scholar] [CrossRef]
- Palsson-McDermott, E.M.; Curtis, A.M.; Goel, G.; Lauterbach, M.A.; Sheedy, F.J.; Gleeson, L.E.; van den Bosch, M.W.; Quinn, S.R.; Domingo-Fernandez, R.; Johnston, D.G. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 2015, 21, 65–80. [Google Scholar] [CrossRef]
- Tesi, R. MDSC; the most important cell you have never heard of. Trends Pharmacol. Sci. 2019, 40, 4–7. [Google Scholar]
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef]
- Tcyganov, E.; Mastio, J.; Chen, E.; Gabrilovich, D.I. Plasticity of myeloid-derived suppressor cells in cancer. Curr. Opin. Immunol. 2018, 51, 76–82. [Google Scholar] [CrossRef]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Qiu, W.; Liu, Q.; Qian, M.; Wang, S.; Zhang, Z.; Gao, X.; Chen, Z.; Xue, H.; Li, G. Immunosuppressive effects of hypoxia-induced glioma exosomes through myeloid-derived suppressor cells via the miR-10a/Rora and miR-21/Pten Pathways. Oncogene 2018, 37, 4239–4259. [Google Scholar] [CrossRef]
- Guo, X.; Qiu, W.; Wang, J.; Liu, Q.; Qian, M.; Wang, S.; Zhang, Z.; Gao, X.; Chen, Z.; Guo, Q. Glioma exosomes mediate the expansion and function of myeloid-derived suppressor cells through microRNA-29a/Hbp1 and microRNA-92a/Prkar1a pathways. Int. J. Cancer 2019, 144, 3111–3126. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Janji, B.; Hu, S.; Wu, J.C.; Martelli, F.; Bronte, V.; Chouaib, S. Tumor-Promoting Effects of Myeloid-Derived Suppressor Cells Are Potentiated by Hypoxia-Induced Expression of miR-210. Cancer Res. 2015, 75, 3771–3787. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lai, L.; Chen, Q.; Song, Y.; Xu, S.; Ma, F.; Wang, X.; Wang, J.; Yu, H.; Cao, X.; et al. MicroRNA-494 is required for the accumulation and functions of tumor-expanded myeloid-derived suppressor cells via targeting of PTEN. J. Immunol. 2012, 188, 5500–5510. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, L.; Fan, J.; Ye, C.; Dominguez, D.; Zhang, Y.; Curiel, T.J.; Fang, D.; Kuzel, T.M.; Zhang, B. Host miR155 promotes tumor growth through a myeloid-derived suppressor cell-dependent mechanism. Cancer Res. 2015, 75, 519–531. [Google Scholar] [CrossRef]
- Chun, E.; Lavoie, S.; Michaud, M.; Gallini, C.A.; Kim, J.; Soucy, G.; Odze, R.; Glickman, J.N.; Garrett, W.S. CCL2 promotes colorectal carcinogenesis by enhancing polymorphonuclear myeloid-derived suppressor cell population and function. Cell Rep. 2015, 12, 244–257. [Google Scholar] [CrossRef]
- Zhang, Y.; Qu, D.; Sun, J.; Zhao, L.; Wang, Q.; Shao, Q.; Kong, B.; Qu, X. Human trophoblast cells induced MDSCs from peripheral blood CD14+ myelomonocytic cells via elevated levels of CCL2. Cell. Mol. Immunol. 2016, 13, 615–627. [Google Scholar] [CrossRef]
- Karakasheva, T.A.; Waldron, T.J.; Eruslanov, E.; Kim, S.-B.; Lee, J.-S.; O’Brien, S.; Hicks, P.D.; Basu, D.; Singhal, S.; Malavasi, F. CD38-expressing myeloid-derived suppressor cells promote tumor growth in a murine model of esophageal cancer. Cancer Res. 2015, 75, 4074–4085. [Google Scholar] [CrossRef]
- Merad, M.; Sathe, P.; Helft, J.; Miller, J.; Mortha, A. The dendritic cell lineage: Ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu. Rev. Immunol. 2013, 31, 563–604. [Google Scholar] [CrossRef]
- Mellman, I. Dendritic cells: Master regulators of the immune response. Cancer Immunol. Res. 2013, 1, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Schlitzer, A.; Sivakamasundari, V.; Chen, J.; Sumatoh, H.R.; Schreuder, J.; Lum, J.; Malleret, B.; Zhang, S.; Larbi, A.; Zolezzi, F.; et al. Identification of cDC1- and cDC2-committed DC progenitors reveals early lineage priming at the common DC progenitor stage in the bone marrow. Nat. Immunol. 2015, 16, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Mildner, A.; Jung, S. Development and function of dendritic cell subsets. Immunity 2014, 40, 642–656. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Mujal, A.M.; Pollack, J.L.; Combes, A.J.; Hardison, E.A.; Barry, K.C.; Tsui, J.; Ruhland, M.K.; Kersten, K.; Abushawish, M.A. Unleashing type-2 dendritic cells to drive protective antitumor CD4+ T cell immunity. Cell 2019, 177, 556–571.e516. [Google Scholar] [CrossRef]
- Reizis, B. Regulation of plasmacytoid dendritic cell development. Curr. Opin. Immunol. 2010, 22, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Vermi, W.; Soncini, M.; Melocchi, L.; Sozzani, S.; Facchetti, F. Plasmacytoid dendritic cells and cancer. J. Leukoc. Biol. 2011, 90, 681–690. [Google Scholar] [CrossRef]
- Yu, S.; Liu, C.; Su, K.; Wang, J.; Liu, Y.; Zhang, L.; Li, C.; Cong, Y.; Kimberly, R.; Grizzle, W.E. Tumor exosomes inhibit differentiation of bone marrow dendritic cells. J. Immunol. 2007, 178, 6867–6875. [Google Scholar] [CrossRef]
- Maus, R.L.; Jakub, J.W.; Nevala, W.K.; Christensen, T.A.; Noble-Orcutt, K.; Sachs, Z.; Hieken, T.J.; Markovic, S.N. Human melanoma-derived extracellular vesicles regulate dendritic cell maturation. Front. Immunol. 2017, 8, 358. [Google Scholar] [CrossRef]
- Pyfferoen, L.; Mestdagh, P.; Vergote, K.; De Cabooter, N.; Vandesompele, J.; Lambrecht, B.N.; Vermaelen, K.Y. Lung tumours reprogram pulmonary dendritic cell immunogenicity at the microRNA level. Int. J. Cancer 2014, 135, 2868–2877. [Google Scholar] [CrossRef]
- Rosenberger, C.M.; Podyminogin, R.L.; Navarro, G.; Zhao, G.W.; Askovich, P.S.; Weiss, M.J.; Aderem, A. miR-451 regulates dendritic cell cytokine responses to influenza infection. J. Immunol. 2012, 189, 5965–5975. [Google Scholar] [CrossRef]
- Omata, N.; Yasutomi, M.; Yamada, A.; Iwasaki, H.; Mayumi, M.; Ohshima, Y. Monocyte chemoattractant protein-1 selectively inhibits the acquisition of CD40 ligand-dependent IL-12-producing capacity of monocyte-derived dendritic cells and modulates Th1 immune response. J. Immunol. 2002, 169, 4861–4866. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Batista, I.A.; Quintas, S.T.; Melo, S.A. The Interplay of Exosomes and NK Cells in Cancer Biology. Cancers 2021, 13, 473. [Google Scholar] [CrossRef]
- Sarkar, S.; Germeraad, W.T.; Rouschop, K.M.; Steeghs, E.M.; van Gelder, M.; Bos, G.M.; Wieten, L. Hypoxia induced impairment of NK cell cytotoxicity against multiple myeloma can be overcome by IL-2 activation of the NK cells. PLoS ONE 2013, 8, e64835. [Google Scholar] [CrossRef]
- Baginska, J.; Viry, E.; Berchem, G.; Poli, A.; Noman, M.Z.; van Moer, K.; Medves, S.; Zimmer, J.; Oudin, A.; Niclou, S.P.; et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc. Natl. Acad. Sci. USA 2013, 110, 17450–17455. [Google Scholar] [CrossRef]
- Berchem, G.; Noman, M.Z.; Bosseler, M.; Paggetti, J.; Baconnais, S.; Le Cam, E.; Nanbakhsh, A.; Moussay, E.; Mami-Chouaib, F.; Janji, B.; et al. Hypoxic tumor-derived microvesicles negatively regulate NK cell function by a mechanism involving TGF-beta and miR23a transfer. Oncoimmunology 2016, 5, e1062968. [Google Scholar] [CrossRef]
- Abbas, A.K. Basic Immunology, 4th ed.; Elsevier Saunders: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Kim, H.-J.; Cantor, H. CD4 T-cell subsets and tumor immunity: The helpful and the not-so-helpful. Cancer Immunol. Res. 2014, 2, 91–98. [Google Scholar] [CrossRef]
- Knochelmann, H.M.; Dwyer, C.J.; Bailey, S.R.; Amaya, S.M.; Elston, D.M.; Mazza-McCrann, J.M.; Paulos, C.M. When worlds collide: Th17 and Treg cells in cancer and autoimmunity. Cell. Mol. Immunol. 2018, 15, 458–469. [Google Scholar] [CrossRef]
- Lanitis, E.; Dangaj, D.; Irving, M.; Coukos, G. Mechanisms regulating T-cell infiltration and activity in solid tumors. Ann. Oncol. 2017, 28, xii18–xii32. [Google Scholar] [CrossRef]
- Geginat, J.; Paroni, M.; Maglie, S.; Alfen, J.S.; Kastirr, I.; Gruarin, P.; De Simone, M.; Pagani, M.; Abrignani, S. Plasticity of human CD4 T cell subsets. Front. Immunol. 2014, 5, 630. [Google Scholar] [CrossRef]
- Ye, S.B.; Zhang, H.; Cai, T.T.; Liu, Y.N.; Ni, J.J.; He, J.; Peng, J.Y.; Chen, Q.Y.; Mo, H.Y.; Zhang, X.S. Exosomal miR-24-3p impedes T-cell function by targeting FGF11 and serves as a potential prognostic biomarker for nasopharyngeal carcinoma. J. Pathol. 2016, 240, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Rossi, R.L.; Rossetti, G.; Wenandy, L.; Curti, S.; Ripamonti, A.; Bonnal, R.J.; Birolo, R.S.; Moro, M.; Crosti, M.C.; Gruarin, P.; et al. Distinct microRNA signatures in human lymphocyte subsets and enforcement of the naive state in CD4+ T cells by the microRNA miR-125b. Nat. Immunol. 2011, 12, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Zhu, S.; Dai, D.; Liu, Z.; Li, D.; Li, B.; Gagliani, N.; Zheng, Y.; Tang, Y.; Weirauch, M.T.; et al. MiR-125a targets effector programs to stabilize Treg-mediated immune homeostasis. Nat. Commun. 2015, 6, 7096. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, S.; Li, Z.; Wang, H.; Li, Z.; Hu, Y.; Chen, H.; Zhang, X.; Cui, L.; Zhang, J.; et al. miR-125b-5p and miR-99a-5p downregulate human gammadelta T-cell activation and cytotoxicity. Cell. Mol. Immunol. 2019, 16, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Ramchandani, D.; Mittal, V. Thrombospondin in tumor microenvironment. In Tumor Microenvironment; Springer: Berlin/Heidelberg, Germany, 2020; pp. 133–147. [Google Scholar]
- Dang, E.V.; Barbi, J.; Yang, H.Y.; Jinasena, D.; Yu, H.; Zheng, Y.; Bordman, Z.; Fu, J.; Kim, Y.; Yen, H.R.; et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 2011, 146, 772–784. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.Z.; Wang, R.; Huang, G.; Vogel, P.; Neale, G.; Green, D.R.; Chi, H. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 2011, 208, 1367–1376. [Google Scholar] [CrossRef]
- Wang, H.; Flach, H.; Onizawa, M.; Wei, L.; McManus, M.T.; Weiss, A. Negative regulation of Hif1a expression and TH17 differentiation by the hypoxia-regulated microRNA miR-210. Nat. Immunol. 2014, 15, 393–401. [Google Scholar] [CrossRef]
- Damasceno, L.E.A.; Prado, D.S.; Veras, F.P.; Fonseca, M.M.; Toller-Kawahisa, J.E.; Rosa, M.H.; Públio, G.A.; Martins, T.V.; Ramalho, F.S.; Waisman, A. PKM2 promotes Th17 cell differentiation and autoimmune inflammation by fine-tuning STAT3 activation. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Hsu, Y.L.; Hung, J.Y.; Chang, W.A.; Lin, Y.S.; Pan, Y.C.; Tsai, P.H.; Wu, C.Y.; Kuo, P.L. Hypoxic lung cancer-secreted exosomal miR-23a increased angiogenesis and vascular permeability by targeting prolyl hydroxylase and tight junction protein ZO-1. Oncogene 2017, 36, 4929–4942. [Google Scholar] [CrossRef]
- Kore, R.A.; Edmondson, J.L.; Jenkins, S.V.; Jamshidi-Parsian, A.; Dings, R.P.; Reyna, N.S.; Griffin, R.J. Hypoxia-derived exosomes induce putative altered pathways in biosynthesis and ion regulatory channels in glioblastoma cells. Biochem. Biophys. Rep. 2018, 14, 104–113. [Google Scholar] [CrossRef]
- Kim, M.; Park, Y.; Kwon, Y.; Kim, Y.; Byun, J.; Jeong, M.S.; Kim, H.U.; Jung, H.S.; Mun, J.Y.; Jeoung, D. MiR-135-5p-p62 Axis Regulates Autophagic Flux, Tumorigenic Potential, and Cellular Interactions Mediated by Extracellular Vesicles During Allergic Inflammation. Front. Immunol. 2019, 10, 738. [Google Scholar] [CrossRef] [PubMed]
- Rong, L.; Li, R.; Li, S.; Luo, R. Immunosuppression of breast cancer cells mediated by transforming growth factor-β in exosomes from cancer cells. Oncol. Lett. 2016, 11, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Horie, K.; Kawakami, K.; Fujita, Y.; Sugaya, M.; Kameyama, K.; Mizutani, K.; Deguchi, T.; Ito, M. Exosomes expressing carbonic anhydrase 9 promote angiogenesis. Biochem. Biophys. Res. Commun. 2017, 492, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Chen, L.; Chen, G.; Hu, C.; Jiang, S.; Sevilla, J.; Wan, Y.; Sampson, J.H.; Zhu, B.; Li, Q.J. Targeting miR-23a in CD8+ cytotoxic T lymphocytes prevents tumor-dependent immunosuppression. J. Clin. Investig. 2014, 124, 5352–5367. [Google Scholar] [CrossRef] [PubMed]
- Kallies, A.; Xin, A.; Belz, G.T.; Nutt, S.L. Blimp-1 transcription factor is required for the differentiation of effector CD8(+) T cells and memory responses. Immunity 2009, 31, 283–295. [Google Scholar] [CrossRef]
- Rutishauser, R.L.; Martins, G.A.; Kalachikov, S.; Chandele, A.; Parish, I.A.; Meffre, E.; Jacob, J.; Calame, K.; Kaech, S.M. Transcriptional repressor Blimp-1 promotes CD8(+) T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity 2009, 31, 296–308. [Google Scholar] [CrossRef]
- Calcinotto, A.; Filipazzi, P.; Grioni, M.; Iero, M.; De Milito, A.; Ricupito, A.; Cova, A.; Canese, R.; Jachetti, E.; Rossetti, M.; et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. 2012, 72, 2746–2756. [Google Scholar] [CrossRef]
- Chafe, S.C.; McDonald, P.C.; Saberi, S.; Nemirovsky, O.; Venkateswaran, G.; Burugu, S.; Gao, D.; Delaidelli, A.; Kyle, A.H.; Baker, J.H. Targeting hypoxia-induced carbonic anhydrase IX enhances immune-checkpoint blockade locally and systemically. Cancer Immunol. Res. 2019, 7, 1064–1078. [Google Scholar] [CrossRef]
- Wu, H.; Estrella, V.; Beatty, M.; Abrahams, D.; El-Kenawi, A.; Russell, S.; Ibrahim-Hashim, A.; Longo, D.L.; Reshetnyak, Y.K.; Moshnikova, A.; et al. T-cells produce acidic niches in lymph nodes to suppress their own effector functions. Nat. Commun. 2020, 11, 4113. [Google Scholar] [CrossRef]
- Chi, H. Regulation and function of mTOR signalling in T cell fate decisions. Nat. Rev. Immunol. 2012, 12, 325–338. [Google Scholar] [CrossRef]
- Pollizzi, K.N.; Powell, J.D. Regulation of T cells by mTOR: The known knowns and the known unknowns. Trends Immunol. 2015, 36, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Giatromanolaki, A.; Harris, A.L.; Banham, A.H.; Contrafouris, C.A.; Koukourakis, M.I. Carbonic anhydrase 9 (CA9) expression in non-small-cell lung cancer: Correlation with regulatory FOXP3+ T-cell tumour stroma infiltration. Br. J. Cancer 2020, 122, 1205–1210. [Google Scholar] [CrossRef] [PubMed]
- Panigrahi, G.K.; Ramteke, A.; Birks, D.; Abouzeid Ali, H.E.; Venkataraman, S.; Agarwal, C.; Vibhakar, R.; Miller, L.D.; Agarwal, R.; Abd Elmageed, Z.Y.; et al. Exosomal microRNA profiling to identify hypoxia-related biomarkers in prostate cancer. Oncotarget 2018, 9, 13894–13910. [Google Scholar] [CrossRef]
- Sheu, B.-C.; Hsu, S.-M.; Ho, H.-N.; Lien, H.-C.; Huang, S.-C.; Lin, R.-H. A novel role of metalloproteinase in cancer-mediated immunosuppression. Cancer Res. 2001, 61, 237–242. [Google Scholar]
- Dou, R.; Nishihara, R.; Cao, Y.; Hamada, T.; Mima, K.; Masuda, A.; Masugi, Y.; Shi, Y.; Gu, M.; Li, W.; et al. MicroRNA let-7, T Cells, and Patient Survival in Colorectal Cancer. Cancer Immunol. Res. 2016, 4, 927–935. [Google Scholar] [CrossRef]
- Hu, X.P.; Xie, Q.; Chen, C.F.; Zhang, W.; Yu, B. Let-7a Inhibits T-Cell Proliferation and IFN-gamma Secretion by Down-Regulating STAT3 Expression in Patients with Psoriasis. Cell. Physiol. Biochem. 2017, 42, 115–125. [Google Scholar] [CrossRef]
- Scully, T.; Scott, C.D.; Firth, S.M.; Sedger, L.M.; Pintar, J.E.; Twigg, S.M.; Baxter, R.C. Enhancement of mammary tumour growth by IGFBP-3 involves impaired T cell accumulation. Endocr.-Relat. Cancer 2018, 25, 111–122. [Google Scholar] [CrossRef]
- Marcheteau, E.; Farge, T.; Pérès, M.; Labrousse, G.; Tenet, J.; Delmas, S.; Chusseau, M.; Duprez-Paumier, R.; Franchet, C.; Dalenc, F. Thrombospondin-1 Silencing Improves Lymphocyte Infiltration in Tumors and Response to Anti-PD-1 in Triple-Negative Breast Cancer. Cancers 2021, 13, 4059. [Google Scholar] [CrossRef]
- de Arao Tan, I.; Ricciardelli, C.; Russell, D.L. The metalloproteinase ADAMTS1: A comprehensive review of its role in tumorigenic and metastatic pathways. Int. J. Cancer 2013, 133, 2263–2276. [Google Scholar] [CrossRef]
- Rodríguez-Baena, F.J.; Redondo-García, S.; Peris-Torres, C.; Martino-Echarri, E.; Fernández-Rodríguez, R.; del Carmen Plaza-Calonge, M.; Anderson, P.; Rodríguez-Manzaneque, J.C. ADAMTS1 protease is required for a balanced immune cell repertoire and tumour inflammatory response. Sci. Rep. 2018, 8, 13103. [Google Scholar] [CrossRef]
- Ricciardelli, C.; Frewin, K.M.; de Arao Tan, I.; Williams, E.D.; Opeskin, K.; Pritchard, M.A.; Ingman, W.V.; Russell, D.L. The ADAMTS1 protease gene is required for mammary tumor growth and metastasis. Am. J. Pathol. 2011, 179, 3075–3085. [Google Scholar] [CrossRef]
- Hope, C.; Foulcer, S.; Jagodinsky, J.; Chen, S.X.; Jensen, J.L.; Patel, S.; Leith, C.; Maroulakou, I.; Callander, N.; Miyamoto, S. Immunoregulatory roles of versican proteolysis in the myeloma microenvironment. Blood J. Am. Soc. Hematol. 2016, 128, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Terabe, M.; Berzofsky, J.A. The immunoregulatory role of type I and type II NKT cells in cancer and other diseases. Cancer Immunol. Immunother. 2014, 63, 199–213. [Google Scholar] [CrossRef]
- Tang, B.; Wu, W.; Wei, X.; Li, Y.; Ren, G.; Fan, W. Activation of glioma cells generates immune tolerant NKT cells. J. Biol. Chem. 2014, 289, 34595–34600. [Google Scholar] [CrossRef]
- Flynn, N.J.; Somasundaram, R.; Arnold, K.M.; Sims-Mourtada, J. The Multifaceted Roles of B Cells in Solid Tumors: Emerging Treatment Opportunities. Target Oncol. 2017, 12, 139–152. [Google Scholar] [CrossRef]
- Mok, Y.; Schwierzeck, V.; Thomas, D.C.; Vigorito, E.; Rayner, T.F.; Jarvis, L.B.; Prosser, H.M.; Bradley, A.; Withers, D.R.; Martensson, I.L.; et al. MiR-210 is induced by Oct-2, regulates B cells, and inhibits autoantibody production. J. Immunol. 2013, 191, 3037–3048. [Google Scholar] [CrossRef]
- Gururajan, M.; Haga, C.L.; Das, S.; Leu, C.M.; Hodson, D.; Josson, S.; Turner, M.; Cooper, M.D. MicroRNA 125b inhibition of B cell differentiation in germinal centers. Int. Immunol. 2010, 22, 583–592. [Google Scholar] [CrossRef]
- Cichocki, F.; Felices, M.; McCullar, V.; Presnell, S.R.; Al-Attar, A.; Lutz, C.T.; Miller, J.S. Cutting edge: MicroRNA-181 promotes human NK cell development by regulating Notch signaling. J. Immunol. 2011, 187, 6171–6175. [Google Scholar] [CrossRef]
- Ishitani, T.; Hirao, T.; Suzuki, M.; Isoda, M.; Ishitani, S.; Harigaya, K.; Kitagawa, M.; Matsumoto, K.; Itoh, M. Nemo-like kinase suppresses Notch signalling by interfering with formation of the Notch active transcriptional complex. Nat. Cell Biol. 2010, 12, 278–285. [Google Scholar] [CrossRef]
- Felices, M.; Ankarlo, D.E.; Lenvik, T.R.; Nelson, H.H.; Blazar, B.R.; Verneris, M.R.; Miller, J.S. Notch signaling at later stages of NK cell development enhances KIR expression and functional maturation. J. Immunol. 2014, 193, 3344–3354. [Google Scholar] [CrossRef]
- Beck, R.C.; Padival, M.; Yeh, D.; Ralston, J.; Cooke, K.R.; Lowe, J.B. The Notch ligands Jagged2, Delta1, and Delta4 induce differentiation and expansion of functional human NK cells from CD34+ cord blood hematopoietic progenitor cells. Biol. Blood Marrow Transplant. 2009, 15, 1026–1037. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, K.; Suzuki, T.; Koyama, N.; Kumano, K.; Nakahara, F.; Matsumoto, A.; Yokoyama, Y.; Sakata-Yanagimoto, M.; Masuda, S.; Takahashi, T.; et al. Notch activation induces the generation of functional NK cells from human cord blood CD34-positive cells devoid of IL-15. J. Immunol. 2009, 182, 6168–6178. [Google Scholar] [CrossRef] [PubMed]
- Trotta, R.; Chen, L.; Ciarlariello, D.; Josyula, S.; Mao, C.; Costinean, S.; Yu, L.; Butchar, J.P.; Tridandapani, S.; Croce, C.M.; et al. miR-155 regulates IFN-gamma production in natural killer cells. Blood 2012, 119, 3478–3485. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.A.; So, A.Y.; Sinha, N.; Gibson, W.S.; Taganov, K.D.; O’Connell, R.M.; Baltimore, D. MicroRNA-125b potentiates macrophage activation. J. Immunol. 2011, 187, 5062–5068. [Google Scholar] [CrossRef]
- Honma, K.; Udono, H.; Kohno, T.; Yamamoto, K.; Ogawa, A.; Takemori, T.; Kumatori, A.; Suzuki, S.; Matsuyama, T.; Yui, K. Interferon regulatory factor 4 negatively regulates the production of proinflammatory cytokines by macrophages in response to LPS. Proc. Natl. Acad. Sci. USA 2005, 102, 16001–16006. [Google Scholar] [CrossRef]
- Gerloff, D.; Lutzkendorf, J.; Moritz, R.K.C.; Wersig, T.; Mader, K.; Muller, L.P.; Sunderkotter, C. Melanoma-Derived Exosomal miR-125b-5p Educates Tumor Associated Macrophages (TAMs) by Targeting Lysosomal Acid Lipase A (LIPA). Cancers 2020, 12, 464. [Google Scholar] [CrossRef]
- Cai, X.; Yin, Y.; Li, N.; Zhu, D.; Zhang, J.; Zhang, C.Y.; Zen, K. Re-polarization of tumor-associated macrophages to pro-inflammatory M1 macrophages by microRNA-155. J. Mol. Cell Biol. 2012, 4, 341–343. [Google Scholar] [CrossRef]
- Martinez-Nunez, R.T.; Louafi, F.; Sanchez-Elsner, T. The interleukin 13 (IL-13) pathway in human macrophages is modulated by microRNA-155 via direct targeting of interleukin 13 receptor alpha1 (IL13Ralpha1). J. Biol. Chem. 2011, 286, 1786–1794. [Google Scholar] [CrossRef]
- Zonari, E.; Pucci, F.; Saini, M.; Mazzieri, R.; Politi, L.S.; Gentner, B.; Naldini, L. A role for miR-155 in enabling tumor-infiltrating innate immune cells to mount effective antitumor responses in mice. Blood 2013, 122, 243–252. [Google Scholar] [CrossRef]
- Huffaker, T.B.; Lee, S.H.; Tang, W.W.; Wallace, J.A.; Alexander, M.; Runtsch, M.C.; Larsen, D.K.; Thompson, J.; Ramstead, A.G.; Voth, W.P.; et al. Antitumor immunity is defective in T cell-specific microRNA-155-deficient mice and is rescued by immune checkpoint blockade. J. Biol. Chem. 2017, 292, 18530–18541. [Google Scholar] [CrossRef]
- Dudda, J.C.; Salaun, B.; Ji, Y.; Palmer, D.C.; Monnot, G.C.; Merck, E.; Boudousquie, C.; Utzschneider, D.T.; Escobar, T.M.; Perret, R.; et al. MicroRNA-155 is required for effector CD8+ T cell responses to virus infection and cancer. Immunity 2013, 38, 742–753. [Google Scholar] [CrossRef] [PubMed]
- Willms, E.; Cabanas, C.; Mager, I.; Wood, M.J.A.; Vader, P. Extracellular Vesicle Heterogeneity: Subpopulations, Isolation Techniques, and Diverse Functions in Cancer Progression. Front. Immunol. 2018, 9, 738. [Google Scholar] [CrossRef] [PubMed]
- Greening, D.W.; Simpson, R.J. Understanding extracellular vesicle diversity—Current status. Expert Rev. Proteom. 2018, 15, 887–910. [Google Scholar] [CrossRef]
- Van Deun, J.; Mestdagh, P.; Sormunen, R.; Cocquyt, V.; Vermaelen, K.; Vandesompele, J.; Bracke, M.; De Wever, O.; Hendrix, A. The impact of disparate isolation methods for extracellular vesicles on downstream RNA profiling. J. Extracell. Vesicles 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Konoshenko, M.Y.; Lekchnov, E.A.; Vlassov, A.V.; Laktionov, P.P. Isolation of Extracellular Vesicles: General Methodologies and Latest Trends. BioMed Res. Int. 2018, 2018, 8545347. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.D.; Shah, S. Methods of isolating extracellular vesicles impact down-stream analyses of their cargoes. Methods 2015, 87, 3–10. [Google Scholar] [CrossRef]
- Coumans, F.A.W.; Brisson, A.R.; Buzas, E.I.; Dignat-George, F.; Drees, E.E.E.; El-Andaloussi, S.; Emanueli, C.; Gasecka, A.; Hendrix, A.; Hill, A.F.; et al. Methodological Guidelines to Study Extracellular Vesicles. Circ. Res. 2017, 120, 1632–1648. [Google Scholar] [CrossRef]
- Paolini, L.; Zendrini, A.; Di Noto, G.; Busatto, S.; Lottini, E.; Radeghieri, A.; Dossi, A.; Caneschi, A.; Ricotta, D.; Bergese, P. Residual matrix from different separation techniques impacts exosome biological activity. Sci. Rep. 2016, 6, 23550. [Google Scholar] [CrossRef]
- Brennan, K.; Martin, K.; FitzGerald, S.P.; O’Sullivan, J.; Wu, Y.; Blanco, A.; Richardson, C.; Mc Gee, M.M. A comparison of methods for the isolation and separation of extracellular vesicles from protein and lipid particles in human serum. Sci. Rep. 2020, 10, 1039. [Google Scholar] [CrossRef]
- Stranska, R.; Gysbrechts, L.; Wouters, J.; Vermeersch, P.; Bloch, K.; Dierickx, D.; Andrei, G.; Snoeck, R. Comparison of membrane affinity-based method with size-exclusion chromatography for isolation of exosome-like vesicles from human plasma. J. Transl. Med. 2018, 16, 1. [Google Scholar] [CrossRef]
- Mol, E.A.; Goumans, M.J.; Doevendans, P.A.; Sluijter, J.P.G.; Vader, P. Higher functionality of extracellular vesicles isolated using size-exclusion chromatography compared to ultracentrifugation. Nanomedicine 2017, 13, 2061–2065. [Google Scholar] [CrossRef] [PubMed]
- Umezu, T.; Tadokoro, H.; Azuma, K.; Yoshizawa, S.; Ohyashiki, K.; Ohyashiki, J.H. Exosomal miR-135b shed from hypoxic multiple myeloma cells enhances angiogenesis by targeting factor-inhibiting HIF-1. Blood 2014, 124, 3748–3757. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, Y.; Wada, H.; Eguchi, H.; Gotoh, K.; Kobayashi, S.; Kinoshita, M.; Kubo, M.; Hayashi, K.; Iwagami, Y.; Yamada, D.; et al. Exosomal miR-155 Derived from Hepatocellular Carcinoma Cells Under Hypoxia Promotes Angiogenesis in Endothelial Cells. Dig. Dis. Sci. 2019, 64, 792–802. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Min, Z.; Zhou, Z.; Linhong, M.; Tao, R.; Yan, L.; Song, H. Hypoxia-induced exosomes promote hepatocellular carcinoma proliferation and metastasis via miR-1273f transfer. Exp. Cell Res. 2019, 385, 111649. [Google Scholar] [CrossRef] [PubMed]
- Walbrecq, G.; Lecha, O.; Gaigneaux, A.; Fougeras, M.R.; Philippidou, D.; Margue, C.; Tetsi Nomigni, M.; Bernardin, F.; Dittmar, G.; Behrmann, I.; et al. Hypoxia-Induced Adaptations of miRNomes and Proteomes in Melanoma Cells and Their Secreted Extracellular Vesicles. Cancers 2020, 12, 692. [Google Scholar] [CrossRef]
- Wozniak, M.; Peczek, L.; Czernek, L.; Duchler, M. Analysis of the miRNA Profiles of Melanoma Exosomes Derived Under Normoxic and Hypoxic Culture Conditions. Anticancer Res. 2017, 37, 6779–6789. [Google Scholar] [CrossRef]
- Svensson, K.J.; Kucharzewska, P.; Christianson, H.C.; Sköld, S.; Löfstedt, T.; Johansson, M.C.; Mörgelin, M.; Bengzon, J.; Ruf, W.; Belting, M. Hypoxia triggers a proangiogenic pathway involving cancer cell microvesicles and PAR-2–mediated heparin-binding EGF signaling in endothelial cells. Proc. Natl. Acad. Sci. USA 2011, 108, 13147–13152. [Google Scholar] [CrossRef]
- Huang, Z.; Feng, Y. Exosomes derived from hypoxic colorectal cancer cells promote angiogenesis through Wnt4-induced β-catenin signaling in endothelial cells. Oncol. Res. 2017, 25, 651. [Google Scholar] [CrossRef]
- Huang, Z.; Yang, M.; Li, Y.; Yang, F.; Feng, Y. Exosomes derived from hypoxic colorectal cancer cells transfer Wnt4 to normoxic cells to elicit a prometastatic phenotype. Int. J. Biol. Sci. 2018, 14, 2094. [Google Scholar] [CrossRef]
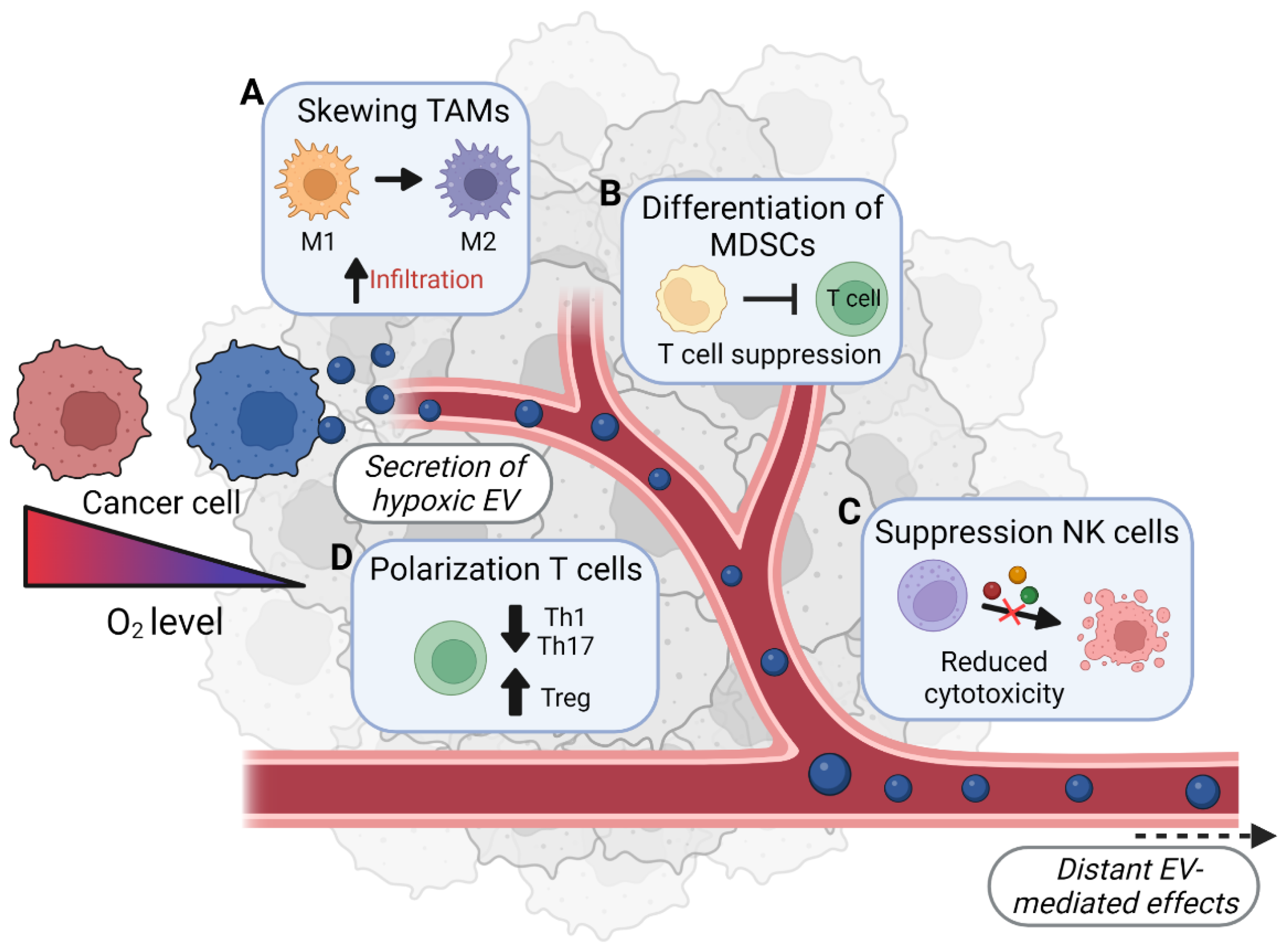
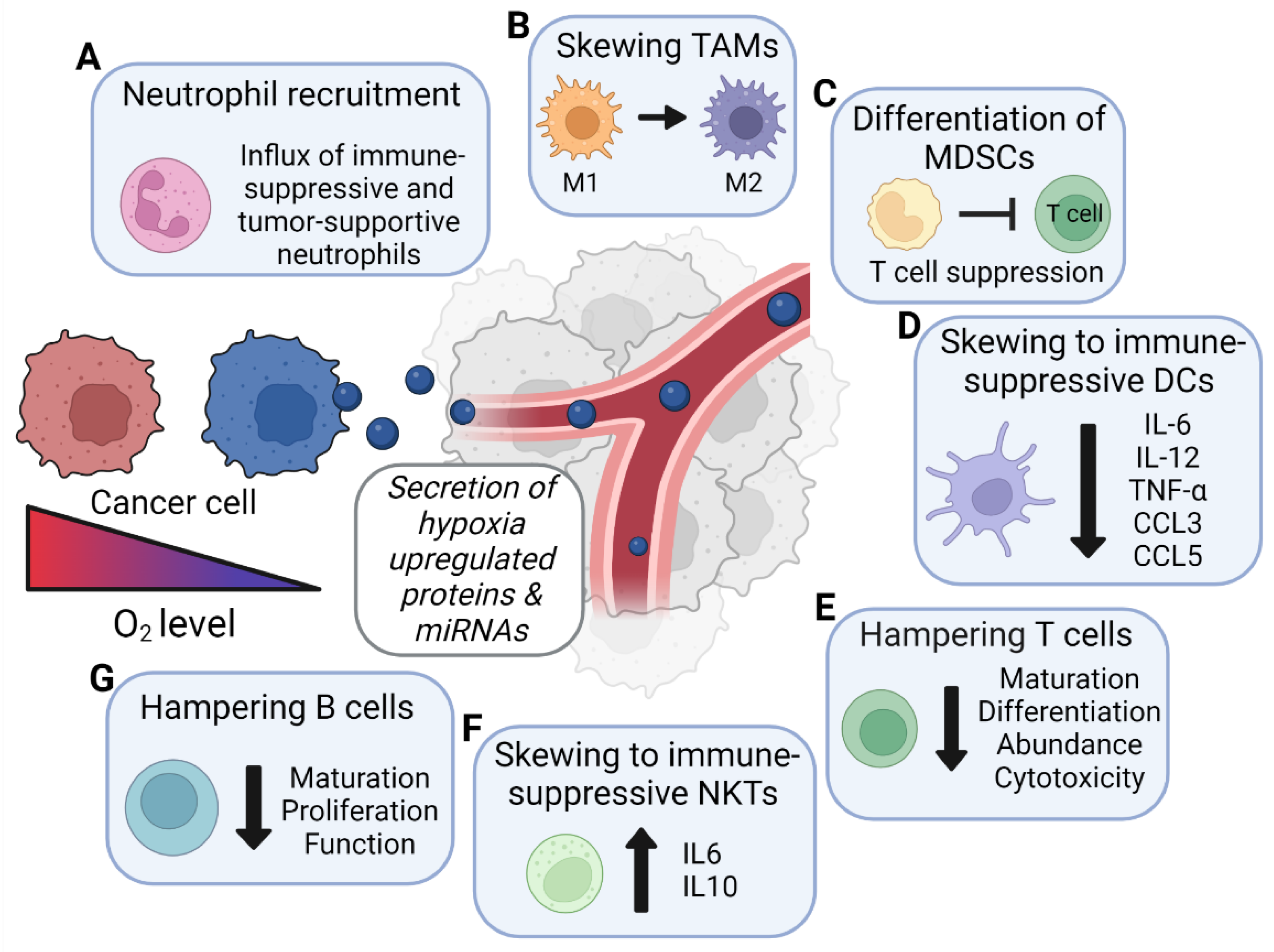
| Factor | Effect on Neutrophils | Proven to Be EV-Mediated? 1 |
|---|---|---|
| CXCL8 (IL-8) [28,29] | Enhances recruitment to tumors through binding with CXCR1 and/or CXCR2. Reduces apoptosis and enhances proliferation via activation of MAPK signaling. | No |
| MIR-451 [30] | Represses recruitment to tumors via inhibition of MAPK signaling. | No |
| Factor | Effect on Dendritic Cells | Proven to Be EV-Mediated? 1 |
|---|---|---|
| MIR-301 [90] | Reduces production of the pro-inflammatory cytokines IL-6, IL-12, and TNF-α. Expression of miR-301 in DC represses the release of IFN-γ from DC-primed CD8+ and CD4+ responder cells. | No |
| MIR-451 [91] | Reduces production of the pro-inflammatory cytokines IL-6, CCL3/MIP1a, CCL5/RANTES, and TNF-α. | No |
| CCL2 [92] | Reduces production of pro-inflammatory IL-12, hampering effective T-cell-mediated toxicity. | No |
| Factor | Effect on NK Cells | Proven to Be EV-Mediated? 1 |
|---|---|---|
| TGF-β [97] | Inhibits NK cell function by decreasing surface expression of the activating receptor NKG2D and decreasing IFN-γ production. | Yes |
| MIR-23a [97] | Decreases the percentage of degranulating NK cells. | Yes |
| Factor | Effect on NKT Cells | Proven to Be EV-Mediated? 1 |
|---|---|---|
| MIR-92a [137] | Induces immunosuppressive NKT cells with reduced antitumor effects via increased IL-6 and IL-10 expression. | No |
| Factor | Effect on B Cells | Proven to Be EV-Mediated? 1 |
|---|---|---|
| MIR-210 [139] | Impairs B-cell proliferation. Reduces antibody production. | No |
| MIR-125 [73,140] | Prevents B cells’ maturation and release from the bone marrow. | No |
| Factor | Immune-Stimulating Effect |
|---|---|
| MIR-181 | Enhances the development of NK cells. |
| MIR-155 | Enhances antitumor reactions in NK cells. Reduces the capacity of macrophages to respond to M2-inducing signals. Pushes macrophages towards the M1 phenotype. Modulates CD8+ T cells’ responsiveness to IL-2, IL-7, and IL-15 stimulation. |
| miRNA | Cancer Type | Reported Culturing Conditions | Reported EV Isolation Method |
|---|---|---|---|
| MIR-10 MIR-21 MIR-125 [73] | Glioma | 1% O2, 48 h 10% EV-depleted FCS | Differential centrifugation (300× g 10 min, 2.000× g 10 min, 10,000× g 30 min) 0.22 μm filtration, ultracentrifugation (2 × 100,000× g 70 min). |
| MIR-21-3p MIR-125b-5p MIR-181d-5p [48] | Epithelial ovarian | 1% O2, 24 h 10% EV-depleted FCS (100,000 g, 20 h) | Differential centrifugation (1000× g 10 min, 3000× g 30 min, Total Exosome Isolation Reagent (Life Technologies). |
| MIR-23a [112] | Lung | 1% O2, 24 h 1% EV-free serum (Life Technologies) | Total Exosome Isolation Reagent (from cells) (Life Technologies). |
| MIR-24-3p [103] | Nasopharyngeal carcinoma | 0.1% O2, 48 h | Differential centrifugation (300× g 10 min, 1.200× g 20 min, 10,000× g 30 min, 4 °C), 0.22 μm filtration, ultracentrifugation (2 × 100,000× g 3 h). |
| MIR-92a MIR-127 MIR-143 MIR-181 MIR-204 MIR-292 MIR-335 MIR-433 MIR-451 MIR-542 MIR-547a MIR-885 [126] | Prostate | 1% O2, 72 h | Differential centrifugation at low speed (unspecified), ultracentrifugation at 30,000 RPM (type 70.1Ti fixed-angle rotor, L-80 Ultracentrifuge, Beckman Coulter). |
| MIR-135b [165] | Multiple myeloma | 1% O2, 24 h serum-free medium | Centrifugation (3000× g 15 min), 0.22 μm PVDF filtration, ExoQuick Exosome Precipitation Solution (System Biosciences, Mountain View, CA). |
| MIR-155 [166] | Hepatocellular carcinoma | 1% O2, 24–72 h—CoCl2 100 µM, 48 h 10% EV-depleted FCS (120,000 g overnight, 0.22 µm filtration) | Centrifugation (3000× g 15 min), 0.22 μm PVDF filtration, ExoQuick Exosome Precipitation Solution (System Biosciences, Mountain View, CA). |
| MIR-210 [61] | Leukemia | 1% O2, 24 h serum-free medium | Centrifugation (3000× g 15 min), 0.22 μm PVDF filtration, ExoQuick Exosome Precipitation Solution (System Biosciences, Mountain View, CA). |
| MIR301a [46] | Pancreas | 1% O2 10% EV depleted FCS | Differential centrifugation (300× g 10 min, 2000× g 10 min, 10,000× g 30 min, ultracentrifugation (100,000× g 70 min) OR ExoQuick Exosome Precipitation Solution. |
| MIR-940 [47] | Epithelial ovarian | 1% O2, 72 h 10% EV-depleted FCS (100,000 g, 20 h) | Centrifugation (2.500 RPM 30 min), Total Exosome Isolation Reagent (Invitrogen). |
| MIR-1246 [44] | Glioma | 1% O2 10% EV depleted FCS | Differential centrifugation (300× g 10 min, 2000× g 10 min, 10,000× g 30 min), 0.22 μm filtration, ultracentrifugation (2 × 100,000× g 70 min). |
| MIR-1273f [167] | Hepatocellular carcinoma | 1% O2, 48 h 10% EV-free FCS | Differential centrifugation (300× g 10 min, 2000× g 10 min, 10,000× g 30 min), ultracentrifugation (100,000× g 70 min). |
| MIR-1290 [168] | Melanoma | 1% O2, 72 h serum-free medium | Differential centrifugation (400× g 10 min, 2000× g 30 min), ultracentrifugation (110,000× g 70 min), flotation on an OptiPrep cushion (100,000× g 70 min), PBS wash, ultracentrifugation (110,000× g 70 min). |
| MIR-135a MIR-494 MIR-513a MIR-575 MIR-1233-1 MIR-4463 MIR-4497 MIR-4498 MIR-4530 MIR-4721 MIR-4728 MIR-4741 MIR-4763 MIR-6087 MIR-6132 [169] | Melanoma | 1% O2 serum-free medium | Differential centrifugation (300× g 4 min, 10,000× g 30 min), ultracentrifugation (2 × 100,000× g 2.5 h). |
| Let-7a [42] | Melanoma | <0.5% O2, 24 h 5% chemically defined medium (protein-free serum replacement) | Centrifugation (1.200× g 30 min), 300 kDa MWCO centrifugation at 4000 g, centrifugation (12,000× g 30 min), flotation on sucrose cushion (5.5% sucrose, 100,000× g 15 h). |
| Protein | Cancer Type | Reported Culturing Conditions | Reported EV Isolation Method |
|---|---|---|---|
| ADAMTS1 [113] | Glioblastoma | <0.5% O2, 8–24 h, serum-free DMEM medium | CM centrifugation at 300× g for 10 min, 10,000× g for 30 min, and twice at 100,000× g for 2 h. |
| CCL2 (MCP1) [42] | Melanoma | <0.5% O2, 24 h 5% chemically defined medium (protein-free serum replacement) | CM centrifugation at 1.200× g for 30 min, 300 kDa MWCO centrifugation at 4000 g, 12,000× g for 30 min, and 100,000× g on a 5.5% sucrose pad for 15 h. |
| CSF-1 Ferritin heavy chain Ferritin light chain [42] | Melanoma | <0.5% O2, 24 h 5% chemically defined medium (protein-free serum replacement) | CM centrifugation at 1200× g for 30 min, 300 kDa MWCO centrifugation at 4000 g, 12,000× g for 30 min, and 100,000× g on a 5.5% sucrose pad for 15 h. |
| IGFBP1 IGFBP3 CXCL8 (IL-8) [27] | Glioma | 1% O2, 48 h DMEM supplemented with 1% BSA | CM centrifugation at 300× g for 5 min, 16,500× g for 30 min, and 100,000× g for 2 h, and 2x PBS wash at 100,000× g for 2 h. |
| LOX [113] | Glioblastoma | <0.5% O2, 8–24 h serum-free DMEM | CM centrifugation at 300× g for 10 min, 10,000× g for 30 min, and twice at 100,000× g for 2 h. |
| Macrophage migration inhibitory factor (MIF) [42] | Melanoma | <0.5% O2, 24 h 5% chemically defined medium (protein-free serum replacement) | CM centrifugation at 1.200× g for 30 min, 300 kDa MWCO centrifugation at 4000 g, 12,000× g for 30 min, and 100,000× g on a 5.5% sucrose pad for 15 h. |
| PRMT5 [168] | Melanoma | 1% O2, 72 h serum-free medium | CM centrifugation at 400× g for 10 min, 2000× g for 30 min, 100,000× g for 70 min, and 100,000× g for 70 min on an OptiPrep cushion. |
| TF [170] | Glioblastoma | 1% O2, 30 min–48 h serum-free medium supplemented with 1% BSA (wt/vol). | CM centrifugation at 300× g for 10 min, 16,500× g for 20 min, 100,000× g for 2 h, and PBS washed at 100,000× g for 70 min. |
| TGF-β [97,114,115] | Park: Melanoma Berchem: Lung Rong: Breast | Park: <0.5% O2, 24 h 5% chemically defined medium (protein-free serum replacement). Berchem: 0.1% O2, 48 h exosome-depleted FBS. Rong: 1% O2, 4 days serum-free medium | Park: CM centrifugation at 1.200× g for 30 min, 300 kDa MWCO centrifugation at 4000× g, 12,000× g for 30 min, and 100,000× g on a 5.5% sucrose pad for 15 h. Berchem: CM centrifugation at 400× g for 5 min, 2.500× g for 20 min, 4.500 for 20 min, and 10,000× g for 1 h. Rong: CM centrifugation at 500× g for 2 × 10 min, 2000× g for 20 min, 10,000× g for 30 min, and 100,000× g for 1 h. |
| TSP-1 VEGF [113] | Glioblastoma | <0.5% O2, 8–24 h serum-free DMEM | CM centrifugation at 300× g for 10 min, 10,000× g for 30 min, and twice at 100,000× g for 2 h. |
| CAIX [116] | Renal-cell carcinoma | 1% O2 or 200 μM CoCl2 advanced DMEM or advanced RPMI | CM centrifugation at 2000× g for 10 min and 12,000× g for 30 min; 0.22 μm PVDF filtration, and 70 min at 110,000 g, followed by density gradient centrifugation. Second method: Isolation by immunocapture Dynabeads conjugated with murine monoclonal anti-CD9 antibody. |
| Wnt4 [171,172] | Colorectal | 250 μM Cocl2, 48 h exosome-depleted FBS | CM centrifugation at 1000 g for 10 min and 3000× g for 30 min. Added to Total Exosome Isolation Kit overnight and centrifuged at 10,000× g for 1 h. |
| MTA1 [42] | Melanoma | <0.5% O2, 24 h 5% chemically defined medium (protein-free serum replacement) | CM centrifugation at 1.200× g for 30 min, 300 kDa MWCO centrifugation at 4000 g, 12,000× g for 30 min, and 100,000× g on a 5.5% sucrose pad for 15 h. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beaumont, J.E.J.; Beelen, N.A.; Wieten, L.; Rouschop, K.M.A. The Immunomodulatory Role of Hypoxic Tumor-Derived Extracellular Vesicles. Cancers 2022, 14, 4001. https://doi.org/10.3390/cancers14164001
Beaumont JEJ, Beelen NA, Wieten L, Rouschop KMA. The Immunomodulatory Role of Hypoxic Tumor-Derived Extracellular Vesicles. Cancers. 2022; 14(16):4001. https://doi.org/10.3390/cancers14164001
Chicago/Turabian StyleBeaumont, Joel E. J., Nicky A. Beelen, Lotte Wieten, and Kasper M. A. Rouschop. 2022. "The Immunomodulatory Role of Hypoxic Tumor-Derived Extracellular Vesicles" Cancers 14, no. 16: 4001. https://doi.org/10.3390/cancers14164001
APA StyleBeaumont, J. E. J., Beelen, N. A., Wieten, L., & Rouschop, K. M. A. (2022). The Immunomodulatory Role of Hypoxic Tumor-Derived Extracellular Vesicles. Cancers, 14(16), 4001. https://doi.org/10.3390/cancers14164001