MYCN and Metabolic Reprogramming in Neuroblastoma



Abstract
:Simple Summary
Abstract
1. Introduction
2. MYCN and Neuroblastoma
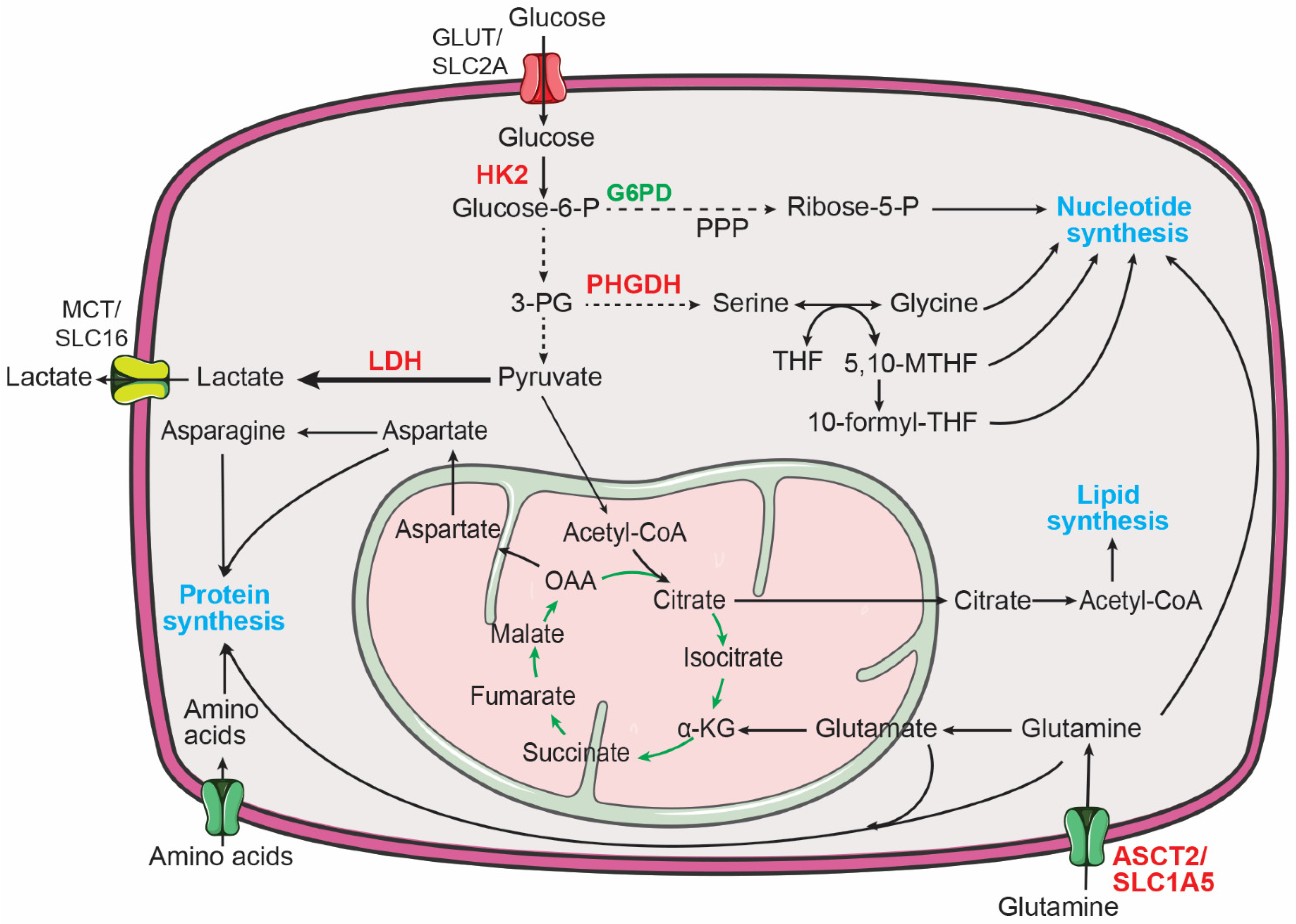
3. Metabolic Reprogramming in Neuroblastoma
3.1. Aerobic Glycolysis in Neuroblastoma
3.2. Reprogramming of Amino Acid Metabolism in Neuroblastoma
3.2.1. Glutamine Metabolism
3.2.2. Serine and Glycine Metabolism
3.2.3. Cysteine Metabolism
3.3. Reprogramming of Nucleotide Metabolism in Neuroblastoma
3.3.1. Purine Metabolism
3.3.2. Pyrimidine Metabolism
3.4. Reprogramming of Lipid Metabolism in Neuroblastoma
3.4.1. Fatty Acid Metabolism
3.4.2. Cholesterol Metabolism
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Schulze, A.; Harris, A.L. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 2012, 491, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Reyes, I.; Chandel, N.S. Cancer metabolism: Looking forward. Nat. Rev. Cancer 2021, 21, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Zhu, J.; Thompson, C.B. The hallmarks of cancer metabolism: Still emerging. Cell Metab. 2022, 34, 355–377. [Google Scholar] [CrossRef]
- Brodeur, G.M. Neuroblastoma: Biological insights into a clinical enigma. Nat. Rev. Cancer 2003, 3, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Cheung, N.K.; Dyer, M.A. Neuroblastoma: Developmental biology, cancer genomics and immunotherapy. Nat. Rev. Cancer 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Marshall, G.M.; Carter, D.R.; Cheung, B.B.; Liu, T.; Mateos, M.K.; Meyerowitz, J.G.; Weiss, W.A. The prenatal origins of cancer. Nat. Rev. Cancer 2014, 14, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Maris, J.M.; Hogarty, M.D.; Bagatell, R.; Cohn, S.L. Neuroblastoma. Lancet 2007, 369, 2106–2120. [Google Scholar] [CrossRef]
- Anderson, D.J. Cellular and molecular biology of neural crest cell lineage determination. Trends Genet. 1997, 13, 276–280. [Google Scholar] [CrossRef]
- Francis, N.J.; Landis, S.C. Cellular and molecular determinants of sympathetic neuron development. Annu. Rev. Neurosci. 1999, 22, 541–566. [Google Scholar] [CrossRef]
- Cohn, S.L.; Pearson, A.D.J.; London, W.B.; Monclair, T.; Ambros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iehara, T.; Machin, D.; et al. The International Neuroblastoma Risk Group (INRG) Classification System: An INRG Task Force Report. J. Clin. Oncol. 2009, 27, 289–297. [Google Scholar] [CrossRef] [Green Version]
- Pinto, N.R.; Applebaum, M.A.; Volchenboum, S.L.; Matthay, K.K.; London, W.B.; Ambros, P.F.; Nakagawara, A.; Berthold, F.; Schleiermacher, G.; Park, J.R.; et al. Advances in Risk Classification and Treatment Strategies for Neuroblastoma. J. Clin. Oncol. 2015, 33, 3008–3017. [Google Scholar] [CrossRef]
- Newman, E.A.; Abdessalam, S.; Aldrink, J.H.; Austin, M.; Heaton, T.E.; Bruny, J.; Ehrlich, P.; Dasgupta, R.; Baertschiger, R.M.; Lautz, T.B.; et al. Update on neuroblastoma. J. Pediatric Surg. 2019, 54, 383–389. [Google Scholar] [CrossRef]
- Schwab, M.; Alitalo, K.; Klempnauer, K.H.; Varmus, H.E.; Bishop, J.M.; Gilbert, F.; Brodeur, G.; Goldstein, M.; Trent, J. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature 1983, 305, 245–248. [Google Scholar] [CrossRef]
- Kohl, N.E.; Kanda, N.; Schreck, R.R.; Bruns, G.; Latt, S.A.; Gilbert, F.; Alt, F.W. Transposition and amplification of oncogene-related sequences in human neuroblastomas. Cell 1983, 35, 359–367. [Google Scholar] [CrossRef]
- Brodeur, G.M.; Seeger, R.C.; Schwab, M.; Varmus, H.E.; Bishop, J.M. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science 1984, 224, 1121–1124. [Google Scholar] [CrossRef]
- Seeger, R.C.; Brodeur, G.M.; Sather, H.; Dalton, A.; Siegel, S.E.; Wong, K.Y.; Hammond, D. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N. Engl. J. Med. 1985, 313, 1111–1116. [Google Scholar] [CrossRef]
- Rickman, D.S.; Schulte, J.H.; Eilers, M. The Expanding World of N-MYC-Driven Tumors. Cancer Discov. 2018, 8, 150–163. [Google Scholar] [CrossRef]
- Weiss, W.A.; Aldape, K.; Mohapatra, G.; Feuerstein, B.G.; Bishop, J.M. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 1997, 16, 2985–2995. [Google Scholar] [CrossRef]
- Moore, H.C.; Wood, K.M.; Jackson, M.S.; Lastowska, M.A.; Hall, D.; Imrie, H.; Redfern, C.P.; Lovat, P.E.; Ponthan, F.; O’Toole, K.; et al. Histological profile of tumours from MYCN transgenic mice. J. Clin. Pathol. 2008, 61, 1098–1103. [Google Scholar] [CrossRef]
- Teitz, T.; Stanke, J.J.; Federico, S.; Bradley, C.L.; Brennan, R.; Zhang, J.; Johnson, M.D.; Sedlacik, J.; Inoue, M.; Zhang, Z.M.; et al. Preclinical models for neuroblastoma: Establishing a baseline for treatment. PLoS ONE 2011, 6, e19133. [Google Scholar] [CrossRef]
- Hansford, L.M.; Thomas, W.D.; Keating, J.M.; Burkhart, C.A.; Peaston, A.E.; Norris, M.D.; Haber, M.; Armati, P.J.; Weiss, W.A.; Marshall, G.M. Mechanisms of embryonal tumor initiation: Distinct roles for MycN expression and MYCN amplification. Proc. Natl. Acad. Sci. USA 2004, 101, 12664–12669. [Google Scholar] [CrossRef] [PubMed]
- Alam, G.; Cui, H.; Shi, H.; Yang, L.; Ding, J.; Mao, L.; Maltese, W.A.; Ding, H.F. MYCN promotes the expansion of Phox2B-positive neuronal progenitors to drive neuroblastoma development. Am. J. Pathol. 2009, 175, 856–866. [Google Scholar] [CrossRef] [PubMed]
- Pattyn, A.; Morin, X.; Cremer, H.; Goridis, C.; Brunet, J.F. The homeobox gene Phox2b is essential for the development of autonomic neural crest derivatives. Nature 1999, 399, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Althoff, K.; Beckers, A.; Bell, E.; Nortmeyer, M.; Thor, T.; Sprüssel, A.; Lindner, S.; De Preter, K.; Florin, A.; Heukamp, L.C.; et al. A Cre-conditional MYCN-driven neuroblastoma mouse model as an improved tool for preclinical studies. Oncogene 2015, 34, 3357–3368. [Google Scholar] [CrossRef]
- Olsen, R.R.; Otero, J.H.; García-López, J.; Wallace, K.; Finkelstein, D.; Rehg, J.E.; Yin, Z.; Wang, Y.D.; Freeman, K.W. MYCN induces neuroblastoma in primary neural crest cells. Oncogene 2017, 36, 5075–5082. [Google Scholar] [CrossRef]
- Zhu, S.; Lee, J.S.; Guo, F.; Shin, J.; Perez-Atayde, A.R.; Kutok, J.L.; Rodig, S.J.; Neuberg, D.S.; Helman, D.; Feng, H.; et al. Activated ALK collaborates with MYCN in neuroblastoma pathogenesis. Cancer Cell 2012, 21, 362–373. [Google Scholar] [CrossRef]
- Ruiz-Perez, M.V.; Henley, A.B.; Arsenian-Henriksson, M. The MYCN Protein in Health and Disease. Genes 2017, 8, 113. [Google Scholar] [CrossRef]
- Gessi, M.; von Bueren, A.O.; Treszl, A.; Mühlen, A.Z.; Hartmann, W.; Warmuth-Metz, M.; Rutkowski, S.; Pietsch, T. MYCN amplification predicts poor outcome for patients with supratentorial primitive neuroectodermal tumors of the central nervous system. Neuro-Oncol. 2014, 16, 924–932. [Google Scholar] [CrossRef]
- Pfister, S.; Remke, M.; Benner, A.; Mendrzyk, F.; Toedt, G.; Felsberg, J.; Wittmann, A.; Devens, F.; Gerber, N.U.; Joos, S.; et al. Outcome Prediction in Pediatric Medulloblastoma Based on DNA Copy-Number Aberrations of Chromosomes 6q and 17q and the MYC and MYCN Loci. J. Clin. Oncol. 2009, 27, 1627–1636. [Google Scholar] [CrossRef]
- Williams, R.D.; Chagtai, T.; Alcaide-German, M.; Apps, J.; Wegert, J.; Popov, S.; Vujanic, G.; van Tinteren, H.; van den Heuvel-Eibrink, M.M.; Kool, M.; et al. Multiple mechanisms of MYCN dysregulation in Wilms tumour. Oncotarget 2015, 6, 7232–7243. [Google Scholar] [CrossRef] [Green Version]
- Rushlow, D.E.; Mol, B.M.; Kennett, J.Y.; Yee, S.; Pajovic, S.; Theriault, B.L.; Prigoda-Lee, N.L.; Spencer, C.; Dimaras, H.; Corson, T.W.; et al. Characterisation of retinoblastomas without RB1 mutations: Genomic, gene expression, and clinical studies. Lancet Oncol. 2013, 14, 327–334. [Google Scholar] [CrossRef]
- Lee, J.K.; Phillips, J.W.; Smith, B.A.; Park, J.W.; Stoyanova, T.; McCaffrey, E.F.; Baertsch, R.; Sokolov, A.; Meyerowitz, J.G.; Mathis, C.; et al. N-Myc Drives Neuroendocrine Prostate Cancer Initiated from Human Prostate Epithelial Cells. Cancer Cell 2016, 29, 536–547. [Google Scholar] [CrossRef]
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; MacDonald, T.Y.; Wang, Y.; Sheikh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011, 1, 487–495. [Google Scholar] [CrossRef]
- Dardenne, E.; Beltran, H.; Benelli, M.; Gayvert, K.; Berger, A.; Puca, L.; Cyrta, J.; Sboner, A.; Noorzad, Z.; MacDonald, T.; et al. N-Myc Induces an EZH2-Mediated Transcriptional Program Driving Neuroendocrine Prostate Cancer. Cancer Cell 2016, 30, 563–577. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Hodgson, J.G.; Yeh, R.F.; Ray, A.; Wang, N.J.; Smirnov, I.; Yu, M.; Hariono, S.; Silber, J.; Feiler, H.S.; Gray, J.W.; et al. Comparative analyses of gene copy number and mRNA expression in glioblastoma multiforme tumors and xenografts. Neuro-Oncol. 2009, 11, 477–487. [Google Scholar] [CrossRef]
- Kim, Y.H.; Girard, L.; Giacomini, C.P.; Wang, P.; Hernandez-Boussard, T.; Tibshirani, R.; Minna, J.D.; Pollack, J.R. Combined microarray analysis of small cell lung cancer reveals altered apoptotic balance and distinct expression signatures of MYC family gene amplification. Oncogene 2006, 25, 130–138. [Google Scholar] [CrossRef]
- Nau, M.M.; Brooks, B.J., Jr.; Carney, D.N.; Gazdar, A.F.; Battey, J.F.; Sausville, E.A.; Minna, J.D. Human small-cell lung cancers show amplification and expression of the N-myc gene. Proc. Natl. Acad. Sci. USA 1986, 83, 1092–1096. [Google Scholar] [CrossRef]
- Sos, M.L.; Dietlein, F.; Peifer, M.; Schottle, J.; Balke-Want, H.; Muller, C.; Koker, M.; Richters, A.; Heynck, S.; Malchers, F.; et al. A framework for identification of actionable cancer genome dependencies in small cell lung cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 17034–17039. [Google Scholar] [CrossRef]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Weiss, W.A. Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 2013, 3, a014415. [Google Scholar] [CrossRef]
- Charron, J.; Malynn, B.A.; Fisher, P.; Stewart, V.; Jeannotte, L.; Goff, S.P.; Robertson, E.J.; Alt, F.W. Embryonic lethality in mice homozygous for a targeted disruption of the N-myc gene. Genes Dev. 1992, 6, 2248–2257. [Google Scholar] [CrossRef]
- Davis, A.C.; Wims, M.; Spotts, G.D.; Hann, S.R.; Bradley, A. A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev. 1993, 7, 671–682. [Google Scholar] [CrossRef]
- Sawai, S.; Shimono, A.; Wakamatsu, Y.; Palmes, C.; Hanaoka, K.; Kondoh, H. Defects of embryonic organogenesis resulting from targeted disruption of the N-myc gene in the mouse. Development 1993, 117, 1445–1455. [Google Scholar] [CrossRef]
- Downs, K.M.; Martin, G.R.; Bishop, J.M. Contrasting patterns of myc and N-myc expression during gastrulation of the mouse embryo. Genes Dev. 1989, 3, 860–869. [Google Scholar] [CrossRef]
- Zimmerman, K.A.; Yancopoulos, G.D.; Collum, R.G.; Smith, R.K.; Kohl, N.E.; Denis, K.A.; Nau, M.M.; Witte, O.N.; Toran-Allerand, D.; Gee, C.E.; et al. Differential expression of myc family genes during murine development. Nature 1986, 319, 780–783. [Google Scholar] [CrossRef]
- Malynn, B.A.; de Alboran, I.M.; O′Hagan, R.C.; Bronson, R.; Davidson, L.; DePinho, R.A.; Alt, F.W. N-myc can functionally replace c-myc in murine development, cellular growth, and differentiation. Genes Dev. 2000, 14, 1390–1399. [Google Scholar] [CrossRef]
- Grandori, C.; Cowley, S.M.; James, L.P.; Eisenman, R.N. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu. Rev. Cell Dev. Biol. 2000, 16, 653–699. [Google Scholar] [CrossRef]
- Cavalieri, F.; Goldfarb, M. N-myc proto-oncogene expression can induce DNA replication in Balb/c 3T3 fibroblasts. Oncogene 1988, 2, 289–291. [Google Scholar]
- Lutz, W.; Stöhr, M.; Schürmann, J.; Wenzel, A.; Löhr, A.; Schwab, M. Conditional expression of N-myc in human neuroblastoma cells increases expression of alpha-prothymosin and ornithine decarboxylase and accelerates progression into S-phase early after mitogenic stimulation of quiescent cells. Oncogene 1996, 13, 803–812. [Google Scholar]
- Bell, E.; Premkumar, R.; Carr, J.; Lu, X.; Lovat, P.E.; Kees, U.R.; Lunec, J.; Tweddle, D.A. The role of MYCN in the failure of MYCN amplified neuroblastoma cell lines to G1 arrest after DNA damage. Cell Cycle 2006, 5, 2639–2647. [Google Scholar] [CrossRef] [PubMed]
- Hatton, B.A.; Knoepfler, P.S.; Kenney, A.M.; Rowitch, D.H.; de Alborán, I.M.; Olson, J.M.; Eisenman, R.N. N-myc Is an Essential Downstream Effector of Shh Signaling during both Normal and Neoplastic Cerebellar Growth. Cancer Res. 2006, 66, 8655–8661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knoepfler, P.S.; Cheng, P.F.; Eisenman, R.N. N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes Dev. 2002, 16, 2699–2712. [Google Scholar] [CrossRef] [PubMed]
- Kress, T.R.; Sabo, A.; Amati, B. MYC: Connecting selective transcriptional control to global RNA production. Nat. Rev. Cancer 2015, 15, 593–607. [Google Scholar] [CrossRef]
- Blackwell, T.K.; Huang, J.; Ma, A.; Kretzner, L.; Alt, F.W.; Eisenman, R.N.; Weintraub, H. Binding of myc proteins to canonical and noncanonical DNA sequences. Mol. Cell Biol. 1993, 13, 5216–5224. [Google Scholar] [CrossRef]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef]
- Chipumuro, E.; Marco, E.; Christensen, C.L.; Kwiatkowski, N.; Zhang, T.; Hatheway, C.M.; Abraham, B.J.; Sharma, B.; Yeung, C.; Altabef, A.; et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell 2014, 159, 1126–1139. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Warburg, O. The Metabolism of Carcinoma Cells. J. Cancer Res. 1925, 9, 148–163. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef]
- Shulkin, B.L.; Mitchell, D.S.; Ungar, D.R.; Prakash, D.; Dole, M.G.; Castle, V.P.; Hernandez, R.J.; Koeppe, R.A.; Hutchinson, R.J. Neoplasms in a pediatric population: 2-[F-18]-fluoro-2-deoxy-D-glucose PET studies. Radiology 1995, 194, 495–500. [Google Scholar] [CrossRef]
- Levy, A.G.; Zage, P.E.; Akers, L.J.; Ghisoli, M.L.; Chen, Z.; Fang, W.; Kannan, S.; Graham, T.; Zeng, L.; Franklin, A.R.; et al. The combination of the novel glycolysis inhibitor 3-BrOP and rapamycin is effective against neuroblastoma. Investig. New Drugs 2012, 30, 191–199. [Google Scholar] [CrossRef]
- Qing, G.; Skuli, N.; Mayes, P.A.; Pawel, B.; Martinez, D.; Maris, J.M.; Simon, M.C. Combinatorial Regulation of Neuroblastoma Tumor Progression by N-Myc and Hypoxia Inducible Factor HIF-1α. Cancer Res. 2010, 70, 10351–10361. [Google Scholar] [CrossRef]
- Oliynyk, G.; Ruiz-Perez, M.V.; Sainero-Alcolado, L.; Dzieran, J.; Zirath, H.; Gallart-Ayala, H.; Wheelock, C.E.; Johansson, H.J.; Nilsson, R.; Lehtio, J.; et al. MYCN-enhanced Oxidative and Glycolytic Metabolism Reveals Vulnerabilities for Targeting Neuroblastoma. iScience 2019, 21, 188–204. [Google Scholar] [CrossRef]
- Tjaden, B.; Baum, K.; Marquardt, V.; Simon, M.; Trajkovic-Arsic, M.; Kouril, T.; Siebers, B.; Lisec, J.; Siveke, J.T.; Schulte, J.H.; et al. N-Myc-induced metabolic rewiring creates novel therapeutic vulnerabilities in neuroblastoma. Sci. Rep. 2020, 10, 7157. [Google Scholar] [CrossRef]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, Metabolism, and Cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef]
- Li, H.; Yang, F.; Hu, A.; Wang, X.; Fang, E.; Chen, Y.; Li, D.; Song, H.; Wang, J.; Guo, Y.; et al. Therapeutic targeting of circ-CUX1/EWSR1/MAZ axis inhibits glycolysis and neuroblastoma progression. EMBO Mol. Med. 2019, 11, e10835. [Google Scholar] [CrossRef]
- Song, H.; Li, D.; Wang, X.; Fang, E.; Yang, F.; Hu, A.; Wang, J.; Guo, Y.; Liu, Y.; Li, H.; et al. HNF4A-AS1/hnRNPU/CTCF axis as a therapeutic target for aerobic glycolysis and neuroblastoma progression. J. Hematol. Oncol. 2020, 13, 24. [Google Scholar] [CrossRef]
- Markert, C.L. Lactate dehydrogenase. Biochemistry and function of lactate dehydrogenase. Cell Biochem. Funct. 1984, 2, 131–134. [Google Scholar] [CrossRef]
- Storey, K.B. Comparative enzymology—New insights from studies of an “old” enzyme, lactate dehydrogenase. Comp. Biochem. Physiol. Part. B Biochem. Mol. Biol. 2016, 199, 13–20. [Google Scholar] [CrossRef]
- Dorneburg, C.; Fischer, M.; Barth, T.F.E.; Mueller-Klieser, W.; Hero, B.; Gecht, J.; Carter, D.R.; de Preter, K.; Mayer, B.; Christner, L.; et al. LDHA in Neuroblastoma Is Associated with Poor Outcome and Its Depletion Decreases Neuroblastoma Growth Independent of Aerobic Glycolysis. Clin. Cancer Res. 2018, 24, 5772–5783. [Google Scholar] [CrossRef] [Green Version]
- Bhutia, Y.D.; Babu, E.; Ramachandran, S.; Ganapathy, V. Amino Acid Transporters in Cancer and Their Relevance to “Glutamine Addiction”: Novel Targets for the Design of a New Class of Anticancer Drugs. Cancer Res. 2015, 75, 1782–1788. [Google Scholar] [CrossRef]
- Yue, M.; Jiang, J.; Gao, P.; Liu, H.; Qing, G. Oncogenic MYC Activates a Feedforward Regulatory Loop Promoting Essential Amino Acid Metabolism and Tumorigenesis. Cell Rep. 2017, 21, 3819–3832. [Google Scholar] [CrossRef]
- Choi, B.-H.; Coloff, J.L. The Diverse Functions of Non-Essential Amino Acids in Cancer. Cancers 2019, 11, 675. [Google Scholar] [CrossRef]
- Kilberg, M.S.; Shan, J.; Su, N. ATF4-dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol. Metab. 2009, 20, 436–443. [Google Scholar] [CrossRef]
- Ameri, K.; Harris, A.L. Activating transcription factor 4. Int. J. Biochem. Cell Biol. 2008, 40, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef]
- Zhao, E.; Ding, J.; Xia, Y.; Liu, M.; Ye, B.; Choi, J.H.; Yan, C.; Dong, Z.; Huang, S.; Zha, Y.; et al. KDM4C and ATF4 Cooperate in Transcriptional Control of Amino Acid Metabolism. Cell Rep. 2016, 14, 506–519. [Google Scholar] [CrossRef]
- Xia, Y.; Ye, B.; Ding, J.; Yu, Y.; Alptekin, A.; Thangaraju, M.; Prasad, P.D.; Ding, Z.C.; Park, E.J.; Choi, J.H.; et al. Metabolic Reprogramming by MYCN Confers Dependence on the Serine-Glycine-One-Carbon Biosynthetic Pathway. Cancer Res. 2019, 79, 3837–3850. [Google Scholar] [CrossRef]
- Ding, J.; Li, T.; Wang, X.; Zhao, E.; Choi, J.H.; Yang, L.; Zha, Y.; Dong, Z.; Huang, S.; Asara, J.M.; et al. The histone H3 methyltransferase G9A epigenetically activates the serine-glycine synthesis pathway to sustain cancer cell survival and proliferation. Cell Metab. 2013, 18, 896–907. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Xia, Y.; Ding, J.; Ye, B.; Zhao, E.; Choi, J.H.; Alptekin, A.; Yan, C.; Dong, Z.; Huang, S.; et al. Transcriptional Profiling Reveals a Common Metabolic Program in High-Risk Human Neuroblastoma and Mouse Neuroblastoma Sphere-Forming Cells. Cell Rep. 2016, 17, 609–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eagle, H.; Oyama, V.I.; Levy, M.; Horton, C.L.; Fleischman, R. The growth response of mammalian cells in tissue culture to L-glutamine and L-glutamic acid. J. Biol. Chem. 1956, 218, 607–616. [Google Scholar] [CrossRef]
- Kovacevic, Z.; McGivan, J.D. Mitochondrial metabolism of glutamine and glutamate and its physiological significance. Physiol. Rev. 1983, 63, 547–605. [Google Scholar] [CrossRef]
- Baggetto, L.G. Deviant energetic metabolism of glycolytic cancer cells. Biochimie 1992, 74, 959–974. [Google Scholar] [CrossRef]
- Souba, W.W. Glutamine and cancer. Ann. Surg. 1993, 218, 715–728. [Google Scholar] [CrossRef]
- Smith, R.J.; Wilmore, D.W. Glutamine nutrition and requirements. J. Parenter. Enter. Nutr. 1990, 14, 94S–99S. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Cheng, T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef]
- Yang, L.; Venneti, S.; Nagrath, D. Glutaminolysis: A Hallmark of Cancer Metabolism. Annu. Rev. Biomed. Eng. 2017, 19, 163–194. [Google Scholar] [CrossRef]
- Zhang, J.; Pavlova, N.N.; Thompson, C.B. Cancer cell metabolism: The essential role of the nonessential amino acid, glutamine. EMBO J. 2017, 36, 1302–1315. [Google Scholar] [CrossRef]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef]
- Qing, G.; Li, B.; Vu, A.; Skuli, N.; Walton, Z.E.; Liu, X.; Mayes, P.A.; Wise, D.R.; Thompson, C.B.; Maris, J.M.; et al. ATF4 regulates MYC-mediated neuroblastoma cell death upon glutamine deprivation. Cancer Cell 2012, 22, 631–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, P.; Yue, M.; Xiao, D.; Xiu, R.; Gan, L.; Liu, H.; Qing, G. ATF4 and N-Myc coordinate glutamine metabolism in MYCN-amplified neuroblastoma cells through ASCT2 activation. J. Pathol. 2015, 235, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Liu, L.; Chen, X.; Shen, Y.; Lian, G.; Shah, N.; Davidoff, A.M.; Yang, J.; Wang, R. MYCN drives glutaminolysis in neuroblastoma and confers sensitivity to an ROS augmenting agent. Cell Death Dis. 2018, 9, 220. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Ren, P.; Su, H.; Yue, M.; Xiu, R.; Hu, Y.; Liu, H.; Qing, G. Myc promotes glutaminolysis in human neuroblastoma through direct activation of glutaminase 2. Oncotarget 2015, 6, 40655–40666. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Ding, J.; Zhu, S.; Alptekin, A.; Dong, Z.; Yan, C.; Zha, Y.; Ding, H.-F. Therapeutic targeting of both dihydroorotate dehydrogenase and nucleoside transport in MYCN-amplified neuroblastoma. Cell Death Dis. 2021, 12, 821. [Google Scholar] [CrossRef]
- Lockart, R.Z., Jr.; Eagle, H. Requirements for growth of single human cells. Science 1959, 129, 252–254. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Vousden, K.H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 2016, 16, 650–662. [Google Scholar] [CrossRef]
- Locasale, J.W. Serine, glycine and one-carbon units: Cancer metabolism in full circle. Nat. Rev. Cancer 2013, 13, 572–583. [Google Scholar] [CrossRef]
- Reina-Campos, M.; Diaz-Meco, M.T.; Moscat, J. The complexity of the serine glycine one-carbon pathway in cancer. J. Cell Biol. 2019, 219, e201907022. [Google Scholar] [CrossRef]
- Geeraerts, S.L.; Heylen, E.; De Keersmaecker, K.; Kampen, K.R. The ins and outs of serine and glycine metabolism in cancer. Nat. Metab. 2021, 3, 131–141. [Google Scholar] [CrossRef]
- Shinkai, Y.; Tachibana, M. H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev. 2011, 25, 781–788. [Google Scholar] [CrossRef] [Green Version]
- Peters, A.H.F.M.; Kubicek, S.; Mechtler, K.; O’Sullivan, R.J.; Derijck, A.A.H.A.; Perez-Burgos, L.; Kohlmaier, A.; Opravil, S.; Tachibana, M.; Shinkai, Y.; et al. Partitioning and Plasticity of Repressive Histone Methylation States in Mammalian Chromatin. Mol. Cell 2003, 12, 1577–1589. [Google Scholar] [CrossRef]
- Rice, J.C.; Briggs, S.D.; Ueberheide, B.; Barber, C.M.; Shabanowitz, J.; Hunt, D.F.; Shinkai, Y.; Allis, C.D. Histone Methyltransferases Direct Different Degrees of Methylation to Define Distinct Chromatin Domains. Mol. Cell 2003, 12, 1591–1598. [Google Scholar] [CrossRef]
- Tachibana, M.; Sugimoto, K.; Nozaki, M.; Ueda, J.; Ohta, T.; Ohki, M.; Fukuda, M.; Takeda, N.; Niida, H.; Kato, H.; et al. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 2002, 16, 1779–1791. [Google Scholar] [CrossRef]
- Mosammaparast, N.; Shi, Y. Reversal of histone methylation: Biochemical and molecular mechanisms of histone demethylases. Annu. Rev. Biochem. 2010, 79, 155–179. [Google Scholar] [CrossRef]
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone lysine methylation dynamics: Establishment, regulation, and biological impact. Mol. Cell 2012, 48, 491–507. [Google Scholar] [CrossRef]
- Berry, W.L.; Janknecht, R. KDM4/JMJD2 Histone Demethylases: Epigenetic Regulators in Cancer Cells. Cancer Res. 2013, 73, 2936–2942. [Google Scholar] [CrossRef]
- Labuschagne, C.F.; van den Broek, N.J.; Mackay, G.M.; Vousden, K.H.; Maddocks, O.D. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep. 2014, 7, 1248–1258. [Google Scholar] [CrossRef]
- Kim, D.; Fiske, B.P.; Birsoy, K.; Freinkman, E.; Kami, K.; Possemato, R.L.; Chudnovsky, Y.; Pacold, M.E.; Chen, W.W.; Cantor, J.R.; et al. SHMT2 drives glioma cell survival in ischaemia but imposes a dependence on glycine clearance. Nature 2015, 520, 363–367. [Google Scholar] [CrossRef]
- Alptekin, A.; Ye, B.; Yu, Y.; Poole, C.J.; van Riggelen, J.; Zha, Y.; Ding, H.F. Glycine decarboxylase is a transcriptional target of MYCN required for neuroblastoma cell proliferation and tumorigenicity. Oncogene 2019, 38, 7504–7520. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wu, Z.; Dai, Z.; Yang, Y.; Wang, J.; Wu, G. Glycine metabolism in animals and humans: Implications for nutrition and health. Amino Acids 2013, 45, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Tibbetts, A.S.; Appling, D.R. Compartmentalization of Mammalian folate-mediated one-carbon metabolism. Annu. Rev. Nutr. 2010, 30, 57–81. [Google Scholar] [CrossRef] [PubMed]
- Narisawa, A.; Komatsuzaki, S.; Kikuchi, A.; Niihori, T.; Aoki, Y.; Fujiwara, K.; Tanemura, M.; Hata, A.; Suzuki, Y.; Relton, C.L.; et al. Mutations in genes encoding the glycine cleavage system predispose to neural tube defects in mice and humans. Hum. Mol. Genet. 2012, 21, 1496–1503. [Google Scholar] [CrossRef]
- Conter, C.; Rolland, M.O.; Cheillan, D.; Bonnet, V.; Maire, I.; Froissart, R. Genetic heterogeneity of the GLDC gene in 28 unrelated patients with glycine encephalopathy. J. Inherit. Metab. Dis. 2006, 29, 135–142. [Google Scholar] [CrossRef]
- Pai, Y.J.; Leung, K.Y.; Savery, D.; Hutchin, T.; Prunty, H.; Heales, S.; Brosnan, M.E.; Brosnan, J.T.; Copp, A.J.; Greene, N.D. Glycine decarboxylase deficiency causes neural tube defects and features of non-ketotic hyperglycinemia in mice. Nat. Commun. 2015, 6, 6388. [Google Scholar] [CrossRef]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and Thioredoxin Antioxidant Pathways Synergize to Drive Cancer Initiation and Progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef]
- Bonifácio, V.D.B.; Pereira, S.A.; Serpa, J.; Vicente, J.B. Cysteine metabolic circuitries: Druggable targets in cancer. Br. J. Cancer 2021, 124, 862–879. [Google Scholar] [CrossRef]
- Bannai, S. Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J. Biol. Chem. 1986, 261, 2256–2263. [Google Scholar] [CrossRef]
- Sato, H.; Tamba, M.; Ishii, T.; Bannai, S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J. Biol. Chem. 1999, 274, 11455–11458. [Google Scholar] [CrossRef]
- Floros, K.V.; Cai, J.; Jacob, S.; Kurupi, R.; Fairchild, C.K.; Shende, M.; Coon, C.M.; Powell, K.M.; Belvin, B.R.; Hu, B.; et al. MYCN-Amplified Neuroblastoma Is Addicted to Iron and Vulnerable to Inhibition of the System Xc-/Glutathione Axis. Cancer Res. 2021, 81, 1896–1908. [Google Scholar] [CrossRef]
- Alborzinia, H.; Florez, A.F.; Kreth, S.; Bruckner, L.M.; Yildiz, U.; Gartlgruber, M.; Odoni, D.I.; Poschet, G.; Garbowicz, K.; Shao, C.; et al. MYCN mediates cysteine addiction and sensitizes neuroblastoma to ferroptosis. Nat. Cancer 2022, 3, 471–485. [Google Scholar] [CrossRef]
- Fasullo, M.; Endres, L. Nucleotide salvage deficiencies, DNA damage and neurodegeneration. Int. J. Mol. Sci. 2015, 16, 9431–9449. [Google Scholar] [CrossRef]
- Pedley, A.M.; Benkovic, S.J. A New View into the Regulation of Purine Metabolism: The Purinosome. Trends Biochem. Sci. 2017, 42, 141–154. [Google Scholar] [CrossRef]
- Lane, A.N.; Fan, T.W. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 2015, 43, 2466–2485. [Google Scholar] [CrossRef]
- Hartman, S.C.; Buchanan, J.M. The Biosynthesis of the Purines. In Ergebnisse der Physiologie Biologischen Chemie und Experimentellen Pharmakologie. Ergebnisse der Physiologie, Biologischen Chemie und Experimentellen Pharmakologie; Springer: Berlin/Heidelberg, Germany, 1959; Volume 50, pp. 75–121. [Google Scholar] [CrossRef]
- Cheung, C.H.Y.; Hsu, C.L.; Tsuei, C.Y.; Kuo, T.T.; Huang, C.T.; Hsu, W.M.; Chung, Y.H.; Wu, H.Y.; Hsu, C.C.; Huang, H.C.; et al. Combinatorial targeting of MTHFD2 and PAICS in purine synthesis as a novel therapeutic strategy. Cell Death Dis. 2019, 10, 786. [Google Scholar] [CrossRef]
- Evans, D.R.; Guy, H.I. Mammalian Pyrimidine Biosynthesis: Fresh Insights into an Ancient Pathway. J. Biol. Chem. 2004, 279, 33035–33038. [Google Scholar] [CrossRef]
- Okesli, A.; Khosla, C.; Bassik, M.C. Human pyrimidine nucleotide biosynthesis as a target for antiviral chemotherapy. Curr. Opin. Biotechnol. 2017, 48, 127–134. [Google Scholar] [CrossRef]
- Jennings, W.; Epand, R.M. CDP-diacylglycerol, a critical intermediate in lipid metabolism. Chem. Phys. Lipids 2020, 230, 104914. [Google Scholar] [CrossRef]
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab. 2020, 32, 341–352. [Google Scholar] [CrossRef]
- Röhrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef]
- Kazantzis, M.; Stahl, A. Fatty acid transport proteins, implications in physiology and disease. Biochim. Biophys. Acta—Mol. Cell Biol. Lipids 2012, 1821, 852–857. [Google Scholar] [CrossRef]
- Gimeno, R.E. Fatty acid transport proteins. Curr. Opin. Lipidol. 2007, 18, 271–276. [Google Scholar] [CrossRef]
- Tao, L.; Mohammad, M.A.; Milazzo, G.; Moreno-Smith, M.; Patel, T.D.; Zorman, B.; Badachhape, A.; Hernandez, B.E.; Wolf, A.B.; Zeng, Z.; et al. MYCN-driven fatty acid uptake is a metabolic vulnerability in neuroblastoma. Nat. Commun. 2022, 13, 3728. [Google Scholar] [CrossRef]
- Comerford, S.A.; Huang, Z.; Du, X.; Wang, Y.; Cai, L.; Witkiewicz, A.K.; Walters, H.; Tantawy, M.N.; Fu, A.; Manning, H.C.; et al. Acetate dependence of tumors. Cell 2014, 159, 1591–1602. [Google Scholar] [CrossRef]
- Mashimo, T.; Pichumani, K.; Vemireddy, V.; Hatanpaa, K.J.; Singh, D.K.; Sirasanagandla, S.; Nannepaga, S.; Piccirillo, S.G.; Kovacs, Z.; Foong, C.; et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 2014, 159, 1603–1614. [Google Scholar] [CrossRef]
- Pietrocola, F.; Galluzzi, L.; Pedro, J.M.B.-S.; Madeo, F.; Kroemer, G. Acetyl coenzyme A: A central metabolite and second messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef]
- Schug, Z.T.; Voorde, J.V.; Gottlieb, E. The metabolic fate of acetate in cancer. Nat. Rev. Cancer 2016, 16, 708–717. [Google Scholar] [CrossRef]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Carroll, P.A.; Diolaiti, D.; McFerrin, L.; Gu, H.; Djukovic, D.; Du, J.; Cheng, P.F.; Anderson, S.; Ulrich, M.; Hurley, J.B.; et al. Deregulated Myc Requires MondoA/Mlx for Metabolic Reprogramming and Tumorigenesis. Cancer Cell 2015, 27, 271–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, C.V. Web of the Extended Myc Network Captures Metabolism for Tumorigenesis. Cancer Cell 2015, 27, 160–162. [Google Scholar] [CrossRef] [PubMed]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Rinaldo, P.; Matern, D.; Bennett, M.J. Fatty acid oxidation disorders. Annu. Rev. Physiol. 2002, 64, 477–502. [Google Scholar] [CrossRef]
- Bartlett, K.; Eaton, S. Mitochondrial beta-oxidation. Eur. J. Biochem. 2004, 271, 462–469. [Google Scholar] [CrossRef]
- Zirath, H.; Frenzel, A.; Oliynyk, G.; Segerström, L.; Westermark, U.K.; Larsson, K.; Persson, M.M.; Hultenby, K.; Lehtiö, J.; Einvik, C.; et al. MYC inhibition induces metabolic changes leading to accumulation of lipid droplets in tumor cells. Proc. Natl. Acad. Sci. USA 2013, 110, 10258–10263. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. Regulation of the mevalonate pathway. Nature 1990, 343, 425–430. [Google Scholar] [CrossRef]
- Mullen, P.J.; Yu, R.; Longo, J.; Archer, M.C.; Penn, L.Z. The interplay between cell signalling and the mevalonate pathway in cancer. Nat. Rev. Cancer 2016, 16, 718–731. [Google Scholar] [CrossRef]
- McTaggart, S.J. Isoprenylated proteins. Cell Mol. Life Sci. 2006, 63, 255–267. [Google Scholar] [CrossRef]
- Stefely, J.A.; Pagliarini, D.J. Biochemistry of Mitochondrial Coenzyme Q Biosynthesis. Trends Biochem. Sci. 2017, 42, 824–843. [Google Scholar] [CrossRef]
- Hederstedt, L. Heme A biosynthesis. Biochim. Biophys. Acta 2012, 1817, 920–927. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, J.L.; DeBose-Boyd, R.A.; Brown, M.S. Protein Sensors for Membrane Sterols. Cell 2006, 124, 35–46. [Google Scholar] [CrossRef]
- Espenshade, P.J.; Hughes, A.L. Regulation of sterol synthesis in eukaryotes. Annu. Rev. Genet. 2007, 41, 401–427. [Google Scholar] [CrossRef]
- Rawson, R.B. The SREBP pathway—Insights from Insigs and insects. Nat. Rev. Mol. Cell Biol. 2003, 4, 631–640. [Google Scholar] [CrossRef]
- Harayama, T.; Riezman, H. Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Biol. 2018, 19, 281–296. [Google Scholar] [CrossRef]
- Gruenbacher, G.; Thurnher, M. Mevalonate metabolism in cancer. Cancer Lett. 2015, 356, 192–196. [Google Scholar] [CrossRef]
- Silvente-Poirot, S.; Poirot, M. Cancer. Cholesterol and cancer, in the balance. Science 2014, 343, 1445–1446. [Google Scholar] [CrossRef]
- Moon, S.-H.; Huang, C.-H.; Houlihan, S.L.; Regunath, K.; Freed-Pastor, W.A.; Morris, J.P.; Tschaharganeh, D.F.; Kastenhuber, E.R.; Barsotti, A.M.; Culp-Hill, R.; et al. p53 Represses the Mevalonate Pathway to Mediate Tumor Suppression. Cell 2019, 176, 564–580.e19. [Google Scholar] [CrossRef]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef]
- Longo, J.; van Leeuwen, J.E.; Elbaz, M.; Branchard, E.; Penn, L.Z. Statins as Anticancer Agents in the Era of Precision Medicine. Clin. Cancer Res. 2020, 26, 5791–5800. [Google Scholar] [CrossRef]
- Pott, S.; Lieb, J.D. What are super-enhancers? Nat. Genet. 2015, 47, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; George, R.E. Super-Enhancer-Driven Transcriptional Dependencies in Cancer. Trends Cancer 2017, 3, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Gartlgruber, M.; Sharma, A.K.; Quintero, A.; Dreidax, D.; Jansky, S.; Park, Y.-G.; Kreth, S.; Meder, J.; Doncevic, D.; Saary, P.; et al. Super enhancers define regulatory subtypes and cell identity in neuroblastoma. Nat. Cancer 2021, 2, 114–128. [Google Scholar] [CrossRef] [PubMed]
- van Groningen, T.; Koster, J.; Valentijn, L.J.; Zwijnenburg, D.A.; Akogul, N.; Hasselt, N.E.; Broekmans, M.; Haneveld, F.; Nowakowska, N.E.; Bras, J.; et al. Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nat. Genet. 2017, 49, 1261–1266. [Google Scholar] [CrossRef]
- Olsen, R.R.; Mary-Sinclair, M.N.; Yin, Z.; Freeman, K.W. Antagonizing Bcl-2 family members sensitizes neuroblastoma and Ewing’s sarcoma to an inhibitor of glutamine metabolism. PLoS ONE 2015, 10, e0116998. [Google Scholar] [CrossRef]
- Anderson, N.M.; Qin, X.; Finan, J.M.; Lam, A.; Athoe, J.; Missiaen, R.; Skuli, N.; Kennedy, A.; Saini, A.S.; Tao, T.; et al. Metabolic Enzyme DLST Promotes Tumor Aggression and Reveals a Vulnerability to OXPHOS Inhibition in High-Risk Neuroblastoma. Cancer Res. 2021, 81, 4417–4430. [Google Scholar] [CrossRef]
- Laszlo, J.; Humphreys, S.R.; Goldin, A. Effects of glucose analogues (2-deoxy-D-glucose, 2-deoxy-D-galactose) on experimental tumors. J. Natl. Cancer Inst. 1960, 24, 267–281. [Google Scholar]
- Landau, B.R.; Laszlo, J.; Stengle, J.; Burk, D. Certain metabolic and pharmacologic effects in cancer patients given infusions of 2-deoxy-D-glucose. J. Natl. Cancer Inst. 1958, 21, 485–494. [Google Scholar]
- Zheng, M.; Wu, C.; Yang, K.; Yang, Y.; Liu, Y.; Gao, S.; Wang, Q.; Li, C.; Chen, L.; Li, H. Novel selective hexokinase 2 inhibitor Benitrobenrazide blocks cancer cells growth by targeting glycolysis. Pharmacol. Res. 2021, 164, 105367. [Google Scholar] [CrossRef]
- Purkey, H.E.; Robarge, K.; Chen, J.; Chen, Z.; Corson, L.B.; Ding, C.Z.; DiPasquale, A.G.; Dragovich, P.S.; Eigenbrot, C.; Evangelista, M.; et al. Cell Active Hydroxylactam Inhibitors of Human Lactate Dehydrogenase with Oral Bioavailability in Mice. ACS Med. Chem. Lett. 2016, 7, 896–901. [Google Scholar] [CrossRef]
- Rellinger, E.J.; Craig, B.T.; Alvarez, A.L.; Dusek, H.L.; Kim, K.W.; Qiao, J.; Chung, D.H. FX11 inhibits aerobic glycolysis and growth of neuroblastoma cells. Surgery 2017, 161, 747–752. [Google Scholar] [CrossRef]
- Deck, L.M.; Royer, R.E.; Chamblee, B.B.; Hernandez, V.M.; Malone, R.R.; Torres, J.E.; Hunsaker, L.A.; Piper, R.C.; Makler, M.T.; Jagt, D.L.V. Selective Inhibitors of Human Lactate Dehydrogenases and Lactate Dehydrogenase from the Malarial Parasite Plasmodium falciparum. J. Med. Chem. 1998, 41, 3879–3887. [Google Scholar] [CrossRef]
- Manerba, M.; Vettraino, M.; Fiume, L.; Di Stefano, G.; Sartini, A.; Giacomini, E.; Buonfiglio, R.; Roberti, M.; Recanatini, M. Galloflavin (CAS 568-80-9): A novel inhibitor of lactate dehydrogenase. ChemMedChem 2012, 7, 311–317. [Google Scholar] [CrossRef]
- Lukey, M.J.; Cluntun, A.A.; Katt, W.P.; Lin, M.-c.J.; Druso, J.E.; Ramachandran, S.; Erickson, J.W.; Le, H.H.; Wang, Z.-E.; Blank, B.; et al. Liver-Type Glutaminase GLS2 Is a Druggable Metabolic Node in Luminal-Subtype Breast Cancer. Cell Rep. 2019, 29, 76–88.e7. [Google Scholar] [CrossRef]
- Pacold, M.E.; Brimacombe, K.R.; Chan, S.H.; Rohde, J.M.; Lewis, C.A.; Swier, L.J.; Possemato, R.; Chen, W.W.; Sullivan, L.B.; Fiske, B.P.; et al. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat. Chem. Biol. 2016, 12, 452–458. [Google Scholar] [CrossRef]
- Mullarky, E.; Lucki, N.C.; Zavareh, R.B.; Anglin, J.L.; Gomes, A.P.; Nicolay, B.N.; Wong, J.C.Y.; Christen, S.; Takahashi, H.; Singh, P.K.; et al. Identification of a small molecule inhibitor of 3-phosphoglycerate dehydrogenase to target serine biosynthesis in cancers. Proc. Natl. Acad. Sci. USA 2016, 113, 1778–1783. [Google Scholar] [CrossRef]
- Wang, Q.; Liberti, M.V.; Liu, P.; Deng, X.; Liu, Y.; Locasale, J.W.; Lai, L. Rational Design of Selective Allosteric Inhibitors of PHGDH and Serine Synthesis with Anti-tumor Activity. Cell Chem. Biol. 2017, 24, 55–65. [Google Scholar] [CrossRef]
- Rodriguez, A.E.; Ducker, G.S.; Billingham, L.K.; Martinez, C.A.; Mainolfi, N.; Suri, V.; Friedman, A.; Manfredi, M.G.; Weinberg, S.E.; Rabinowitz, J.D.; et al. Serine Metabolism Supports Macrophage IL-1β Production. Cell Metab. 2019, 29, 1003–1011.e4. [Google Scholar] [CrossRef]
- García-Cañaveras, J.C.; Lancho, O.; Ducker, G.S.; Ghergurovich, J.M.; Xu, X.; da Silva-Diz, V.; Minuzzo, S.; Indraccolo, S.; Kim, H.; Herranz, D.; et al. SHMT inhibition is effective and synergizes with methotrexate in T-cell acute lymphoblastic leukemia. Leukemia 2021, 35, 377–388. [Google Scholar] [CrossRef]
- Dekhne, A.S.; Shah, K.; Ducker, G.S.; Katinas, J.M.; Wong-Roushar, J.; Nayeen, M.J.; Doshi, A.; Ning, C.; Bao, X.; Frühauf, J.; et al. Novel Pyrrolo[3,2-d]pyrimidine Compounds Target Mitochondrial and Cytosolic One-Carbon Metabolism with Broad-spectrum Antitumor Efficacy. Mol. Cancer Ther. 2019, 18, 1787–1799. [Google Scholar] [CrossRef]
- Pinkus, L.M. Glutamine binding sites. Methods Enzymol. 1977, 46, 414–427. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, Y.; Lian, X.; Li, F.; Wang, C.; Zhu, F.; Qiu, Y.; Chen, Y. Therapeutic target database update 2022: Facilitating drug discovery with enriched comparative data of targeted agents. Nucleic Acids Res. 2022, 50, D1398–D1407. [Google Scholar] [CrossRef] [PubMed]
- Jayaram, H.N.; Dion, R.L.; Glazer, R.I.; Johns, D.G.; Robins, R.K.; Srivastava, P.C.; Cooney, D.A. Initial studies on the mechanism of action of a new oncolytic thiazole nucleoside, 2-β-d-ribofuranosylthiazole-4-carboxamide NSC 286193). Biochem. Pharmacol. 1982, 31, 2371–2380. [Google Scholar] [CrossRef]
- Murase, M.; Iwamura, H.; Komatsu, K.; Saito, M.; Maekawa, T.; Nakamura, T.; Yokokawa, T.; Shimada, Y. Lack of cross-resistance to FF-10501, an inhibitor of inosine-5′-monophosphate dehydrogenase, in azacitidine-resistant cell lines selected from SKM-1 and MOLM-13 leukemia cell lines. Pharmacol. Res. Perspect. 2016, 4, e00206. [Google Scholar] [CrossRef]
- Davis, J.P.; Cain, G.A.; Pitts, W.J.; Magolda, R.L.; Copeland, R.A. The immunosuppressive metabolite of leflunomide is a potent inhibitor of human dihydroorotate dehydrogenase. Biochemistry 1996, 35, 1270–1273. [Google Scholar] [CrossRef]
- Zhu, S.; Yan, X.; Xiang, Z.; Ding, H.F.; Cui, H. Leflunomide reduces proliferation and induces apoptosis in neuroblastoma cells in vitro and in vivo. PLoS ONE 2013, 8, e71555. [Google Scholar] [CrossRef]
- Chen, S.-F.; Ruben, R.L.; Dexter, D.L. Mechanism of Action of the Novel Anticancer Agent 6-Fluoro-2-(2′-fluoro-1,1′-biphenyl-4-yl)-3-methyl-4-quinolinecarboxylic Acid Sodium Salt (NSC 368390): Inhibition of de Novo Pyrimidine Nucleotide Biosynthesis. Cancer Res. 1986, 46, 5014–5019. [Google Scholar]
- Peters, G.J.; Sharma, S.L.; Laurensse, E.; Pinedo, H.M. Inhibition of pyrimidine de novo synthesis by DUP-785 (NSC 368390). Investig. New Drugs 1987, 5, 235–244. [Google Scholar] [CrossRef]
- Deans, R.M.; Morgens, D.W.; Ökesli, A.; Pillay, S.; Horlbeck, M.A.; Kampmann, M.; Gilbert, L.A.; Li, A.; Mateo, R.; Smith, M.; et al. Parallel shRNA and CRISPR-Cas9 screens enable antiviral drug target identification. Nat. Chem. Biol. 2016, 12, 361–366. [Google Scholar] [CrossRef]
- Sandoval, A.; Chokshi, A.; Jesch, E.D.; Black, P.N.; Dirusso, C.C. Identification and characterization of small compound inhibitors of human FATP2. Biochem. Pharmacol. 2010, 79, 990–999. [Google Scholar] [CrossRef]
- Ahowesso, C.; Black, P.N.; Saini, N.; Montefusco, D.; Chekal, J.; Malosh, C.; Lindsley, C.W.; Stauffer, S.R.; DiRusso, C.C. Chemical inhibition of fatty acid absorption and cellular uptake limits lipotoxic cell death. Biochem. Pharmacol. 2015, 98, 167–181. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolic Pathway | MYCN Target | Inhibitor |
|---|---|---|
| Glycolysis | HK2 [64] | 2-Deoxy-D-Glucose (2-DG) [168,169] Benitrobenrazide [170] |
| LDHA [64] | GNE-140 [171] FX11 [172,173] Galloflavin [174] | |
| Glutamine metabolism | GLS2 [95] | Compound 968 [175] |
| Serine-Glycine synthesis | PHGDH [80] | NCT503 [176] CBR-5884 [177] PKUMDL-WQ-2101 [178] PH-755 [179] |
| SHMT2 [80] | SHIN2 [180] AGF347 [181] | |
| Nucleotide synthesis | Glutamine amidotransferases (PPAT, PFAS, GMPS, CAD, and CTPS1/2) [96] | 6-diazo-5-oxo-L-norleucine (DON) [182] |
| PPAT [96] | 6-Mercaptopurine [183] | |
| IMPDH [96] | VX-944 [184] Tiazofurin [184] FF-10501 [185] | |
| DHODH [96] | Leflunomide [186,187] Brequinar [188,189] GSK983 [190] | |
| Lipid metabolism | FATP2 (SLC27A2) [136] | Lipofermata [191,192] Grassofermata (CB5) [136,191] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bansal, M.; Gupta, A.; Ding, H.-F. MYCN and Metabolic Reprogramming in Neuroblastoma. Cancers 2022, 14, 4113. https://doi.org/10.3390/cancers14174113
Bansal M, Gupta A, Ding H-F. MYCN and Metabolic Reprogramming in Neuroblastoma. Cancers. 2022; 14(17):4113. https://doi.org/10.3390/cancers14174113
Chicago/Turabian StyleBansal, Mohit, Anamika Gupta, and Han-Fei Ding. 2022. "MYCN and Metabolic Reprogramming in Neuroblastoma" Cancers 14, no. 17: 4113. https://doi.org/10.3390/cancers14174113
APA StyleBansal, M., Gupta, A., & Ding, H.-F. (2022). MYCN and Metabolic Reprogramming in Neuroblastoma. Cancers, 14(17), 4113. https://doi.org/10.3390/cancers14174113