
Performance of the Use of Genetic Information to Assess the Risk of Colorectal Cancer in the Basque Population

 , , and
, , and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Recruitment
2.2. Genotyping and Imputation
2.3. Genetic Analyses
2.3.1. Admixture Analysis
2.3.2. Genome-Wide Association Study
2.3.3. Mendelian Randomization Analyses
2.3.4. Polygenic Risk Scores
3. Results
3.1. Genome-Wide Association Studies
3.2. Mendelian Randomization
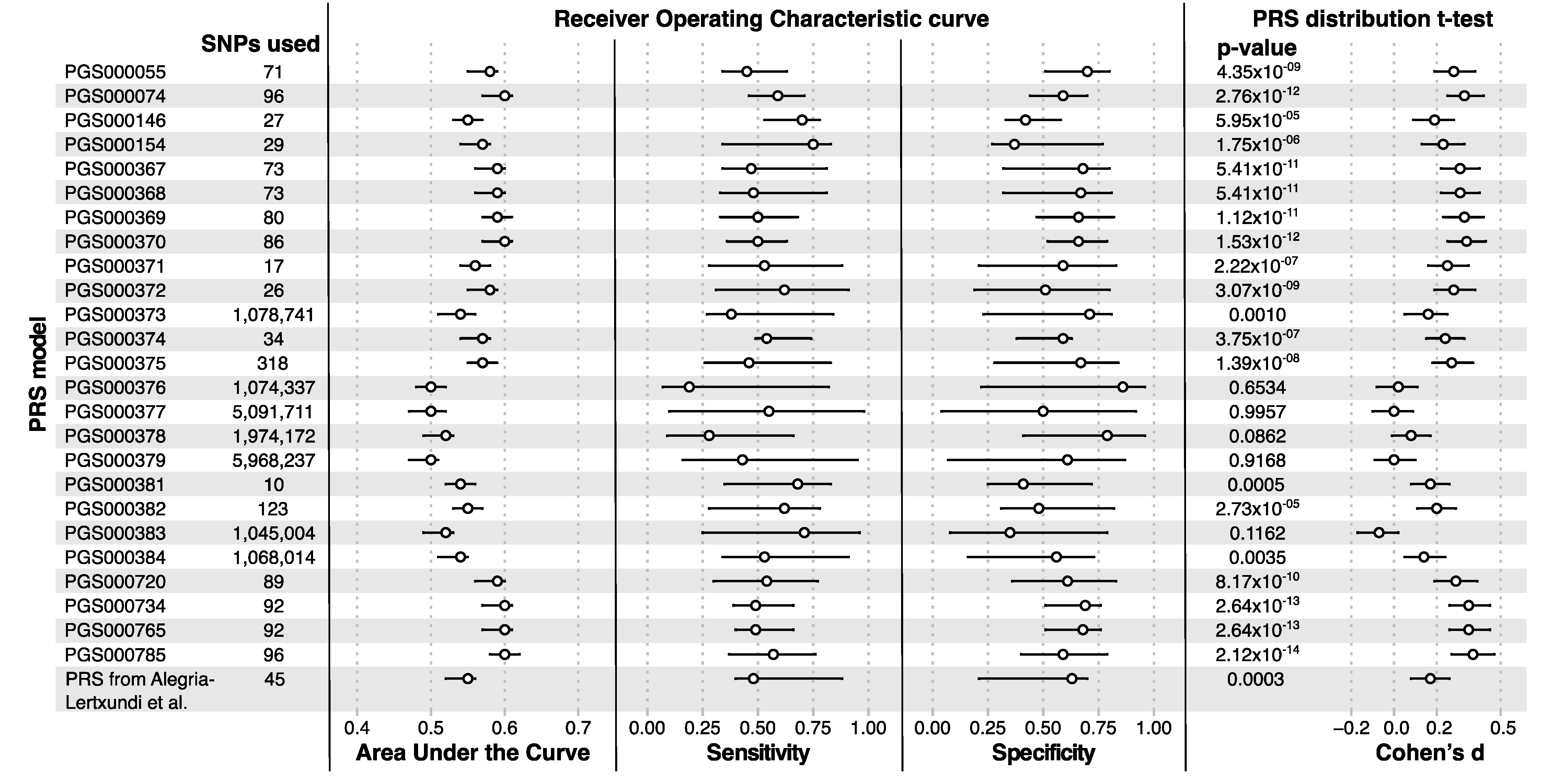
3.3. Polygenic Risk Scores
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stewart, B.; Wild, C. World Cancer Report 2014; IARC Publications: Lyon, France, 2014; ISBN 978-92-832-0443-5. [Google Scholar]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Lasry, A.; Zinger, A.; Ben-Neriah, Y. Inflammatory networks underlying colorectal cancer. Nat. Immunol. 2016, 17, 230–240. [Google Scholar] [CrossRef]
- Cross, A.J.; Ferrucci, L.M.; Risch, A.; Graubard, B.I.; Ward, M.H.; Park, Y.; Hollenbeck, A.R.; Schatzkin, A.; Sinha, R. A large prospective study of meat consumption and colorectal cancer risk: An investigation of potential mechanisms underlying this association. Cancer Res. 2010, 70, 2406–2414. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.H.; Yu, J. Gut microbiota in colorectal cancer: Mechanisms of action and clinical applications. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 690–704. [Google Scholar] [CrossRef]
- Levin, B.; Lieberman, D.A.; McFarland, B.; Smith, R.A.; Brooks, D.; Andrews, K.S.; Dash, C.; Giardiello, F.M.; Glick, S.; Levin, T.R.; et al. Screening and surveillance for the early detection of colorectal cancer and adenomatous polyps, 2008: A joint guideline from the American Cancer Society, the US Multi-Society Task Force on Colorectal Cancer, and the American College of Radiology. CA. Cancer J. Clin. 2008, 58, 130–160. [Google Scholar] [CrossRef] [PubMed]
- Abulí, A.; Fernández-Rozadilla, C.; Alonso-Espinaco, V.; Muñoz, J.; Gonzalo, V.; Bessa, X.; González, D.; Clofent, J.; Cubiella, J.; Morillas, J.D.; et al. Case-control study for colorectal cancer genetic susceptibility in EPICOLON: Previously identified variants and mucins. BMC Cancer 2011, 11, 339. [Google Scholar] [CrossRef]
- Abulí, A.; Bessa, X.; González, J.R.; Ruizponte, C.; Cáceres, A.; Muñoz, J.; Gonzalo, V.; Balaguer, F.; Fernándezrozadilla, C.; González, D.; et al. Susceptibility genetic variants associated with colorectal cancer risk correlate with cancer phenotype. Gastroenterology 2010, 139, 788–796.e6. [Google Scholar] [CrossRef] [PubMed]
- Burns, M.B.; Montassier, E.; Abrahante, J.; Priya, S.; Niccum, D.E.; Khoruts, A.; Starr, T.K.; Knights, D.; Blekhman, R. Colorectal cancer mutational profiles correlate with defined microbial communities in the tumor microenvironment. PLoS Genet. 2018, 14, 1–24. [Google Scholar] [CrossRef]
- Law, P.J.; Timofeeva, M.; Fernandez-Rozadilla, C.; Broderick, P.; Studd, J.; Fernandez-Tajes, J.; Farrington, S.; Svinti, V.; Palles, C.; Orlando, G.; et al. Association analyses identify 31 new risk loci for colorectal cancer susceptibility. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Rodriguez-broadbent, H.; Law, P.J.; Sud, A.; Palin, K.; Tuupanen, S.; Kaasinen, E.; Sarin, A.; Ripatti, S.; Eriksson, J.G. Mendelian randomisation implicates hyperlipidaemia as a risk factor for colorectal cancer. Int. J. Cancer 2017, 140, 2701–2708. [Google Scholar] [CrossRef] [Green Version]
- Cornish, A.J.; Law, P.J.; Timofeeva, M.; Palin, K.; Farrington, S.M.; Palles, C.; Jenkins, M.A.; Casey, G.; Brenner, H.; Chang-Claude, J.; et al. Modifiable pathways for colorectal cancer: A mendelian randomisation analysis. Lancet Gastroenterol. Hepatol. 2020, 5, 55–62. [Google Scholar] [CrossRef]
- Garcia-Etxebarria, K.; Clos-Garcia, M.; Telleria, O.; Nafría, B.; Alonso, C.; Iruarrizaga-Lejarreta, M.; Franke, A.; Crespo, A.; Iglesias, A.; Cubiella, J.; et al. Interplay between genome, metabolome and microbiome in colorectal cancer. Cancers 2021, 13, 6216. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez-Sanz, G.; Diéz-Villanueva, A.; Alonso, M.H.; Rodríguez-Moranta, F.; Pérez-Gómez, B.; Bustamante, M.; Martin, V.; Llorca, J.; Amiano, P.; Ardanaz, E.; et al. Risk Model for Colorectal Cancer in Spanish Population Using Environmental and Genetic Factors: Results from the MCC-Spain study. Sci. Rep. 2017, 7, 43263. [Google Scholar] [CrossRef]
- Thomas, M.; Sakoda, L.C.; Hoffmeister, M.; Rosenthal, E.A.; Lee, J.K.; van Duijnhoven, F.J.B.; Platz, E.A.; Wu, A.H.; Dampier, C.H.; de la Chapelle, A.; et al. Genome-wide Modeling of Polygenic Risk Score in Colorectal Cancer Risk. Am. J. Hum. Genet. 2020, 107, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Alegria-Lertxundi, I.; Aguirre, C.; Bujanda, L.; Fernández, F.J.; Polo, F.; Ordovás, J.M.; Carmen Etxezarraga, M.; Zabalza, I.; Larzabal, M.; Portillo, I.; et al. Single nucleotide polymorphisms associated with susceptibility for development of colorectal cancer: Case-control study in a Basque population. PLoS ONE 2019, 14, e0225779. [Google Scholar] [CrossRef]
- Günther, T.; Valdiosera, C.; Malmström, H.; Ureña, I.; Rodriguez-Varela, R.; Sverrisdóttir, Ó.O.; Daskalaki, E.A.; Skoglund, P.; Naidoo, T.; Svensson, E.M.; et al. Ancient genomes link early farmers from Atapuerca in Spain to modern-day Basques. Proc. Natl. Acad. Sci. USA 2015, 112, 11917–11922. [Google Scholar] [CrossRef]
- Olalde, I.; Mallick, S.; Patterson, N.; Rohland, N.; Villalba-Mouco, V.; Silva, M.; Dulias, K.; Edwards, C.J.; Gandini, F.; Pala, M.; et al. The genomic history of the Iberian Peninsula over the past 8000 years. Science (80-) 2019, 363, 1230–1234. [Google Scholar] [CrossRef]
- Garcia-Etxebarria, K.; Merino, O.; Gaite-Reguero, A.; Rodrigues, P.M.; Herrarte, A.; Etxart, A.; Ellinghaus, D.; Alonso-Galan, H.; Franke, A.; Marigorta, U.M.; et al. Local genetic variation of inflammatory bowel disease in Basque population and its effect in risk prediction. Sci. Rep. 2022, 12, 3386. [Google Scholar] [CrossRef] [PubMed]
- Abraham, G.; Qiu, Y.; Inouye, M. FlashPCA2: Principal component analysis of Biobank-scale genotype datasets. Bioinformatics 2017, 33, 2776–2778. [Google Scholar] [CrossRef] [PubMed]
- Loh, P.R.; Danecek, P.; Palamara, P.F.; Fuchsberger, C.; Reshef, Y.A.; Finucane, H.K.; Schoenherr, S.; Forer, L.; McCarthy, S.; Abecasis, G.R.; et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat. Genet. 2016, 48, 1443–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durbin, R. Efficient haplotype matching and storage using the positional Burrows-Wheeler transform (PBWT). Bioinformatics 2014, 30, 1266–1272. [Google Scholar] [CrossRef]
- McCarthy, S.; Das, S.; Kretzschmar, W.; Delaneau, O.; Wood, A.R.; Teumer, A.; Kang, H.M.; Fuchsberger, C.; Danecek, P.; Sharp, K.; et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 2016, 48, 1279–1283. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.A.M.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Buniello, A.; Macarthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef]
- Hemani, G.; Zheng, J.; Elsworth, B.; Wade, K.H.; Haberland, V.; Baird, D.; Laurin, C.; Burgess, S.; Bowden, J.; Langdon, R.; et al. The MR-base platform supports systematic causal inference across the human phenome. Elife 2018, 7, 1–29. [Google Scholar] [CrossRef]
- Zhu, Z.; Zheng, Z.; Zhang, F.; Wu, Y.; Trzaskowski, M.; Maier, R.; Robinson, M.R.; McGrath, J.J.; Visscher, P.M.; Wray, N.R.; et al. Causal associations between risk factors and common diseases inferred from GWAS summary data. Nat. Commun. 2018, 9, 224. [Google Scholar] [CrossRef]
- R Development Core Team. R: A Language and Eviroment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2008. [Google Scholar]
- Kurilshikov, A.; Medina-Gomez, C.; Bacigalupe, R.; Radjabzadeh, D.; Wang, J.; Demirkan, A.; Le Roy, C.I.; Raygoza Garay, J.A.; Finnicum, C.T.; Liu, X.; et al. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat. Genet. 2021, 53, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Thompson, S.G. Avoiding bias from weak instruments in Mendelian randomization studies. Int. J. Epidemiol. 2011, 40, 755–764. [Google Scholar] [CrossRef]
- Lambert, S.A.; Gil, L.; Jupp, S.; Ritchie, S.C.; Xu, Y.; Buniello, A.; McMahon, A.; Abraham, G.; Chapman, M.; Parkinson, H.; et al. The Polygenic Score Catalog as an open database for reproducibility and systematic evaluation. Nat. Genet. 2021, 53, 420–425. [Google Scholar] [CrossRef]
- Jia, G.; Lu, Y.; Wen, W.; Long, J.; Liu, Y.; Tao, R.; Li, B.; Denny, J.C.; Shu, X.-O.; Zheng, W. Evaluating the Utility of Polygenic Risk Scores in Identifying High-Risk Individuals for Eight Common Cancers. JNCI Cancer Spectr. 2020, 4, pkaa021. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.; Jeon, J.; Brenner, H.; Gruber, S.B.; Schoen, R.E.; Berndt, S.I.; Chan, A.T.; Chang-Claude, J.; Du, M.; Gong, J.; et al. A Model to Determine Colorectal Cancer Risk Using Common Genetic Susceptibility Loci. Gastroenterology 2015, 148, 1330–1339.e14. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, L.G.; Patil, S.; Beesley, L.J.; VandeHaar, P.; Salvatore, M.; Ma, Y.; Peng, R.B.; Taliun, D.; Zhou, X.; Mukherjee, B. Cancer PRSweb: An Online Repository with Polygenic Risk Scores for Major Cancer Traits and Their Evaluation in Two Independent Biobanks. Am. J. Hum. Genet. 2020, 107, 815–836. [Google Scholar] [CrossRef]
- Huyghe, J.R.; Bien, S.A.; Harrison, T.A.; Kang, H.M.; Chen, S.; Schmit, S.L.; Conti, D.V.; Qu, C.; Jeon, J.; Edlund, C.K.; et al. Discovery of common and rare genetic risk variants for colorectal cancer. Nat. Genet. 2019, 51, 76–87. [Google Scholar] [CrossRef]
- Kachuri, L.; Graff, R.E.; Smith-Byrne, K.; Meyers, T.J.; Rashkin, S.R.; Ziv, E.; Witte, J.S.; Johansson, M. Pan-cancer analysis demonstrates that integrating polygenic risk scores with modifiable risk factors improves risk prediction. Nat. Commun. 2020, 11, 6084. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Yu, H.; Wu, Y.; Lin, X.; Bao, Q.; Jia, H.; Perschon, C.; Duggan, D.; Helfand, B.T.; Zheng, S.L.; et al. Systematic evaluation of cancer-specific genetic risk score for 11 types of cancer in The Cancer Genome Atlas and Electronic Medical Records and Genomics cohorts. Cancer Med. 2019, 8, 3196–3205. [Google Scholar] [CrossRef]
- Graff, R.E.; Cavazos, T.B.; Thai, K.K.; Kachuri, L.; Rashkin, S.R.; Hoffman, J.D.; Alexeeff, S.E.; Blatchins, M.; Meyers, T.J.; Leong, L.; et al. Cross-cancer evaluation of polygenic risk scores for 16 cancer types in two large cohorts. Nat. Commun. 2021, 12, 970. [Google Scholar] [CrossRef]
- Archambault, A.N.; Su, Y.-R.; Jeon, J.; Thomas, M.; Lin, Y.; Conti, D.V.; Win, A.K.; Sakoda, L.C.; Lansdorp-Vogelaar, I.; Peterse, E.F.P.; et al. Cumulative Burden of Colorectal Cancer–Associated Genetic Variants Is More Strongly Associated with Early-Onset vs Late-Onset Cancer. Gastroenterology 2020, 158, 1274–1286.e12. [Google Scholar] [CrossRef]
- Schmit, S.L.; Edlund, C.K.; Schumacher, F.R.; Gong, J.; Harrison, T.A.; Huyghe, J.R.; Qu, C.; Melas, M.; Van Den Berg, D.J.; Wang, H.; et al. Novel Common Genetic Susceptibility Loci for Colorectal Cancer. JNCI J. Natl. Cancer Inst. 2019, 111, 146–157. [Google Scholar] [CrossRef]
- Dvorak, P.; Pesta, M.; Soucek, P. ABC gene expression profiles have clinical importance and possibly form a new hallmark of cancer. Tumor Biol. 2017, 39, 1010428317699800. [Google Scholar] [CrossRef] [Green Version]
- Hlavata, I.; Mohelnikova-Duchonova, B.; Vaclavikova, R.; Liska, V.; Pitule, P.; Novak, P.; Bruha, J.; Vycital, O.; Holubec, L.; Treska, V.; et al. The role of ABC transporters in progression and clinical outcome of colorectal cancer. Mutagenesis 2012, 27, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, X.; Tseng, Y.; Huang, M.; Luo, F.; Zhang, J.; Liu, J. Distinguishing colorectal adenoma from hyperplastic polyp by WNT2 expression. J. Clin. Lab. Anal. 2021, 35, 1–10. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhu, H.; Li, Q.; Gao, W.; Zang, D.; Su, W.; Yang, R.; Zhong, J. Gene Expression Profiling of Tricarboxylic Acid Cycle and One Carbon Metabolism Related Genes for Prognostic Risk Signature of Colon Carcinoma. Front. Genet. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Chang, L.C.; Hsu, Y.C.; Chiu, H.M.; Ueda, K.; Wu, M.S.; Kao, C.H.; Shen, T.L. Exploration of the Proteomic Landscape of Small Extracellular Vesicles in Serum as Biomarkers for Early Detection of Colorectal Neoplasia. Front. Oncol. 2021, 11, 1–12. [Google Scholar] [CrossRef]
- Frey, M.R.; Edelblum, K.L.; Mullane, M.T.; Liang, D.; Polk, D.B. The ErbB4 Growth Factor Receptor Is Required for Colon Epithelial Cell Survival in the Presence of TNF. Gastroenterology 2009, 136, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Frey, M.R.; Hilliard, V.C.; Mullane, M.T.; Polk, D.B. ErbB4 promotes cyclooxygenase-2 expression and cell survival in colon epithelial cells. Lab. Investig. 2010, 90, 1415–1424. [Google Scholar] [CrossRef]
- Williams, C.S.; Bernard, J.K.; Beckler, M.D.; Almohazey, D.; Washington, M.K.; Smith, J.J.; Frey, M.R. ERBB4 is over-expressed in human colon cancer and enhances cellular transformation. Carcinogenesis 2015, 36, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Wang, H.; Li, Z.; Yan, J.; Guo, Y.; Zhao, W.; Gao, L.; Wang, B.; Jia, Y. HER4 promotes the progression of colorectal cancer by promoting epithelial-mesenchymal transition. Mol. Med. Rep. 2020, 21, 1779–1788. [Google Scholar] [CrossRef]
- Baiocchi, G.; Lopes, A.; Coudry, R.A.; Rossi, B.M.; Soares, F.A.; Aguiar, S.; Guimarães, G.C.; Ferreira, F.O.; Nakagawa, W.T. ErbB family immunohistochemical expression in colorectal cancer patients with higher risk of recurrence after radical surgery. Int. J. Colorectal Dis. 2009, 24, 1059–1068. [Google Scholar] [CrossRef]
- Chu, S.; Wang, H.; Yu, M. A putative molecular network associated with colon cancer metastasis constructed from microarray data. World J. Surg. Oncol. 2017, 15, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, J.A.; Yoon, S.; Park, S.Y.; Lee, J.H.; Hwang, J.E.; Kim, H.; Seo, Y.W.; Cha, Y.J.; Hong, S.P.; Kim, H.; et al. An unconventional KITENIN/ErbB4-mediated downstream signal of EGF upregulates c-Jun and the invasiveness of colorectal cancer cells. Clin. Cancer Res. 2014, 20, 4115–4128. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Li, Z.; Tang, Z.; Li, J.; Lang, X. Long noncoding MAGI2-AS3 promotes colorectal cancer progression through regulating miR-3163/TMEM106B axis. J. Cell. Physiol. 2020, 235, 4824–4833. [Google Scholar] [CrossRef] [PubMed]
- Poursheikhani, A.; Abbaszadegan, M.R.; Nokhandani, N.; Kerachian, M.A. Integration analysis of long non-coding RNA (lncRNA) role in tumorigenesis of colon adenocarcinoma. BMC Med. Genomics 2020, 13, 108. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wu, S.; Li, X.; Yin, Y.; Chen, R. MAGI2-AS3 rs7783388 polymorphism contributes to colorectal cancer risk through altering the binding affinity of the transcription factor GR to the MAGI2-AS3 promoter. J. Clin. Lab. Anal. 2020, 34, 1–11. [Google Scholar] [CrossRef]
- Mlecnik, B.; Bindea, G.; Angell, H.K.; Sasso, M.S.; Obenauf, A.C.; Fredriksen, T.; Lafontaine, L.; Bilocq, A.M.; Kirilovsky, A.; Tosolini, M.; et al. Functional network pipeline reveals genetic determinants associated with in situ lymphocyte proliferation and survival of cancer patients. Sci. Transl. Med. 2014, 6, 228ra37. [Google Scholar] [CrossRef]
- Yang, Q.-X.; Liu, T.; Yang, J.-L.; Liu, F.; Chang, L.; Che, G.-L.; Lai, S.-Y.; Jiang, Y.-M. Low expression of NTF3 is associated with unfavorable prognosis in hepatocellular carcinoma. Int. J. Clin. Exp. Pathol. 2020, 13, 2280–2288. [Google Scholar]
- Liu, R.; Li, R.; Yu, H.; Liu, J.; Zheng, S.; Li, Y.; Ye, L. NTF3 Correlates With Prognosis and Immune Infiltration in Hepatocellular Carcinoma. Front. Med. 2021, 8, 795849. [Google Scholar] [CrossRef]
- Zhao, R.; Liu, Y.; Wu, C.; Li, M.; Wei, Y.; Niu, W.; Yang, J.; Fan, S.; Xie, Y.; Li, H.; et al. BRD7 Promotes Cell Proliferation and Tumor Growth Through Stabilization of c-Myc in Colorectal Cancer. Front. Cell Dev. Biol. 2021, 9, 659392. [Google Scholar] [CrossRef]
- Pelullo, M.; Nardozza, F.; Zema, S.; Quaranta, R.; Nicoletti, C.; Besharat, Z.M.; Felli, M.P.; Cerbelli, B.; d’Amati, G.; Palermo, R.; et al. Kras/ADAM17-Dependent Jag1-ICD Reverse Signaling Sustains Colorectal Cancer Progression and Chemoresistance. Cancer Res. 2019, 79, 5575–5586. [Google Scholar] [CrossRef]
- Kiriyama, K.; Hirohashi, Y.; Torigoe, T.; Kubo, T.; Tamura, Y.; Kanaseki, T.; Takahashi, A.; Nakazawa, E.; Saka, E.; Ragnarsson, C.; et al. Expression and function of FERMT genes in colon carcinoma cells. Anticancer Res. 2013, 33, 167–173. [Google Scholar]
- Huyghe, J.R.; Harrison, T.A.; Bien, S.A.; Hampel, H.; Figueiredo, J.C.; Schmit, S.L.; Conti, D.V.; Chen, S.; Qu, C.; Lin, Y.; et al. Genetic architectures of proximal and distal colorectal cancer are partly distinct. Gut 2021, 70, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Passarelli, M.N.; Newcomb, P.A.; Makar, K.W.; Burnett-Hartman, A.N.; Potter, J.D.; Upton, M.P.; Zhu, L.C.; Rosenfeld, M.E.; Schwartz, S.M.; Rutter, C.M. Blood lipids and colorectal polyps: Testing an etiologic hypothesis using phenotypic measurements and Mendelian randomization. Cancer Causes Control 2015, 26, 467–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Yang, Y.; Wang, F.; Zhang, P.; Shi, C.; Zou, Y.; Qin, H. Obesity and risk of colorectal cancer: A systematic review of prospective studies. PLoS ONE 2013, 8, e53916. [Google Scholar] [CrossRef]
- Saus, E.; Iraola-Guzmán, S.; Willis, J.R.; Brunet-Vega, A.; Gabaldón, T. Microbiome and colorectal cancer: Roles in carcinogenesis and clinical potential. Mol. Aspects Med. 2019, 69, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Flemer, B.; Lynch, D.B.; Brown, J.M.R.; Jeffery, I.B.; Ryan, F.J.; Claesson, M.J.; O’Riordain, M.; Shanahan, F.; O’Toole, P.W. Tumour-associated and non-tumour-associated microbiota in colorectal cancer. Gut 2017, 66, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Miyake, T.; Mori, H.; Yasukawa, D.; Hexun, Z.; Maehira, H.; Ueki, T.; Kojima, M.; Kaida, S.; Iida, H.; Shimizu, T.; et al. The Comparison of Fecal Microbiota in Left-Side and Right-Side Human Colorectal Cancer. Eur. Surg. Res. 2021, 62, 248–254. [Google Scholar] [CrossRef]
- Phipps, O.; Quraishi, M.N.; Dickson, E.A.; Steed, H.; Kumar, A.; Acheson, A.G.; Beggs, A.D.; Brookes, M.J.; Al-Hassi, H.O. Differences in the on-and off-tumor microbiota between right-and left-sided colorectal cancer. Microorganisms 2021, 9, 1108. [Google Scholar] [CrossRef]
- Yang, X.; Pan, Y.; Wu, W.; Qi, Q.; Zhuang, J.; Xu, J.; Han, S. Analysis of prognosis, genome, microbiome, and microbial metabolome in different sites of colorectal cancer. J. Transl. Med. 2019, 17, 1–22. [Google Scholar] [CrossRef]
- Clos-Garcia, M.; Garcia, K.; Alonso, C.; Iruarrizaga-Lejarreta, M.; D’amato, M.; Crespo, A.; Iglesias, A.; Cubiella, J.; Bujanda, L.; Falcón-Pérez, J.M. Integrative analysis of fecal metagenomics and metabolomics in colorectal cancer. Cancers 2020, 12, 1142. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, J.; Zheng, J.; Hu, G.; Wang, J.; Huang, C.; Lou, L.; Wang, X.; Zeng, Y. Mucosal adherent bacterial dysbiosis in patients with colorectal adenomas. Sci. Rep. 2016, 6, 26337. [Google Scholar] [CrossRef]
- Luo, H.; Liu, L.; Zhao, J.-J.; Mi, X.-F.; Wang, Q.-J.; Yu, M. Effects of oxaliplatin on inflammation and intestinal floras in rats with colorectal cancer. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 10542–10549. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Cases | Controls | |
|---|---|---|
| N | 835 | 940 |
| Male (%) | 530 (63.47%) | 631 (67.13%) |
| Female (%) | 305 (36.53%) | 309 (32.87%) |
| Age (SE) | 73.54 (11.38) | 41.53 (11.79) |
| Stage | ||
| 0 | 37 (4.43%) | |
| I | 130 (15.57%) | |
| II | 314 (37.61%) | |
| III | 223 (26.71%) | |
| IV | 105 (12.57%) | |
| Undetermined | 26 (3.11%) | |
| Location | ||
| Right | 170 (20.36%) | |
| Left | 219 (26.23%) | |
| Rectal | 235 (28.14%) | |
| Unspecific | 211 (25.27%) |
| Lead SNP | Position | Gene | A1 | A2 | OR (CI 95%) | p-Value | Freq | Freq EUR |
|---|---|---|---|---|---|---|---|---|
| Colorectal cancer vs. controls | ||||||||
| rs79374732 | 2:212815957 | ERBB4 | T | C | 8.5 (3.4–21.0) | 4.5 × 10−6 | 0.032 | 0.022 |
| rs77317240 | 2:216091445 | Upstream of ABCA12 and ATIC | T | C | 6.4 (3.1–13.2) | 5.8 × 10−7 | 0.039 | 0.024 |
| rs116443146 | 4:142699393 | Downstream of IL15 | G | A | 16.3 (5.0–53.8) | 4.4 × 10−6 | 0.013 | 0.02 |
| rs34931968 | 7:79055118 | MAGI2 | T | G | 29.7 (7.1–124.3) | 3.4 × 10−6 | 0.011 | 0.01 |
| rs1693967 | 16:86289580 | LINC01081 | G | A | 11.4 (4.1–32.1) | 3.9 × 10−6 | 0.017 | 0.024 |
| Right colon cancer vs. controls | ||||||||
| rs3004681 | 1:69054715 | Downstream of DEPDC1 | T | G | 11.8 (4.3–32.7) | 2.0 × 10−6 | 0.062 | 0.073 |
| rs77445470 | 1:226800066 | Downstream of STUM and ITPKB | G | C | 18.5 (5.3–64.5) | 4.8 × 10−6 | 0.044 | 0.055 |
| rs76653793 | 4:47962934 | CNGA1, LOC101927157 | G | T | 21.7 (6.4–73.8) | 7.9 × 10−7 | 0.028 | 0.036 |
| rs142444738 | 4:106095747 | TET2, TET2-AS1 | A | G | 51.1 (9.6–270.9) | 3.8 × 10−6 | 0.011 | 0.005 |
| rs4696337 | 4:153602674 | TMEM154, LOC105377495 | A | C | 35.8 (8.2–156.2) | 2.0 × 10−6 | 0.023 | 0.023 |
| rs139432545 | 4:174624195 | G | A | 48.4 (9.6–244.9) | 2.7 × 10−6 | 0.012 | 0.022 | |
| rs13211079 | 6:36977349 | FGD2 | G | C | 43.9 (9.2–210.2) | 2.2 × 10−6 | 0.019 | 0.012 |
| rs190591066 | 7:89988294 | GTPBP10 | A | G | 40.6 (8.8–186.4) | 1.9 × 10−6 | 0.017 | 0.011 |
| rs75772232 | 8:83689525 | T | C | 15.8 (4.9–51.2) | 4.3 × 10−6 | 0.039 | 0.045 | |
| rs118025264 | 9:119407781 | ASTN2, LOC105376240 | T | C | 25.7 (6.4–102.7) | 4.3 × 10−6 | 0.026 | 0.022 |
| rs16933489 | 12:5572210 | NTF3 | T | C | 34.9 (9.1–133.3) | 2.0 × 10−7 | 0.02 | 0.044 |
| rs78263620 | 18:72995680 | TSHZ1 | T | C | 43.6 (9.2–207.9) | 2.2 × 10−6 | 0.011 | 0.019 |
| rs148452202 | 19:2527577 | GNG7 | A | G | 34.6 (8.3–144.8) | 1.2 × 10−6 | 0.022 | 0.01 |
| rs35914129 | 19:48115566 | BICRA | T | G | 56.2 (11.2–283.0) | 1.0 × 10−6 | 0.013 | 0.009 |
| rs28495197 | 22:36050632 | APOL6 | T | C | 39.9 (9.1–174.2) | 9.4 × 10−7 | 0.023 | 0.017 |
| rs117820381 | 22:40738486 | Downstream of TNRC6B, upstream of ADSL | A | G | 37.0 (8.4–163.1) | 1.8 × 10−6 | 0.013 | 0.028 |
| Left colon cancer vs. controls | ||||||||
| rs112033525 | 2:23176856 | T | G | 39.4 (8.2–189.6) | 4.5 × 10−6 | 0.017 | 0.015 | |
| rs139367040 | 2:173950614 | MAP3K20 | T | C | 33.0 (7.7–142.5) | 2.8 × 10−6 | 0.019 | 0.014 |
| rs72774468 | 9:137697318 | COL5A1 | C | T | 15.1 (5.0–45.3) | 1.3 × 10−6 | 0.035 | 0.051 |
| rs114144417 | 16:48116976 | ABCC12 | T | C | 149.8 (20.2–1112.0) | 9.7 × 10−7 | 0.01 | 0.008 |
| rs17721600 | 17:27268513 | PHF12, LOC101927018 | A | G | 25.9 (6.9–97.7) | 1.6 × 10−6 | 0.037 | 0.053 |
| rs140107269 | 18:1828990 | T | C | 26.8 (6.6–109.2) | 4.4 × 10−6 | 0.023 | 0.027 | |
| rs62093285 | 18:49252189 | A | G | 12.8 (4.3–38.4) | 4.9 × 10−6 | 0.044 | 0.035 | |
| Rectal cancer vs. controls | ||||||||
| rs78144988 | 1:102199388 | LINC01709 | C | T | 54.9 (11.2–268.4) | 7.6 × 10−7 | 0.013 | 0.018 |
| rs13403794 | 2:9785060 | Upstream of YWHAQ and ADAM17 | C | T | 65.5 (12.0–355.9) | 1.3 × 10−6 | 0.012 | 0.021 |
| rs354856 | 2:142433670 | LRP1B, LOC107985779 | C | T | 17.4 (5.5–55.0) | 1.1 × 10−6 | 0.027 | 0.062 |
| rs116443146 | 4:142699393 | Downstream of IL15 | G | A | 40.3 (9.2–176.9) | 9.7 × 10−7 | 0.013 | 0.02 |
| rs72909399 | 6:86581045 | T | G | 74.7 (13.5–414.7) | 8.1 × 10−7 | 0.014 | 0.03 | |
| rs71516114 | 8:784674 | DLGAP2 | C | T | 5.2 (2.6–10.4) | 2.7 × 10−6 | 0.111 | 0.112 |
| rs61848097 | 10:50134508 | WDFY4, LRRC18 | G | A | 8.6 (3.5–21.0) | 2.9 × 10−6 | 0.073 | 0.089 |
| rs77470802 | 14:27547598 | LOC105370420 | G | T | 12.4 (4.2–36.5) | 4.6 × 10−6 | 0.027 | 0.033 |
| rs76799782 | 14:91624544 | DGLUCY | A | G | 18.9 (5.4–65.4) | 3.8 × 10−6 | 0.029 | 0.039 |
| rs141553824 | 16:50380386 | BRD7 | C | T | 45.8 (10.4–202.4) | 4.5 × 10−7 | 0.017 | 0.05 |
| Left colon cancer vs. right colon cancer | ||||||||
| rs4655303 | 1:213834643 | LOC105372912 | T | A | 2.2 (1.6–3.0) | 3.6 × 10−6 | 0.43 | 0.377 |
| rs62005704 | 14:53465150 | Downstream of DDHD1, upstream of FERMT2 | A | G | 0.4 (0.3–0.6) | 9.8 × 10−7 | 0.464 | 0.503 |
| Rectal cancer vs. colon cancer | ||||||||
| rs73171906 | 7:147986529 | CNTNAP2 | T | C | 2.2 (1.6–2.9) | 6.4 × 10−7 | 0.23 | 0.154 |
| rs9773025 | 8:6674458 | XKR5 | G | A | 0.5 (0.3–0.6) | 1.5 × 10−6 | 0.414 | 0.468 |
| rs79619562 | 21:38742422 | DYRK1A | C | T | 2.7 (1.8–4.1) | 1.8 × 10−6 | 0.1 | 0.093 |
| Lead SNP | Position | Gene | A1 | A2 | OR (CI 95%) | p-Value | Freq | Freq EUR |
|---|---|---|---|---|---|---|---|---|
| Colorectal cancer vs inflammatory bowel disease | ||||||||
| rs35493687 | 1:41285292 | KCNQ4 | A | C | 0.4 (0.3–0.6) | 4.2 × 10−6 | 0.122 | 0.147 |
| rs76845271 | 2:73665817 | ALMS1 | T | G | 0.3 (0.2–0.5) | 2.9 × 10−6 | 0.043 | 0.048 |
| rs6738805 | 2:231083171 | SP110 | C | T | 0.4 (0.3–0.6) | 4.6 × 10−7 | 0.135 | 0.128 |
| rs10007784 | 4:81977690 | BMP3 | C | T | 0.5 (0.4–0.7) | 1.8 × 10−6 | 0.228 | 0.222 |
| rs181206673 | 5:25834969 | C | G | 0.3 (0.1–0.5) | 4.1 × 10−6 | 0.039 | 0.0467 | |
| rs72840740 | 6:18745458 | C | T | 0.1 (0.0–0.2) | 1.1 × 10−6 | 0.014 | 0.03 | |
| rs9271365 | 6:32586794 | Downstream of HLA-DRB1 and upstream of HLA-DQA1 | G | T | 1.8 (1.4–2.3) | 2.2 × 10−6 | 0.353 | 0.388 |
| rs951197 | 6:103210765 | C | A | 0.5 (0.4–0.7) | 5.6 × 10−7 | 0.476 | 0.446 | |
| rs1875664 | 8:827824 | DLGAP2 | G | A | 2.3 (1.6–3.3) | 2.8 × 10−6 | 0.128 | 0.161 |
| rs988874 | 10:27684660 | Downstream of PTCHD3 | A | T | 0.5 (0.3–0.6) | 1.6 × 10−6 | 0.174 | 0.157 |
| rs541295 | 15:50056050 | Upstream of ATP8B4 | G | A | 0.2 (0.1–0.4) | 1.8 × 10−8 | 0.055 | 0.022 |
| Colorectal cancer vs. controls + inflammatory bowel disease | ||||||||
| rs7550486 | 1:14777040 | KAZN | C | T | 0.6 (0.5–0.7) | 1.3 × 10−6 | 0.498 | 0.475 |
| rs115681984 | 2:216032071 | Upstream of ABCA12 and ATIC | T | C | 4.2 (2.4–7.1) | 2.6 × 10−7 | 0.034 | 0.026 |
| rs72840741 | 6:18747455 | G | A | 0.1 (0.0–0.2) | 1.8 × 10−6 | 0.014 | 0.03 | |
| rs5002178 | 6:32611590 | HLA-DQA1 | G | A | 0.6 (0.5–0.7) | 6.8 × 10−7 | 0.33 | 0.374 |
| rs951197 | 6:103210765 | C | A | 0.6 (0.5–0.7) | 2.4 × 10−7 | 0.484 | 0.446 | |
| rs1875664 | 8:827824 | DLGAP2 | G | A | 2.2 (1.6–3.0) | 3.24 × 10−7 | 0.124 | 0.161 |
| rs988874 | 10:27684660 | Downstream of PTCHD3 | A | T | 0.5 (0.3–0.6) | 2.0 × 10−6 | 0.171 | 0.157 |
| rs150840049 | 14:59165709 | Downstream of DACT1 | C | T | 0.1 (0.1–0.3) | 2.6 × 10−6 | 0.025 | 0.052 |
| rs541295 | 15:50056050 | Upstream of ATP8B4 | G | A | 0.2 (0.1–0.4) | 5.3 × 10−8 | 0.045 | 0.022 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia-Etxebarria, K.; Etxart, A.; Barrero, M.; Nafria, B.; Segues Merino, N.M.; Romero-Garmendia, I.; Franke, A.; D’Amato, M.; Bujanda, L. Performance of the Use of Genetic Information to Assess the Risk of Colorectal Cancer in the Basque Population. Cancers 2022, 14, 4193. https://doi.org/10.3390/cancers14174193
Garcia-Etxebarria K, Etxart A, Barrero M, Nafria B, Segues Merino NM, Romero-Garmendia I, Franke A, D’Amato M, Bujanda L. Performance of the Use of Genetic Information to Assess the Risk of Colorectal Cancer in the Basque Population. Cancers. 2022; 14(17):4193. https://doi.org/10.3390/cancers14174193
Chicago/Turabian StyleGarcia-Etxebarria, Koldo, Ane Etxart, Maialen Barrero, Beatriz Nafria, Nerea Miren Segues Merino, Irati Romero-Garmendia, Andre Franke, Mauro D’Amato, and Luis Bujanda. 2022. "Performance of the Use of Genetic Information to Assess the Risk of Colorectal Cancer in the Basque Population" Cancers 14, no. 17: 4193. https://doi.org/10.3390/cancers14174193
APA StyleGarcia-Etxebarria, K., Etxart, A., Barrero, M., Nafria, B., Segues Merino, N. M., Romero-Garmendia, I., Franke, A., D’Amato, M., & Bujanda, L. (2022). Performance of the Use of Genetic Information to Assess the Risk of Colorectal Cancer in the Basque Population. Cancers, 14(17), 4193. https://doi.org/10.3390/cancers14174193