Changing Landscape in the Treatment of Adult Acute Lymphoblastic Leukemia (ALL)


Abstract
:Simple Summary
Abstract
1. Introduction
2. Epidemiology, Clinical Presentation
3. Diagnostic, Genetic and Prognostic Factors
4. Measurable Residual Disease (MRD) Monitoring
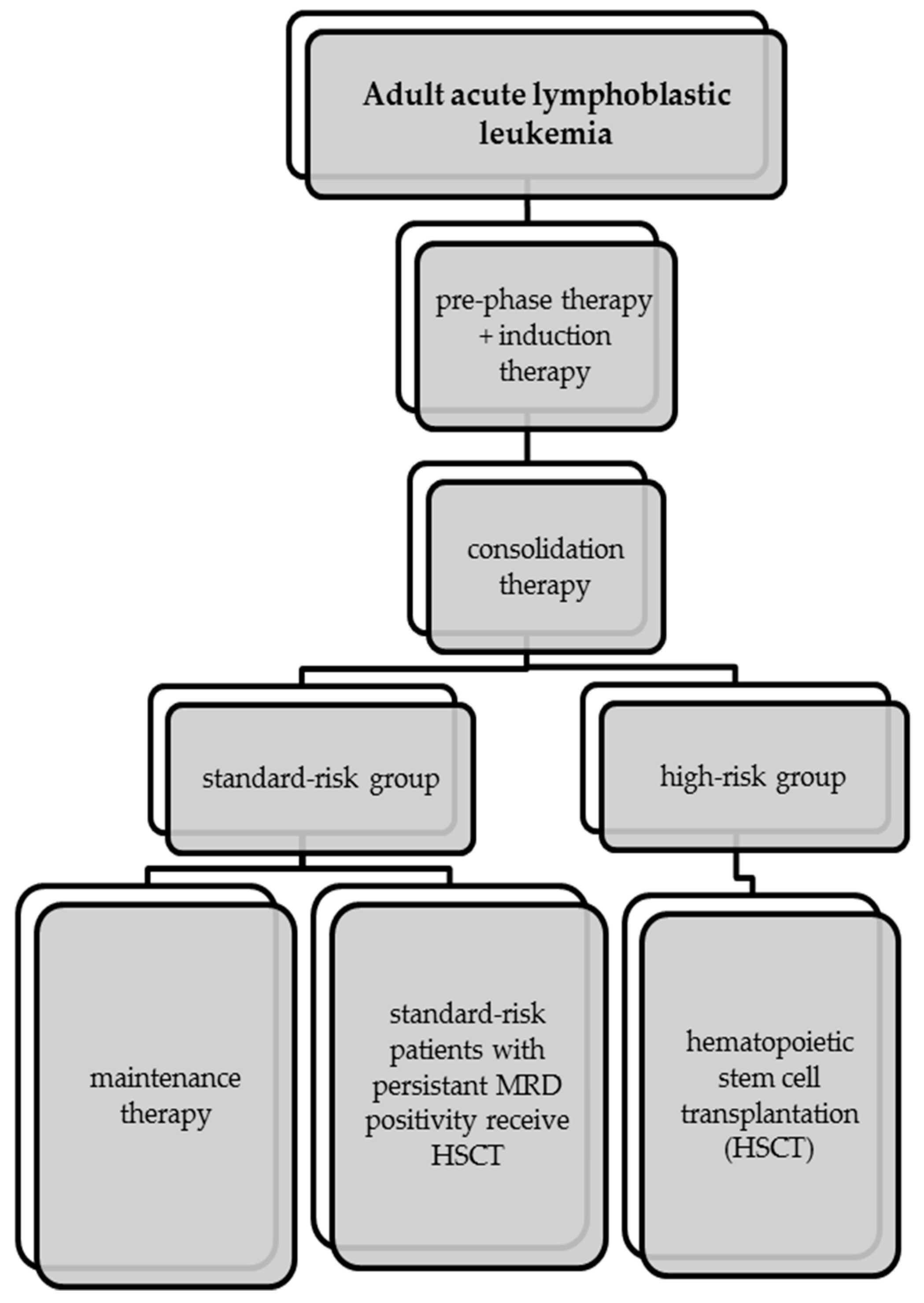
5. First-Line Therapy of Acute Lymphoblastic Leukemia
6. CNS Prophylaxis
7. Antibody-Based Therapy
7.1. Monotherapy with Blinatumomab in Relapsed/Refractory ALL
7.2. Monotherapy with Inotuzumab Ozogamicin in Relapsed/Refractory ALL
7.3. Chemoimmunotherapy
7.4. Chemoimmunotherapy with Inotuzumab Ozogamicin in Relapsed/Refractory ALL
7.5. Chemoimmunotherapy with Blinatumomab in Newly Diagnosed B-Cell ALL
7.6. Chemoimmunotherapy with Rituximab
7.7. Chemoimmunotherapy with Ofatumumab
7.8. Chemoimmunotherapy with Epratuzumab
8. Therapy Opportunities for Older Adults

{kind=link}
| Regimen | Study Population | N | Median Age | CR/CRi Rate | CR Duration | MRD- Negativity | OS Rate | Reference |
|---|---|---|---|---|---|---|---|---|
| Inotuzumab + mini-HCVD ± blinatumomab | Patients aged ≥60 years | 64 | 68 | 98% | 76% (3-year) | 95% | 54% (3-year) | [37] |
| Blinatumomab + POMP | Patients aged >60 years | 31 | 73 | 66% | DFS 56% (1-year) | 92% | 65% (1-year) | [38] |
9. Therapy of T-ALL
10. Therapy of Philadelphia Chromosome-Positive ALL
11. Chimeric Antigen Receptor (CAR) T Cell Therapy
12. Hematopoietic Stem Cell Transplantation
13. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hoelzer, D.; Bassan, R.; Dombret, H.; Ribera, J.; Buske, C. Acute lymphoblastic leukemia in adult patients: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow up. Ann. Hematol. Oncol. 2016, 27, 69–82. [Google Scholar]
- Faderl, S.; O’Brien, S.; Pui, C.H.; Stock, W.; Wetzler, M.; Hoelzer, D.; Kantarjian, H.M. Adult Acute Lymphoblastic Leukemia. Cancer 2010, 116, 1165–1176. [Google Scholar] [CrossRef] [PubMed]
- Inaba, H.; Greaves, M.; Mullighan, C. Acute lymphoblastic leukaemia. Lancet Haematol. 2013, 381, 1943–1955. [Google Scholar] [CrossRef]
- Coccaro, N.; Anelli, L.; Zagaria, A.; Specchia, G.; Albano, F. Next-Generation Sequencing in Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2019, 20, 2929. [Google Scholar] [CrossRef]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5h edition of the World Health Organizaion Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef]
- Moorman, A.V.; Barretta, E.; Butler, E.R.; Ward, E.J.; Twentyman, K.; Kirkwood, A.A.; Enshaei, A.; Schwab, C.; Creasey, T.; Leongamornlert, D.; et al. Prognostic impact of chromosomal abnormalities and copy number alterations in adult B-cell precursor acute lymphoblastic leukaemia: A UKALL14 study. Leukemia 2022, 36, 625–636. [Google Scholar] [CrossRef]
- Paietta, E.; Roberts, K.G.; Wang, V.; Gu, Z.; Buck, G.A.N.; Pei, D.; Cheng, C.; Levine, R.L.; Abdel-Wahab, O.; Cheng, Z.; et al. Molecular classification improves risk assessment in adult BCR-ABL1-negative B-ALL. Blood Adv. 2021, 138, 948–958. [Google Scholar] [CrossRef] [PubMed]
- Shilpa, P.; Hagop, K.; Jabbou, E. Adult Acute Lymphoblastic Leukemia. Mayo Clin. Proc. 2016, 91, 1645–1666. [Google Scholar]
- Kruse, A.; Abdel-Azim, N.; Kim, Y.-M.; Ruan, Y.; Phan, V.; Ogana, H.; Wang, W.; Lee, R.; Gang, E.J.; Khazal, S.; et al. Minimal Residual Disease Detection in Acute Lymphoblastic Leukemia. Int. J. Mol. Med. Sci. 2020, 21, 1054. [Google Scholar] [CrossRef]
- Van Dongen, J.; Van der Velden, V.; Brüggemann, M.; Orfao, A. Minimal residual disease diagnostics in acute lymphoblastic leukemia: Need for sensitive, fast, and standardized technnologies. Blood Adv. 2015, 125, 3996–4009. [Google Scholar] [CrossRef]
- Campana, D. Minimal Residual Disease in Acute Lymphoblastic Leukemia. Blood Adv. 2010, 2010, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Gökbuget, N.; Dombret, H.; Giebel, S.; Bruggemann, M.; Doubek, M.; Foà, R.; Hoelzer, D.; Kim, C.; Martinelli, G.; Parovichnikova, E.; et al. Minimal residual disease level predicts outcome in adults with Ph-negative B-precursor acute lymphoblastic leukemia. Hematology 2019, 24, 337–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brüggemann, M.; Raff, T.; Flohr, T.; Gökbuget, N.; Nakao, M.; Droese, J.; Lüschen, S.; Pott, C.; Ritgen, M.; Scheuring, U.; et al. Clinical significance of minimal residual disease quantification in adult patients with standard-risk acute lymphoblastic leukemia. Blood 2006, 107, 1116–1123. [Google Scholar] [CrossRef] [PubMed]
- Siegel, S.E.; Stock, W.; Johnson, R.H.; Advani, A.; Muffly, L.; Douer, D.; Reed, D.; Lewis, M.; Freyer, D.R.; Shah, B.; et al. Pediatric-Inspired Treatment Regimens for Adolescents and Young Adults with Philadelphia Chromosome-Negative Acute Lymphoblastic Leukemia. JAMA Oncol. 2018, 4, 725–734. [Google Scholar] [CrossRef] [PubMed]
- McNeer, J.; Bleyer, A. Acute lymphoblastic leukemia and lymphoblastic lymphoma in adolescents and young adults. Pediatr. Blood Cancer 2018, 65, e26989. [Google Scholar] [CrossRef]
- Siegel, S.E.; Advani, A.; Seibel, N.; Muffly, L.; Stock, W.; Luger, S.; Shah, B.; DeAngelo, D.; Freyer, D.R.; Douer, D.; et al. Treatment of young adults with Philadelphia-negative acute lymphoblastic leukemia and lymphoblastic lymphoma: Hyper-CVAD vs. pediatric-inspired regimens. Am. J. Hematol. 2018, 93, 1254–1266. [Google Scholar] [CrossRef] [PubMed]
- Larson, R. Managing CNS disease in adults with acute lymphoblastic leukemia. Leuk. Lymphoma 2017, 59, 3–13. [Google Scholar] [CrossRef]
- Jabbour, E.; Thomas, D.; Cortes, J.; Kantarjian, H.; O’Brien, S. Central nervous system prophylaxis in adults with acute lymphoblastic leukemia. Cancer 2010, 116, 2290–2300. [Google Scholar] [CrossRef]
- Del Principe, M.I.; Maurillo, L.; Buccisano, F.; Sconocchia, G.; Cefalo, M.; De Santis, G.; Di Veroli, A.; Ditto, C.; Nasso, D.; Postorino, M.; et al. Central Nervous System Involvement in Adult Acute Lymphoblastic Leukemia: Diagnostic Tools, Prophylaxis, and Therapy. Mediterr. J. Hematol. Infect. Dis. 2014, 6, e2014075. [Google Scholar] [CrossRef]
- Gökbuget, N.; Dombret, H.; Ribera, J.-M.; Fielding, A.K.; Advani, A.; Bassan, R.; Chia, V.; Doubek, M.; Giebel, S.; Hoelzer, D.; et al. International reference analysis of outcomes in adults with B-precursor Ph-negative relapsed/refractory acute lymphoblastic leukemia. Haematologica 2016, 101, 1524–1533. [Google Scholar] [CrossRef]
- Jammal, N.; Chew, S.; Jabbour, E.; Kantarjian, H. Antibody Based Therapy in Relapsed Acute Lymphoblastic Leukemia; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Kantarjian, H.; Stein, A.; Gökbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.-M.; Wei, A.; Dombret, H.; Foà, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Stein, A.S.; Kantarjian, H.; Gökbuget, N.; Bargou, R.; Litzow, M.R.; Rambaldi, A.; Ribera, J.-M.; Zhang, A.; Zimmerman, Z.; Zugmaier, G.; et al. Blinatumomab for Acute Lymphoblastic Leukemia Relapse after Allogeneic Hematopoietic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2019, 25, 1498–1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bueno, C.; Barrera, S.; Bataller, A.; Ortiz-Maldonado, V.; Elliott, N.; O’Byrne, S.; Wang, G.; Rovira, M.; Gutierrez-Agüera, F.; Trincado, J.L.; et al. CD34+CD19−CD22+ B-cell progenitors may underlie phenotypic escape in patients treated with CD19-directed therapies. Blood 2022, 140, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Kantajian, H.; DeAngelo, D.; Stelljes, M.; Martinelli, G.; Liedtke, M. Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblasti Leukemia. N. Engl. J. Med. 2016, 375, 740–753. [Google Scholar] [CrossRef]
- Sasaki, K.; Kantarjian, H.M.; Ravandi, F.; Short, N.J.; Kebriaei, P.; Huang, X.; Rytting, M.E.; Jain, N.; Marina, Y.; Konopleva, G.G.; et al. Sequentil Combination of Low-Intensity Chemotherpy (Mini-hyper-CVD) Plus Inotuzumab Ozogamicin with or without Blinatumomab in Patients with Relapsed/Refractory Philadelphia Chromosome-Negative Acute Lymphoblastic Leukemia (ALL): A Phase 2 Trial. Blood Adv. 2018, 132, 553. [Google Scholar] [CrossRef]
- Advani, A.S.; Moseley, A.; Liedtke, M.; O’Donnell, M.R.; Aldoss, I.; Mims, M.P.; O’Dwyer, K.M.; Othus, M.; Erba, H.P. SWOG 1312 Final Results: A Phase 1 Trial of Inotuzumab in Combination with CVP (Cyclosphosphamide, Vincristine, Prednisone) for Relapsed/Refractory CD22+ Acute Leukemia. Blood Adv. 2019, 134, 227. [Google Scholar] [CrossRef]
- Richard-Carpentier, G.; Kantarjian, H.M.; Short, N.J.; Ravandi, F.; Ferrajoli, A.; Schroeder, H.M.; Garcia-Manero, G.; Bravo, G.M.; Cortes, J.E.; Kwari, B.M.; et al. Updated Results from the Phase II Study of Hyper-CVAD in Sequential Combination with Blinatumomab in Newly Diagnosed Adults with B-Cell Acute Lymphoblastic Leukemia (ALL). Blood Adv. 2019, 134, 3807. [Google Scholar] [CrossRef]
- Maury, S.; Chevret, S.; Thomas, X.; Heim, D. Rituximab in B-Lineage Adult Acute Lymhoblastic Leukemia. N. Engl. J. Med. 2016, 375, 1044–1053. [Google Scholar] [CrossRef]
- Thomas, D.; O’Brien, S.; Faderl, S.; Garcia-Manero, G.; Ferrajoli, A.; Wierda, W. Chemoimmunotherapy with a Modified Hyper-CVAD and Rituximab Regimen Improves Outcome in De Novo Philadelphia Chromosome-Negative Precursor B-Lineage Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2010, 28, 3380–3388. [Google Scholar] [CrossRef]
- Thomas, D.A.; Faderl, S.; O’Brien, S.; Bueso-Ramos, C.; Cortes, J.; Garcia-Manero, G.; Giles, F.J.; Verstovsek, S.; Wierda, W.G.; Pierce, S.A.; et al. Chemoimmunotherapy with hyper-CVAD plus rituximab for the treatment of adult Burkitt and Burkitt-type lymphoma or acute lymphoblastic leukemia. Cancer 2006, 106, 1569–1580. [Google Scholar] [CrossRef]
- Richard-Carpentier, G.; Kantarjian, H.M.; Konopleva, M.Y.; Patel, K.P.; Roberts, K.G.; Gu, Z.; Ravandi, F.; Jain, N.; Short, N.J.; Garcia-Manero, G.; et al. Phase II Study of Hyper-CVAD Regimen in Combination with Ofatumumab (HCVAD-O) As Frontline Therapy for Adult Patients (pts) with CD20-Positive B-Cell Acute Lymphoblastic Leukemia (B-ALL). Blood Adv. 2019, 134, 2577. [Google Scholar] [CrossRef]
- Sasaki, K.; Kantarjian, H.M.; Morita, K.; Short, N.J.; Konopleva, M.; Jain, N.; Ravandi, F.; Garcia-Manero, G.; Wang, S.; Khoury, J.D.; et al. Hyper-CVAD plus ofatumumab versus hyper-CVAD plus rituximab as frontline therapy in adults with Philadelphia chromosome-negative acute lymphoblastic leukemia: A porpensity score analysis. Cancer 2021, 127, 3381–3389. [Google Scholar] [CrossRef]
- Advani, A.S.; McDonough, S.; Coutre, S.; Wood, B.; Radich, J.; Mims, M.; O’Donnell, M.; Elkins, S.; Becker, M.; Othus, M.; et al. SWOG S0910: A Phase 2 Trial of Clofarabine/Cytarabine/Epratuzumab for Relapsed/Refractory Acute Lymphoblastic Leukemia. Br. J. Haematol. 2014, 165, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Gökbuget, N. How I treat older patients with ALL. Blood Adv. 2013, 122, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Ravandi, F.; Short, N.J.; Huang, X.; Jain, N.; Sasaki, K.; Daver, N.; Pemmaraju, N.; Khoury, J.D.; Jorgensen, J.; et al. Inotuzumab ozogamicin in combination with low-intensity chemotherapy for older adults with Philadelphia chromosome-negative acute lymphoblastic leuaemia: A single-arm, phase 2 study. Lancet Oncol. 2018, 19, 240–248. [Google Scholar] [CrossRef]
- Short, N.J.; Kantarjian, H.M.; Ravandi, F.; Huang, X.; Jain, N.; Kadia, T.M.; Khoury, J.D.; Jorgensen, J.L.; Wang, S.A.; Alvarado, Y.; et al. Reduced-intensity Chemotherapy with Mini-Hyper-CVD Plus Inotuzumab Ozogamicin, with or without Blinatumomab, in Older Adults with Newly Diagnosed Philadelphia Chromosome-Negative Acute Lymphoblastic Leukemia: Results form a Phase II Study. ASH Annu. Meet. Expo. 2020, 136, 15–17. [Google Scholar] [CrossRef]
- Advani, A.S.; Moseley, A.; O’Dwyer, K.M.; Wood, B.; Fang, M.; Wieduwilt, M.J.; Aldoss, I.; Park, J.H.; Klisovic, R.; Baer, M.R.; et al. Results of SWOG 1318: A Phase 2 Trial of Blinatumomab Followed By POMP (Prednisone, Vincristine, Methotrexate, 6-Mercaptopurine) Maintenance in Elderly Patients with Newly Diagnosed Philadelphia Chromosome Negative B-Cell Acute Lmyphoblastic Leukemia. Blood Adv. 2018, 132, 33. [Google Scholar] [CrossRef]
- Bardelli, V.; Arniani, S.; Pierini, V.; Di Giacomo, D.; Pierini, T.; Gorello, P.; Mecucci, C.; La Starza, R. T-Cell Acute Lymphoblastic Leukemia: Biomarkers and their Clinical Usefulness. Genes 2021, 12, 1118. [Google Scholar] [CrossRef]
- Raetz, E.; Teachey, D. T-cell acute lymphoblastic leukemia. Hematol. Am. Soc. Hematol. Educ. Program 2016, 2016, 580–588. [Google Scholar] [CrossRef]
- Rakowski, L.A.; Garagiola, D.D.; Li, C.M.; Decker, M.; Caruso, S.; Jones, M.; Kuick, R.; Cierpicki, T.; Maillard, I.; Chiang, M.Y. Convergence of the ZMIZ1 and NOTCH1 pathways at C-MYC in acute T lymphoblastic leukemias. Cancer Res. 2013, 73, 930–941. [Google Scholar] [CrossRef]
- Luo, X.; Tan, H.; Thou, Y.; Xiao, T.; Wang, C.; Li, Y. Notch1 signaling is involved in regulating Foxp3 expression in T-ALL. Cancer Cell Int. 2013, 13, 10–34. [Google Scholar] [CrossRef] [PubMed]
- Nolin, E.; Gans, S.; Llamas, L.; Bandyopadhyay, S.; Brittain, S.M.; Bernasconi-Elias, P.; Carter, K.P.; Loureiro, J.J.; Thomas, J.R.; Schirle, M.; et al. Discoveery of a ZIP7 inhibitor form a Notch pathway screen. Nat. Chem. Biol. 2019, 15, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Pullarkat, V.A.; Lacayo, N.J.; Jabbour, E.; Rubnitz, J.E.; Bajel, A.; Laetsch, T.W.; Leonard, J.; Colace, S.I.; Khaw, S.L.; Fleming, S.A.; et al. Venetoclax and Navitoclax in Combination with Chemotherapy in Patients with Relapsed or Refractory Acute Lymphoblastic Leukemia and Lymphoblastic Lymphoma. Cancer Discov. 2021, 11, 1440–1453. [Google Scholar] [CrossRef] [PubMed]
- Gökbuget, N.; Basara, N.; Baurmann, H.; Beck, J.; Brüggemann, M.; Diedrich, H.; Güldenzoph, B.; Hartung, G.; Horst, H.-A.; Hüttmann, A.; et al. High single-drug activity of nelarabine in relapsed T-lymphoblastic leukemia/lymphoma offers curative option with subsequent stem cell transplantation. Blood Adv. 2011, 118, 3504–3511. [Google Scholar] [CrossRef]
- DeAngelo, D.J.; Yu, D.; Johnson, J.L.; Coutre, S.E.; Stone, R.M.; Stopeck, A.T.; Gockerman, J.P.; Mitchell, B.S.; Appelbaum, F.R.; Larson, R. Nelarabine induces complete remission in adults with relapsed or refractory T-lineage acute lymphoblastic leukemia or lymphoblastic lymphoma: Cancer and Leukemia Group B study 19801. Blood Adv. 2007, 109, 5136–5142. [Google Scholar] [CrossRef]
- Abaza, Y.; Kantarjian, H.M.; Faderl, S.; Jabbour, E.; Jain, N.; Thomas, D.; Kadia, T.; Borthakur, G.; Khoury, J.D.; Burger, J.; et al. Hyper-CVAD plus nelarabine in newly diagnosed adult T-cell acute lymphoblastic leukemia and T-lymphoblastic lymphoma. Am. J. Hematol. Oncol. 2017, 93, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Gavralidis, A.; Brunner, A. Novel Therapies in the Treatment of Adult Acute Lymphoblastic Leukemia. Curr. Hematol. Malig. Rep. 2020, 15, 294–304. [Google Scholar] [CrossRef]
- Daver, N.; Thomas, D.; Ravandi, F.; Cortes, J.; Garris, R.; Jabbour, E.; Garcia-Manero, G.; Borthakur, G.; Kadia, T.; Rytting, M.; et al. Final report of a phase II study of imatinib mesylate with hyper-CVAD for the front-line treatment of adult patients with Philadelphia chromosome-positive acute lymphoblastic leukemia. Haematologica 2015, 100, 653–661. [Google Scholar] [CrossRef]
- Fielding, A.K.; Rowe, J.M.; Buck, G.; Foroni, L.; Gerrard, G.; Litzow, M.R.; Lazarus, H.; Luger, S.M.; Marks, D.I.; McMillan, A.K.; et al. UKALLXII/ECOG2993: Addition of imatinib to a standard treatment regimen enhances long-term outcomes in Philadelphia positive acute lymphoblastic leukemia. Blood Adv. 2014, 123, 843–850. [Google Scholar] [CrossRef]
- Chalandon, Y.; Thomas, X.; Hayette, S.; Cayuela, J.-M.; Abbal, C.; Huguet, F.; Raffoux, E.; Leguay, T.; Rousselot, P.; Lepretre, S.; et al. Randomized study of reduced-intensity chemotherapy combinded with imatinib in adults with Ph-positive acute lymphoblastic leukemia. Blood Adv. 2015, 125, 3711–3719. [Google Scholar] [CrossRef]
- Ravandi, F.; O’Brien, S.M.; Cortes, J.; Thomas, D.M.; Garris, R.; Faderl, S.; Burger, J.A.; Rytting, M.E.; Ferrajoli, A.; Wierda, W.G.; et al. Long-term follow-up of a phase 2 study of chemotherapy plus dasatinib for the initial treatment of patients with Philadelphia chromosome-positive acute lymphoblastic leukemia. Cancer 2015, 121, 4158–4164. [Google Scholar] [CrossRef] [PubMed]
- Rousselot, P.; Coudé, M.M.; Gokbuget, N.; Gambacorti Passerini, C.; Hayette, S.; Cayuela, J.M.; Huguet, F.; Leguay, T.; Chevallier, P.; Salanoubat, C.; et al. Dasatinib and low-intensity chemotherapy in elderly patietns with Philadelphia chromosome-positive ALL. Blood Adv. 2016, 128, 774–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiaretti, S.; Ansuinelli, M.; Vitale, A.; Elia, L.; Matarazzo, M.; Piciocchi, A.; Fazi, P.; Di Raimondo, F.; Santoro, L.; Fabbiano, F.; et al. A multicenter total therapy strategy for de novo adult Philadelphia chromosome positive acute lymphoblastic leukemia patients: Final results of the GIMEMA LAL1509 protocol. Haematologica 2020, 106, 1828–1838. [Google Scholar] [CrossRef]
- Short, N.J.; Kantarjian, H.M.; Ravandi, F.; Huang, X.; Daver, N.G.; Dinardo, M.C.D.; Konopleva, M.Y.; Pemmaraju, N.; Wierda, W.G.; Garcia-Manero, G.; et al. Long-Term Safety and Efficacy of Hyper-CVAD Plus Ponatinib As Frontline Therapy for Adults with Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia. Blood Adv. 2019, 134, 283. [Google Scholar] [CrossRef]
- Rambaldi, A.; Ribera, J.; Kantarjian, H.M.; Dombret, H.; Ottmann, O.G.; Stein, A.S.; Tuglus, C.A.; Zhao, X.; Kim, C.; Martinelli, G. Blinatumomab Compared With Standard of Care for the Treatment of Adult Patients With Relapsed/Refractory Philadelphia Chromosome-Positive B-Precursor Acute Lymphoblastic Leukemia. Cancer 2020, 126, 304–310. [Google Scholar] [CrossRef]
- Assi, R.; Kantarjian, H.; Short, N.J.; Daver, N.; Takahashi, K.; Garcia-Manero, G.; DiNardo, C.; Burger, J.; Cortes, J.; Jain, N.; et al. Safety and Efficacy of Blinatumomab in Combination with a Tyrosine Kinase Inhibitor for the Treatment of Relapsed Philadelphia Chromosome-positive Leukemia. Clin. Lymphoma Myeloma Leuk. 2017, 12, 897–901. [Google Scholar] [CrossRef] [PubMed]
- Foà, R.; Bassan, R.; Vitale, A.; Elia, L.; Piciocchi, A.; Puzzolo, M.-C.; Canichella, M.; Viero, P.; Ferrara, F.; Lunghi, M.; et al. Dasatinib-Blinatumomab for Ph-Positive Acute Lymphoblastic Leukemia in Adults. N. Engl. J. Med. 2020, 383, 1613–1623. [Google Scholar] [CrossRef]
- Short, N.J.; Konopleva, M.; Jabbour, E.; Kadia, T.M.; Daver, N.; Cook, R.; Jain, N.; Ravandi, F. Interim Results of the Phase I/II Study of the Ponatinib, Venotoclax and Dexamethasone for Patients with Relapsed or Refractory Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia. Blood Adv. 2020, 136, 11–12. [Google Scholar] [CrossRef]
- Maude, S.; Shpall, E.; Grupp, S. Chimeric antigen receptor T-cell therapy for ALL. Hematol. Am. Soc. Hematol. Educ. Program 2014, 2014, 559–564. [Google Scholar] [CrossRef]
- Maude, S.; Laetsch, T.; Beuchner, J.; Rives, S. Tisagenlecleucel in Childen and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Park, J.; Riviere, I.; Gonen, M.; Wang, X. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymhoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, A.; Turtle, C. Toxicities of CD19 CAR-T cell immunotherapy. Am. J. Hematol. 2019, 94, S42–S49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, L.M.; Baggott, C.; Prabhu, S.; Pacenta, H.L.; Phillips, C.L.; Rossoff, J.; Stefanski, H.E.; Talano, J.A.; Moskop, A.; Margossian, S.P.; et al. Disease Burden Affects Outcome in Pediatric and Young Adult B-Cell Lymphoblastic Leukemia After Commercial Tisagenlecleucel: A Peditaric Real-World Chimeric Antigen Receptor Consortium Report. J. Clin. Oncol. 2022, 40, 945–955. [Google Scholar] [CrossRef] [PubMed]
- Shah, B.D.; Bishop, M.R.; Oluwole, O.O.; Logan, A.C.; Baer, M.R.; Donnellan, W.B.; O’Dwyer, K.M.; Holmes, H.; Arellano, M.L.; Ghobadi, A.; et al. KTE-X19 anti-CD19 CAR T-cell therapy in adult relapsed/refractory acute lymphoblastic leukemia: ZUMA-3 phase 1 results. Blood Adv. 2021, 138, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Shah, B.D.; Ghobadi, A.; Oluwole, O.; Logan, A.C.; Boissel, N.; Cassaday, R.D.; Leguay, T.; Bishop, M.R.; Topp, M.S.; Tzachanis, D.; et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: Phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet 2021, 398, 491–502. [Google Scholar] [CrossRef]
- Giebel, S.; Marks, D.I.; Boissel, N.; Baron, F.; Chiaretti, S.; Ciceri, F.; Cornelissen, J.J.; Doubek, M.; Esteve, J.; Fielding, A.; et al. Hematopoietic stem cell transpantation for adults with Philadelphia chromosome-negative acute lymphoblastic leukemia in first remission: A position statement of the European Working Group for Adult Acute Lymphoblastic Leukemia (EWALL) and the Acute Leukemia Working Party of the European Society for Blood and Marrow Transplantation (EBMT). Bone Marrow Transpl. 2019, 54, 798–809. [Google Scholar]
- Khan, M.; Elmaaz, A.; Gul, T.; Ingrassia, F.; Cappuzzo, V.; Marceno, R. Stem Cells in Clinical Practice and Tissue Engineering; IntechOpen: London, UK, 2018. [Google Scholar]
- Ribera, J.M.; Oriol, A.; Bethencourt, C.; Parody, R.; Hernández-Rivas, J.M.; Moreno, M.J.; del Potro, E.; Torm, M.; Rivas, C.; Besalduch, J.; et al. Comparison of intensive chemotherapy, allogeneic or autlogous stem cell transplantation as post-remission treatment for adult patients with high-risk acute lymphoblastic leukemia. Results of the PETHEMA ALL-93 trial. Haematologica 2005, 90, 1346–1356. [Google Scholar]
- Goldstone, A.H.; Richards, S.M.; Lazarus, H.M.; Tallman, M.S.; Buck, G.; Fielding, A.K.; Burnett, A.K.; Chopra, R.; Wiernik, P.H.; Foroni, L.; et al. In adults with standard-risk acute lymphoblastic leukemia, the greates benefit is achieved from a matched sibling allogeneic transplantation in first complete remission, and conventional consolidation/maintenance chemotherapy in all patients: Final results of the International ALL Trial (MRC UKALL XII/ ECOG E2993). Blood J. Am. Soc. Hematol. 2008, 111, 1827–1833. [Google Scholar]
- Cornelissen, J.J.; van der Holt, B.; Verhoef, G.E.; van’t Veer, M.B.; van Oers, M.H.; Schouten, H.C.; Ossenkoppele, G.; Sonneveld, P.; Maertens, J.; van Marwijk Kooy, M.; et al. Myeloablative allogeneic versus autologus stem cell transplantatiion in adult patients with acute lymphoblastic leukemia in first remission: A prospective sibling donor verus no-donor comparison. Blood Adv. 2009, 113, 1375–1382. [Google Scholar] [CrossRef]
- Steiner, N.; Brunelli, L.; Hetzenauer, G.; Lindner, B.; Göbel, G.; Rudzki, J.; Peschel, I.; Nevinny-Stickel, M.; Nussbaumer, W.; Mayer, W.; et al. Early autologous and allogeneic peripheral bood stem cell transplantation for adult patietns with acute B and T cell precursor neoplasms: A 12-year single center experience. Ann. Hematol. Oncol. 2021, 100, 809–816. [Google Scholar] [CrossRef]
- Ribera, J.-M.; Morgades, M.; Ciudad, J.; Montesinos, P.; Esteve, J.; Genescà, E.; Barba, P.; Ribera, J.; García-Cadenas, I.; Moreno, M.J.; et al. Chemotherapy or allogeneic transplantation in high-risk Philadelphia chromosome-negative adult lymphoblastic leukemia. Blood Adv. 2021, 137, 1879–1894. [Google Scholar] [CrossRef] [PubMed]

| Subgroup | Immunophenotype | Cytogenetic, Molecular Genetics |
|---|---|---|
| B-Lineage ALL | HLA-DR+, TdT+/−, CD19+, cyCD79a+, cyCD22+ | |
| Pro-B-ALL | CD10- | t(v;11), KMT2A rearrangements |
| c-common-ALL | CD10+ | t(9;22), BCR::ABL1; IKZF1 |
| Premature B-ALL | cyIgM+ | t(1;19)TCF3::PBX1; t(9;22); BCR::ABL1 IKZF1 |
| Mature B-ALL | TdT−, CD34−, sIg+ | t(8;14); MYC rearrangements |
| T-lineage ALL | TdT+/−, cyCD3+, CD7+ | |
| Pro-T-ALL | cCD3+, sCD3−, CD1a−, CD2+, CD5−, CD7+, CD34− | NOTCH1/FBXW7 mutations |
| Pre-T/immature T-ALL | cCD3+, sCD3−, CD1a−, CD2+, CD5+, CD7+, CD34− | NOTCH1/FBXW7 mutations, HOXA, TLX3 |
| Cortical T-ALL | cCD3+, sCD3+/−, CD1a+, CD2+, CD5+, CD7+, CD34− | NOTCH1/FBXW7 mutations, TLX1, NKX2.1./2.2, TLX3, TAL/LMO |
| Early T-cell precursor ALL | cCD3+, sCD3−, CD1a−, CD2+, CD7+, HLA-DR, CD13, CD33, CD34, CD117 | NOTCH1/FBXW7 mutations, HOXA, MEF2C, BCL11B |
| Mature T-ALL | cCD3+, sCD3+, CD1a−, CD2+, CD5+, CD7+, CD34− | NOTCH1/FBXW7 mutations, TAL/LMO |
| Therapy | Target | Antibody Type |
|---|---|---|
| Blinatumomab | CD19 | BiTE |
| Inotuzumab ozogamicin | CD22 | ADC |
| Rituximab | CD20 | mAb |
| Ofatumumab | CD20 | mAb |
| Epratuzumab | CD22 | mAb |
| Regimen | Indication | N | Median Age | CR/CRi Rate | CR Duration | MRD-Negativity | OS Rate | Reference |
|---|---|---|---|---|---|---|---|---|
| Inotuzumab + mini-HCVD ± blinatumomab | R/R ALL | 84 | 35 | 80% | 52% (2-year) | 80% | 39% (2-year) | [26] |
| Inotuzumab + CVP | R/R CD22+ ALL | 48 | 43 | 61% | / | / | 10.9 months (median) | [27] |
| Hyper-CVAD + blinatumomab | Newly diagnosed B-ALL | 27 | 38 | 100% | RFS 76 % | 96% | 89% (1-year) | [28] |
| Hyper-CVAD + rituximab | CD20+, ALL | 209 | 40 | 92% | / | 91% | EFS (2-year) 65% | [29] |
| Standard/ modified Hyper-CVAD + rituximab | B-ALL | 282 | 41 | 95% | 78% | 81% | 60% (3-year) | [30] |
| Hyper-CVAD + rituximab | Newly diagnosed B-ALL | 31 | 46 | 86% | 67% (3-year) | / | 89% | [31] |
| Hyper-CVAD + MTX + cytarabine+ ofatumumab | Newly diagnosed CD20+, B-ALL | 69 | 41 | 98% | / | 65% | 68% (4-year) | [32] |
| Hyper-CVAD + ofatumumab | Newly diagnosed CD20+ B-ALL | 222 | 44 | 93% | / | 93% | 66% (4-year) | [33] |
| Regimen | Study Population | N | Median Age | CR Rate | HSCT Rate | EFS | RFS | OS Rate | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Imatinib + hyper-CVAD | Newly diagnosed Ph + ALL | 54 | 51 | 93% | 30% | 43% (5-year) | 43% (5-year) | 43% (5-year) | [49] |
| Imatinib + intensive chemotherapy | Newly diagnosed Ph + ALL (age 15–65) | 266 | 42 | 92% | 72% | 33% (4-year) | 50% (4-year) | 38% (4-year) | [50] |
| Imatinib + lower-intensity chemotherapy | 268 | 49 | 98% | 62% | 37.1% (5-year) | EFS 37% (5-year) | 46% (5-year) | [51] |
| Regimen | Study Population | N | Median Age | CR Rate | HSCT Rate | EFS | RFS | OS Rate | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Dasatinib + intensive chemotherapy | Ph + ALL | 72 | 55 | 96% | 17% | 27 months (median) | 44% (5-year) | 46% (5-year) | [52] |
| Dasatinib + lower-intensity chemotherapy | Ph + ALL | 71 | 69 | 96% | 10% | 27% (5-year) | EFS 28% (5-year) | 36% (5-year) | [53] |
| Dasatinib + lower-intensity chemotherapy | Ph + ALL | 60 | 42 | 100% | 42% | 48% (5-year) | 49% (3-year) | 58% (3-year) | [54] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Künz, T.; Hauswirth, A.W.; Hetzenauer, G.; Rudzki, J.; Nachbaur, D.; Steiner, N. Changing Landscape in the Treatment of Adult Acute Lymphoblastic Leukemia (ALL). Cancers 2022, 14, 4290. https://doi.org/10.3390/cancers14174290
Künz T, Hauswirth AW, Hetzenauer G, Rudzki J, Nachbaur D, Steiner N. Changing Landscape in the Treatment of Adult Acute Lymphoblastic Leukemia (ALL). Cancers. 2022; 14(17):4290. https://doi.org/10.3390/cancers14174290
Chicago/Turabian StyleKünz, Tina, Alexander W. Hauswirth, Gabriele Hetzenauer, Jakob Rudzki, David Nachbaur, and Normann Steiner. 2022. "Changing Landscape in the Treatment of Adult Acute Lymphoblastic Leukemia (ALL)" Cancers 14, no. 17: 4290. https://doi.org/10.3390/cancers14174290
APA StyleKünz, T., Hauswirth, A. W., Hetzenauer, G., Rudzki, J., Nachbaur, D., & Steiner, N. (2022). Changing Landscape in the Treatment of Adult Acute Lymphoblastic Leukemia (ALL). Cancers, 14(17), 4290. https://doi.org/10.3390/cancers14174290