

Combined Focused Next-Generation Sequencing Assays to Guide Precision Oncology in Solid Tumors: A Retrospective Analysis from an Institutional Molecular Tumor Board
 , , , , add
Show full author list
, , , , add
Show full author list
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Tissue Samples and DNA Extraction
2.3. Library Preparation and Next Generation Sequencing
2.4. Analysis of Genetic Variants
2.5. Variants Interpretation and Therapy Recommendations
2.6. Immunohistochemistry for PD-L1 and MMRD/MSI
2.7. Outcome Assessment
2.8. Statistical Analyses
3. Results
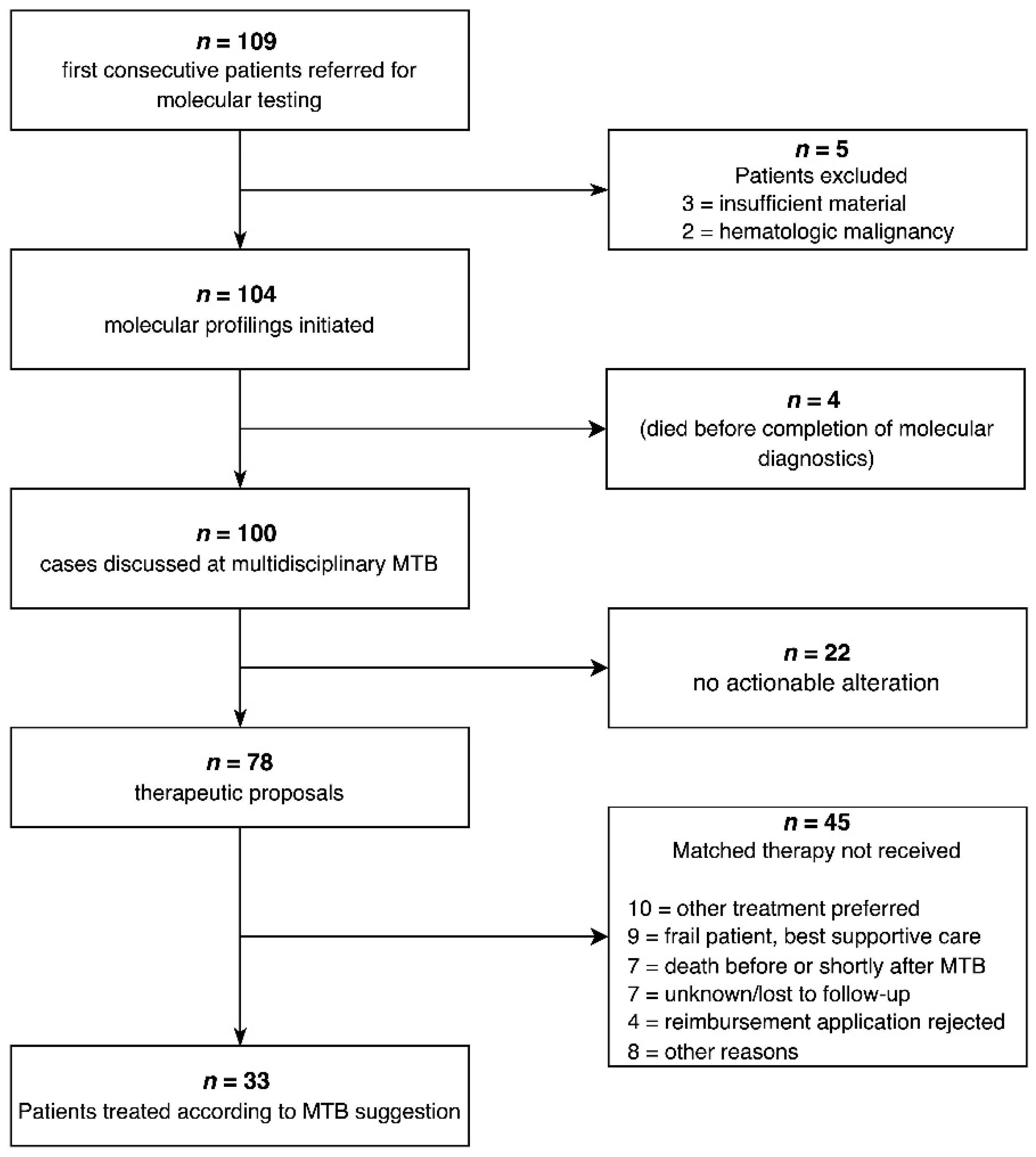
3.1. Clinical Implementation of Precision Oncology—Outline
3.2. Patients Characteristics
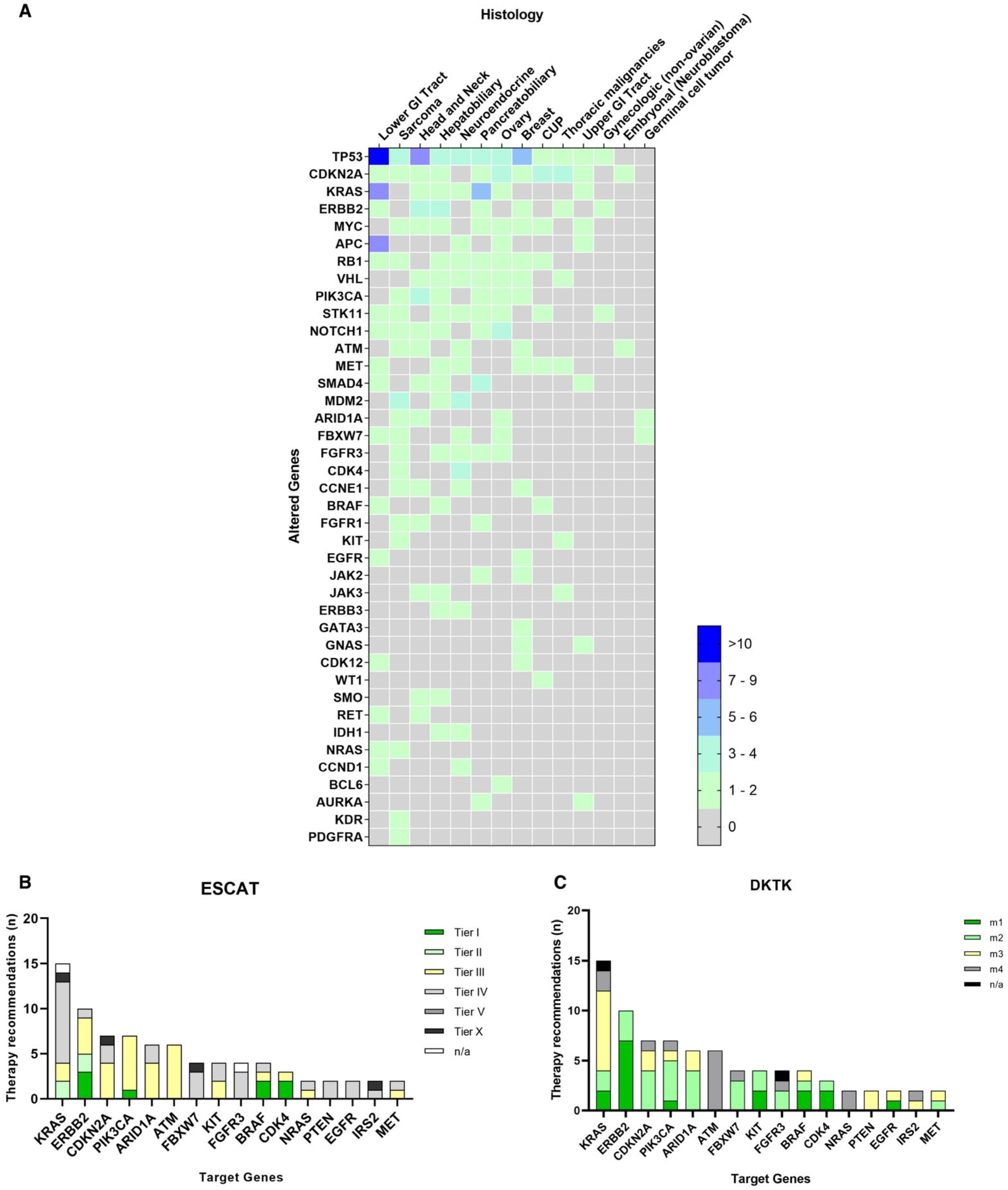
3.3. Molecular Testing and Therapeutic Suggestions
3.4. Outcomes with MTB-Recommended Therapy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rodriguez, H.; Zenklusen, J.C.; Staudt, L.M.; Doroshow, J.H.; Lowy, D.R. The next horizon in precision oncology: Proteogenomics to inform cancer diagnosis and treatment. Cell 2021, 184, 1661–1670. [Google Scholar] [CrossRef] [PubMed]
- Sparano, J.A.; Gray, R.J.; Makower, D.F.; Pritchard, K.I.; Albain, K.S.; Hayes, D.F.; Geyer, C.E., Jr.; Dees, E.C.; Goetz, M.P.; Olson, J.A.; et al. Adjuvant Chemotherapy Guided by a 21-Gene Expression Assay in Breast Cancer. N. Engl. J. Med. 2018, 379, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Vieira, A.; Schmitt, F. An Update on Breast Cancer Multigene Prognostic Tests—Emergent Clinical Biomarkers. Front. Med. 2018, 5, 248. [Google Scholar] [CrossRef] [PubMed]
- Merdan, S.; Subramanian, K.; Ayer, T.; Van Weyenbergh, J.; Chang, A.; Koff, J.L.; Flowers, C. Gene expression profiling-based risk prediction and profiles of immune infiltration in diffuse large B-cell lymphoma. Blood Cancer J. 2021, 11, 2. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of Chemotherapy plus a Monoclonal Antibody against HER2 for Metastatic Breast Cancer That Overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef]
- Bokemeyer, C.; Bondarenko, I.; Makhson, A.; Hartmann, J.T.; Aparicio, J.; de Braud, F.; Donea, S.; Ludwig, H.; Schuch, G.; Stroh, C.; et al. Fluorouracil, Leucovorin, and Oxaliplatin with and Without Cetuximab in the First-Line Treatment of Metastatic Colorectal Cancer. J. Clin. Oncol. 2009, 27, 663–671. [Google Scholar] [CrossRef]
- Solomon, B.J.; Mok, T.; Kim, D.-W.; Wu, Y.-L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; et al. First-Line Crizotinib versus Chemotherapy in ALK-Positive Lung Cancer. N. Engl. J. Med. 2014, 371, 2167–2177. [Google Scholar] [CrossRef]
- Mok, T.; Camidge, D.; Gadgeel, S.; Rosell, R.; Dziadziuszko, R.; Kim, D.-W.; Pérol, M.; Ou, S.-H.; Ahn, J.; Shaw, A.; et al. Updated overall survival and final progression-free survival data for patients with treatment-naive advanced ALK-positive non-small-cell lung cancer in the ALEX study. Ann. Oncol. 2020, 31, 1056–1064. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.L.; Ahn, M.J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or Platinum–Pemetrexed in EGFR T790M–Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef]
- Long, G.V.; Flaherty, K.T.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; De Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/ K-mutant melanoma: Long-term survival and safety analysis of a phase 3 study. Ann. Oncol. 2017, 28, 1631–1639. [Google Scholar] [CrossRef]
- O’Brien, S.G.; Guilhot, F.; Larson, R.A.; Gathmann, I.; Baccarani, M.; Cervantes, F.; Cornelissen, J.J.; Fischer, T.; Hochhaus, A.; Hughes, T.; et al. Imatinib Compared with Interferon and Low-Dose Cytarabine for Newly Diagnosed Chronic-Phase Chronic Myeloid Leukemia. N. Engl. J. Med. 2003, 348, 994–1004. [Google Scholar] [CrossRef]
- Schirrmacher, V. From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment (Review). Int. J. Oncol. 2019, 54, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.P.; Shum, E.; Rajdev, L.; Cheng, H.; Goel, S.; Perez-Soler, R.; Halmos, B. Impact and Diagnostic Gaps of Comprehensive Genomic Profiling in Real-World Clinical Practice. Cancers 2020, 12, 1156. [Google Scholar] [CrossRef] [PubMed]
- Remon, J.; Dienstmann, R. Precision oncology: Separating the wheat from the chaff. ESMO Open 2018, 3, e000446. [Google Scholar] [CrossRef] [PubMed]
- Presley, C.J.; Tang, D.; Soulos, P.R.; Chiang, A.C.; Longtine, J.A.; Adelson, K.B.; Herbst, R.S.; Zhu, W.; Nussbaum, N.C.; Sorg, R.A.; et al. Association of Broad-Based Genomic Sequencing with Survival Among Patients with Advanced Non–Small Cell Lung Cancer in the Community Oncology Setting. JAMA J. Am. Med. Assoc. 2018, 320, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Niogret, J.; Dalens, L.; Truntzer, C.; Chevrier, S.; Favier, L.; Lagrange, A.; Coudert, B.; Fraisse, C.; Foucher, P.; Zouak, A.; et al. Does large NGS panel analysed using exome tumour sequencing improve the management of advanced non-small-cell lung cancers? Lung Cancer 2021, 161, 98–107. [Google Scholar] [CrossRef]
- Cobain, E.F.; Wu, Y.-M.; Vats, P.; Chugh, R.; Worden, F.; Smith, D.C.; Schuetze, S.M.; Zalupski, M.M.; Sahai, V.; Alva, A.; et al. Assessment of Clinical Benefit of Integrative Genomic Profiling in Advanced Solid Tumors. JAMA Oncol. 2021, 7, 525–533. [Google Scholar] [CrossRef]
- Fernandes, G.S.; Marques, D.F.; Girardi, D.M.; Braghiroli, M.I.F.; Coudry, R.A.; Meireles, S.I.; Katz, A.; Hoff, P.M. Next-generation sequencing-based genomic profiling: Fostering innovation in cancer care? Clinics 2017, 72, 588–594. [Google Scholar] [CrossRef]
- Hoefflin, R.; Lazarou, A.; Hess, M.; Reiser, M.; Wehrle, J.; Metzger, P.; Frey, A.; Becker, H.; Aumann, K.; Berner, K.; et al. Transitioning the Molecular Tumor Board from Proof of Concept to Clinical Routine: A German Single-Center Analysis. Cancers 2021, 13, 1151. [Google Scholar] [CrossRef]
- Varnier, R.; Le Saux, O.; Chabaud, S.; Garin, G.; Sohier, E.; Wang, Q.; Paindavoine, S.; Pérol, D.; Baudet, C.; Attignon, V.; et al. Actionable molecular alterations in advanced gynaecologic malignancies: Updated results from the ProfiLER programme. Eur. J. Cancer 2019, 118, 156–165. [Google Scholar] [CrossRef]
- Massard, C.; Michiels, S.; Ferté, C.; Le Deley, M.-C.; Lacroix, L.; Hollebecque, A.; Verlingue, L.; Ileana, E.; Rosellini, S.; Ammari, S.; et al. High-Throughput Genomics and Clinical Outcome in Hard-to-Treat Advanced Cancers: Results of the MOSCATO 01 Trial. Cancer Discov. 2017, 7, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Marquart, J.; Chen, E.Y.; Prasad, V. Estimation of the Percentage of US Patients with Cancer Who Benefit from Genome-Driven Oncology. JAMA Oncol. 2018, 4, 1093–1098. [Google Scholar] [CrossRef] [PubMed]
- Prasad, V. Perspective: The precision-oncology illusion. Nature 2016, 537, S63. [Google Scholar] [CrossRef] [PubMed]
- Pruneri, G.; De Braud, F.; Sapino, A.; Aglietta, M.; Vecchione, A.; Giusti, R.; Marchiò, C.; Scarpino, S.; Baggi, A.; Bonetti, G.; et al. Next-Generation Sequencing in Clinical Practice: Is It a Cost-Saving Alternative to a Single-Gene Testing Approach? PharmacoEconomics—Open 2021, 5, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Schwarze, K.; Buchanan, J.; Fermont, J.M.; Dreau, H.; Tilley, M.W.; Taylor, J.M.; Antoniou, P.; Knight, S.J.L.; Camps, C.; Pentony, M.M.; et al. The complete costs of genome sequencing: A microcosting study in cancer and rare diseases from a single center in the United Kingdom. Genet. Med. 2019, 22, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Christensen, K.D.; Phillips, K.A.; Green, R.C.; Dukhovny, D. Cost Analyses of Genomic Sequencing: Lessons Learned from the MedSeq Project. Value Health 2018, 21, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Schwarze, K.; Buchanan, J.; Taylor, J.C.; Wordsworth, S. Are whole-exome and whole-genome sequencing approaches cost-effective? A systematic review of the literature. Genet. Med. 2018, 20, 1122–1130. [Google Scholar] [CrossRef]
- Damodaran, S.; Berger, M.F.; Roychowdhury, S. Clinical Tumor Sequencing: Opportunities and Challenges for Precision Cancer Medicine. In American Society of Clinical Oncology Educational Book; ASCO: Alexandria, VA, USA, 2015; pp. e175–e182. [Google Scholar] [CrossRef]
- Hong, D.S.; DuBois, S.G.; Kummar, S.; Farago, A.F.; Albert, C.M.; Rohrberg, K.S.; van Tilburg, C.M.; Nagasubramanian, R.; Berlin, J.D.; Federman, N.; et al. Larotrectinib in patients with TRK fusion-positive solid tumours: A pooled analysis of three phase 1/2 clinical trials. Lancet Oncol. 2020, 21, 531–540. [Google Scholar] [CrossRef]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H., Jr.; et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: Prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.-P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair–Deficient Cancer: Results from the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lemery, S.; Keegan, P.; Pazdur, R. First FDA Approval Agnostic of Cancer Site—When a Biomarker Defines the Indication. N. Engl. J. Med. 2017, 377, 1409–1412. [Google Scholar] [CrossRef] [PubMed]
- Mack, E.K.M.; Marquardt, A.; Langer, D.; Ross, P.; Ultsch, A.; Kiehl, M.G.; Mack, H.I.D.; Haferlach, T.; Neubauer, A.; Brendel, C. Comprehensive genetic diagnosis of acute myeloid leukemia by next-generation sequencing. Haematologica 2019, 104, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Wheler, J.J.; Janku, F.; Naing, A.; Li, Y.; Stephen, B.; Zinner, R.; Subbiah, V.; Fu, S.; Karp, D.; Falchook, G.S.; et al. Cancer Therapy Directed by Comprehensive Genomic Profiling: A Single Center Study. Cancer Res. 2016, 76, 3690–3701. [Google Scholar] [CrossRef]
- Bertucci, F.; Gonçalves, A.; Guille, A.; Adelaïde, J.; Garnier, S.; Carbuccia, N.; Billon, E.; Finetti, P.; Sfumato, P.; Monneur, A.; et al. Prospective high-throughput genome profiling of advanced cancers: Results of the PERMED-01 clinical trial. Genome Med. 2021, 13, 87. [Google Scholar] [CrossRef]
- Réda, M.; Richard, C.; Bertaut, A.; Niogret, J.; Collot, T.; Fumet, J.D.; Blanc, J.; Truntzer, C.; Desmoulins, I.; Ladoire, S.; et al. Implementation and use of whole exome sequencing for metastatic solid cancer. eBioMedicine 2020, 51, 102624. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Chakravarty, D.; Gao, J.; Phillips, S.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 2017, 1–16. [Google Scholar] [CrossRef]
- Griffith, M.; Spies, N.C.; Krysiak, K.; McMichael, J.F.; Coffman, A.C.; Danos, A.M.; Ainscough, B.J.; Ramirez, C.A.; Rieke, D.T.; Kujan, L.; et al. CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. Nat. Genet. 2017, 49, 170–174. [Google Scholar] [CrossRef] [Green Version]
- Patterson, S.E.; Liu, R.; Statz, C.M.; Durkin, D.; Lakshminarayana, A.; Mockus, S.M. The clinical trial landscape in oncology and connectivity of somatic mutational profiles to targeted therapies. Hum. Genom. 2016, 10, 4. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2013, 42, D980–D985. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Chitipiralla, S.; Brown, G.R.; Chen, C.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; Kaur, K.; Liu, C.; et al. ClinVar: Improvements to accessing data. Nucleic Acids Res. 2020, 48, D835–D844. [Google Scholar] [CrossRef] [PubMed]
- ClinGen/CGC/VICC SOP for the Classification of Pathogenicity of Somatic Variants in Cancer (Oncogenicity). Available online: https://cancervariants.org/research/standards/onc_path_sop/ (accessed on 27 June 2022).
- Mateo, J.; Chakravarty, D.; Dienstmann, R.; Jezdic, S.; Gonzalez-Perez, A.; Lopez-Bigas, N.; Ng, C.; Bedard, P.; Tortora, G.; Douillard, J.-Y.; et al. A framework to rank genomic alterations as targets for cancer precision medicine: The ESMO Scale for Clinical Actionability of molecular Targets (ESCAT). Ann. Oncol. 2018, 29, 1895–1902. [Google Scholar] [CrossRef] [PubMed]
- Leichsenring, J.; Horak, P.; Kreutzfeldt, S.; Heining, C.; Christopoulos, P.; Volckmar, A.; Neumann, O.; Kirchner, M.; Ploeger, C.; Budczies, J.; et al. Variant classification in precision oncology. Int. J. Cancer 2019, 145, 2996–3010. [Google Scholar] [CrossRef]
- Schildhaus, H.U. Predictive Value of PD-L1 Diagnostics; Pathologe; Springer: Berlin/Heidelberg, Germany, 2018; Volume 39, pp. 498–519. [Google Scholar]
- Mock, A.; Heilig, C.E.; Kreutzfeldt, S.; Huebschmann, D.; Heining, C.; Schröck, E.; Brors, B.; Stenzinger, A.; Jäger, D.; Schlenk, R.; et al. Community-driven development of a modified progression-free survival ratio for precision oncology. ESMO Open 2019, 2019, e000583. [Google Scholar] [CrossRef]
- Colomer, R.; Mondejar, R.; Romero-Laorden, N.; Alfranca, A.; Sanchez-Madrid, F.; Quintela-Fandino, M. When should we order a next generation sequencing test in a patient with cancer? EClinicalMedicine 2020, 25, 100487. [Google Scholar] [CrossRef]
- Ciliberto, G.; Allegretti, M.; Babini, G.; Baldassarre, G.; Botti, G.; Bucci, G.; Buglioni, S.; Calistri, D.; Criscitiello, C.; Curigliano, G.; et al. Linee Guida per l’istituzione e la Gestione dei Molecular Tumor Board Negli Istituti di Alleanza Contro il Cancro; Alliance Against Cancer: Rome, Italy, 2020. [Google Scholar]
- Hoefflin, R.; Geißler, A.L.; Fritsch, R.; Claus, R.; Wehrle, J.; Metzger, P.; Reiser, M.; Mehmed, L.; Fauth, L.; Heiland, D.H.; et al. Personalized Clinical Decision Making Through Implementation of a Molecular Tumor Board: A German Single-Center Experience. JCO Precis. Oncol. 2018, 2, 1–16. [Google Scholar] [CrossRef]
- Heydt, C.; Wölwer, C.B.; Camacho, O.V.; Wagener-Ryczek, S.; Pappesch, R.; Siemanowski, J.; Rehker, J.; Haller, F.; Agaimy, A.; Worm, K.; et al. Detection of gene fusions using targeted next-generation sequencing: A comparative evaluation. BMC Med. Genom. 2021, 14, 62. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.-M.; Lonigro, R.J.; Vats, P.; Cobain, E.; Everett, J.; Cao, X.; Rabban, E.; Kumar-Sinha, C.; Raymond, V.; et al. Integrative clinical genomics of metastatic cancer. Nature 2017, 548, 297–303. [Google Scholar] [CrossRef]
- Van Dijk, E.; van den Bosch, T.; Lenos, K.J.; El Makrini, K.; Nijman, L.E.; van Essen, H.F.B.; Lansu, N.; Boekhout, M.; Hageman, J.H.; Fitzgerald, R.C.; et al. Chromosomal copy number heterogeneity predicts survival rates across cancers. Nat. Commun. 2021, 12, 3188. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and Management of AML in Adults: 2022 ELN Recommendations from an International Expert Panel. 2022. Available online: http://ashpublications.org/blood/article-pdf/doi/10.1182/blood.2022016867/1906555/blood.2022016867.pdf (accessed on 5 July 2022).
- Dalens, L.; Niogret, J.; Kaderbhai, C.G.; Boidot, R. Is There a Role for Large Exome Sequencing in the Management of Metastatic Non-Small Cell Lung Cancer: A Brief Report of Real Life. Front. Oncol. 2022, 12, 694. [Google Scholar] [CrossRef]
- Heinrich, K.; Miller-Phillips, L.; Ziemann, F.; Hasselmann, K.; Rühlmann, K.; Flach, M.; Biro, D.; von Bergwelt-Baildon, M.; Holch, J.; Herold, T.; et al. Lessons learned: The first consecutive 1000 patients of the CCCMunichLMU Molecular Tumor Board. J. Cancer Res. Clin. Oncol. 2022, 7, 1–11. [Google Scholar]
- Chakravarty, D.; Johnson, A.; Sklar, J.; Lindeman, N.I.; Moore, K.; Ganesan, S.; Lovly, C.M.; Perlmutter, J.; Gray, S.W.; Hwang, J.; et al. Somatic Genomic Testing in Patients with Metastatic or Advanced Cancer: ASCO Provisional Clinical Opinion. J. Clin. Oncol. 2022, 40, 1231–1258. [Google Scholar] [CrossRef] [PubMed]
- Mosele, F.; Remon, J.; Mateo, J.; Westphalen, C.; Barlesi, F.; Lolkema, M.; Normanno, N.; Scarpa, A.; Robson, M.; Meric-Bernstam, F.; et al. Recommendations for the use of next-generation sequencing (NGS) for patients with metastatic cancers: A report from the ESMO Precision Medicine Working Group. Ann. Oncol. 2020, 31, 1491–1505. [Google Scholar] [CrossRef]
- Inagaki, C.; Maeda, D.; Hatake, K.; Sato, Y.; Hashimoto, K.; Sakai, D.; Yachida, S.; Nonomura, N.; Satoh, T. Clinical Utility of Next-Generation Sequencing-Based Panel Testing under the Universal Health-Care System in Japan: A Retrospective Analysis at a Single University Hospital. Cancers 2021, 13, 1121. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. feature Counts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Quinlan, A.R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinform. 2014, 47, 11–12. [Google Scholar] [CrossRef]
- Benjaminit, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar]
- Cheung, V.G.; Nowak, N.; Jang, W.; Kirsch, I.R.; Zhao, S.; Chen, X.N.; Furey, T.S.; Kim, U.J.; Kuo, W.L.; Olivier, M.; et al. Integration of cytogenetic landmarks into the draft sequence of the human genome. Nature 2001, 409, 953–958. [Google Scholar] [PubMed]
- An International System for Human Cytogenomic Nomenclature. 2020. Available online: https://www.karger.com/Book/Home/279152 (accessed on 2 August 2022).
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385.e18. [Google Scholar] [PubMed]
- Anai, S.; Takeshita, M.; Ando, N.; Ikematsu, Y.; Mishima, S.; Ishida, K.; Inoue, K. A Case of Lung Adenocarcinoma Resistant to Crizotinib Harboring a Novel EML4-ALK Variant, Exon 6 of EML4 Fused to Exon 18 of ALK. J. Thorac. Oncol. 2016, 11, e126–e128. [Google Scholar] [CrossRef]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E–Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef]
- Okamura, R.; Kato, S.; Lee, S.; Jimenez, R.E.; Sicklick, J.K.; Kurzrock, R. ARID1A alterations function as a biomarker for longer progression-free survival after anti-PD-1/PD-L1 immunotherapy. J. Immunother. Cancer 2020, 8, e000438. [Google Scholar] [CrossRef]
- Bentzien, F.; Zuzow, M.; Heald, N.; Gibson, A.; Shi, Y.; Goon, L.; Yu, P.; Engst, S.; Zhang, W.; Huang, D.; et al. In vitro and in vivo activity of cabozantinib (XL184), an inhibitor of RET, MET, and VEGFR2, in a model of medullary thyroid cancer. Thyroid 2013, 23, 1569–1577. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF -mutant melanoma (COLUMBUS): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef]
- Sabatier, R.; Lopez, M.; Guille, A.; Billon, E.; Carbuccia, N.; Garnier, S.; Adelaide, J.; Extra, J.-M.; Cappiello, M.-A.; Charafe-Jauffret, E.; et al. High Response to Cetuximab in a Patient With EGFR-Amplified Heavily Pretreated Metastatic Triple-Negative Breast Cancer. JCO Precis. Oncol. 2019, 3, 1–8. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, C.; Wang, J.; Chen, D.; Deng, J.; Deng, J.; Fan, J.; Badakhshi, H.; Huang, X.; Zhang, L.; et al. A subset of esophageal squamous cell carcinoma patient-derived xenografts respond to cetuximab, which is predicted by high EGFR expression and amplification. J. Thorac. Dis. 2018, 10, 5328–5338. [Google Scholar] [CrossRef] [PubMed]
- Kiyuna, T.; Murakami, T.; Tome, Y.; Igarashi, K.; Kawaguchi, K.; Miyake, K.; Miyake, M.; Li, Y.; Nelson, S.D.; Dry, S.M.; et al. Doxorubicin-resistant pleomorphic liposarcoma with PDGFRA gene amplification is targeted and regressed by pazopanib in a patient-derived orthotopic xenograft mouse model. Tissue Cell 2018, 53, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Dziadziuszko, R.; Peters, S.; Mok, T.; Noe, J.; Nowicka, M.; Gadgeel, S.M.; Cheema, P.; Pavlakis, N.; de Marinis, F.; et al. Updated Efficacy and Safety Data and Impact of the EML4-ALK Fusion Variant on the Efficacy of Alectinib in Untreated ALK-Positive Advanced Non–Small Cell Lung Cancer in the Global Phase III ALEX Study. J. Thorac. Oncol. 2019, 14, 1233–1243. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N | Percentage (%) or Range | |
|---|---|---|
| Total | 104 | |
| Sex | ||
| Female | 52 | (50) |
| Male | 52 | (50) |
| Age | 57.5 (median) | 22–77 (range) |
| Stage at presentation | ||
| Metastatic disease | 100 | (96.1) |
| Localized | 3 | (2.9) |
| Locally advanced | 1 | (1.0) |
| Initial therapeutic intent | ||
| Primary metastatic, palliative intent | 63 | (60.6) |
| Initially curative intent | 41 | (39.4) |
| Tumor type | ||
| Lower GI tract | 18 | (17.3) |
| Neuroendocrine neoplasms | 13 | (12.5) |
| Sarcoma | 12 | (11.5) |
| Head and neck | 11 | (10.6) |
| Hepatobiliary | 10 | (9.6) |
| CUP | 8 | (7.7) |
| Pancreatobiliary | 7 | (6.7) |
| Breast | 6 | (5.8) |
| Ovary | 6 | (5.8) |
| Thoracic malignancies | 5 | (4.8) |
| Upper GI tract | 3 | (2.9) |
| Gynecologic (non-ovarian) | 2 | (1.9) |
| Germ cell tumor | 2 | (1.9) |
| Embryonal (Neuroblastoma) | 1 | (1.0) |
| Previous lines of treatment | ||
| 0 | 1 | (1.0) |
| 1 | 13 | (12.5) |
| 2 | 33 | (31.7) |
| 3 | 25 | (24.0) |
| >3 | 32 | (30.8) |
| 3 (median) | 0–14 (range) | |
| Diagnosis | Fusion | Clinical Relevance |
|---|---|---|
| CUP | TMPRSS2-ERG | Changed diagnosis from CUP to prostate carcinoma |
| CUP | SEC31A-ALK | Changed diagnosis from CUP to ALK-positive DLBCL |
| CUP | CLDN18-ARHGAP26 | Changed diagnosis from CUP to gastric carcinoma |
| Pancreatic carcinoma | EML4-ALK (previously not characterized transcript—E2;A18) * | Molecular rationale for ALK-inhibitor (Alectinib) |
| Cholangiocarcinoma | FGFR2-ARHGAP24 | Molecular rationale for FGFR-inhibitor (Pemigatinib) |
| Adenoid cystic Carcinoma | MYB-NFIB | Characteristic of adenoid cystic carcinoma, no evidence for direct actionability |
| Diagnosis | Therapeutic Rationale | MTB Recommendation | EL γ ESCAT | EL γ DKTK | Label | PFS2 | PFS1 | PFSr | Outcome |
|---|---|---|---|---|---|---|---|---|---|
| CUP | TMPRSS2-ERG fusion (changed diagnosis from CUP to prostate cancer) | Docetaxel + ADT | n/a | n/a | On | 6 | 3.5 | 1.7 | SD for 6 months |
| Acinic cell carcinoma | ATM del | Niraparib + Carboplatin | III-B | m4 | Off | 12 | n/a | n/a | SD for 12 months |
| CUP | SEC31A-ALK fusion (changed diagnosis from CUP to CD20-negative ALK + DLBCL) | Chemotherapy (CHOEP) | n/a | n/a | On | 23.5 | 10.5 | 2.2 | CR, ongoing at almost-2-year follow-up |
| CRC | BRAF V600E | Encorafenib + Binimetinib + Cetuximab | I-A | m1a | Off | 7.5 | 3 | 2.5 | SD for 7.5 months |
| Sinonasal adenocarcinoma | ARID1A deletion | Pembrolizumab | III-A | m2b | Off | 12 | n/a | n/a | SD for 12 months |
| Steroid cell tumor | RET Y791F | Cabozantinib | IV-A | m3 | Off | 9.5 | 1 | 9.5 | Decrease of Tumor marker and SD for 9.5 months |
| CUP | BRAF V600E | Encorafenib + Binimetinib | III-A | m2a | Off | 18.5 | 3 | 6.2 | PR and SD for 18 months |
| TNBC | EGFR amplification | Cetuximab + Capecitabine | IV-A | m1c | Off | 9.25 | 2 | 4.6 | SD for 9.25 months |
| Sarcoma | KDR, KIT, PDGFRA Amplification | Pazopanib | IV-A | m3 | On | 6.25 | 14 | 0.4 | SD for 6.25 months |
| Pancreatic cancer | EML4-ALK Fusion | Alectinib monotherapy | III-B | m4 | Off | 7.7 | 1 | 7.7 | PR and SD for 7.7 months |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarawneh, T.S.; Rodepeter, F.R.; Teply-Szymanski, J.; Ross, P.; Koch, V.; Thölken, C.; Schäfer, J.A.; Gremke, N.; Mack, H.I.D.; Gold, J.; et al. Combined Focused Next-Generation Sequencing Assays to Guide Precision Oncology in Solid Tumors: A Retrospective Analysis from an Institutional Molecular Tumor Board. Cancers 2022, 14, 4430. https://doi.org/10.3390/cancers14184430
Tarawneh TS, Rodepeter FR, Teply-Szymanski J, Ross P, Koch V, Thölken C, Schäfer JA, Gremke N, Mack HID, Gold J, et al. Combined Focused Next-Generation Sequencing Assays to Guide Precision Oncology in Solid Tumors: A Retrospective Analysis from an Institutional Molecular Tumor Board. Cancers. 2022; 14(18):4430. https://doi.org/10.3390/cancers14184430
Chicago/Turabian StyleTarawneh, Thomas S., Fiona R. Rodepeter, Julia Teply-Szymanski, Petra Ross, Vera Koch, Clemens Thölken, Jonas A. Schäfer, Niklas Gremke, Hildegard I. D. Mack, Judith Gold, and et al. 2022. "Combined Focused Next-Generation Sequencing Assays to Guide Precision Oncology in Solid Tumors: A Retrospective Analysis from an Institutional Molecular Tumor Board" Cancers 14, no. 18: 4430. https://doi.org/10.3390/cancers14184430
APA StyleTarawneh, T. S., Rodepeter, F. R., Teply-Szymanski, J., Ross, P., Koch, V., Thölken, C., Schäfer, J. A., Gremke, N., Mack, H. I. D., Gold, J., Riera-Knorrenschild, J., Wilhelm, C., Rinke, A., Middeke, M., Klemmer, A., Romey, M., Hattesohl, A., Jesinghaus, M., Görg, C., ... Mack, E. K. M. (2022). Combined Focused Next-Generation Sequencing Assays to Guide Precision Oncology in Solid Tumors: A Retrospective Analysis from an Institutional Molecular Tumor Board. Cancers, 14(18), 4430. https://doi.org/10.3390/cancers14184430