

Androgen Receptor Splice Variants Contribute to the Upregulation of DNA Repair in Prostate Cancer

 , , , , , , ,
, , , , , , ,  ,
,
Abstract
Simple Summary
Abstract
1. Introduction
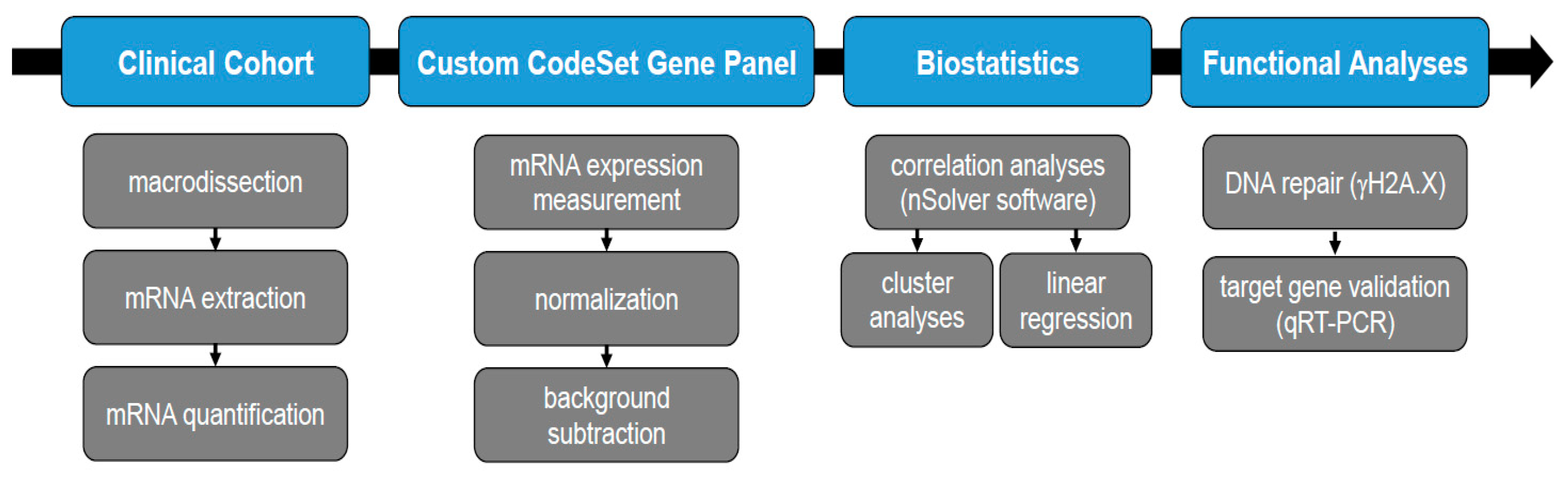
2. Materials and Methods
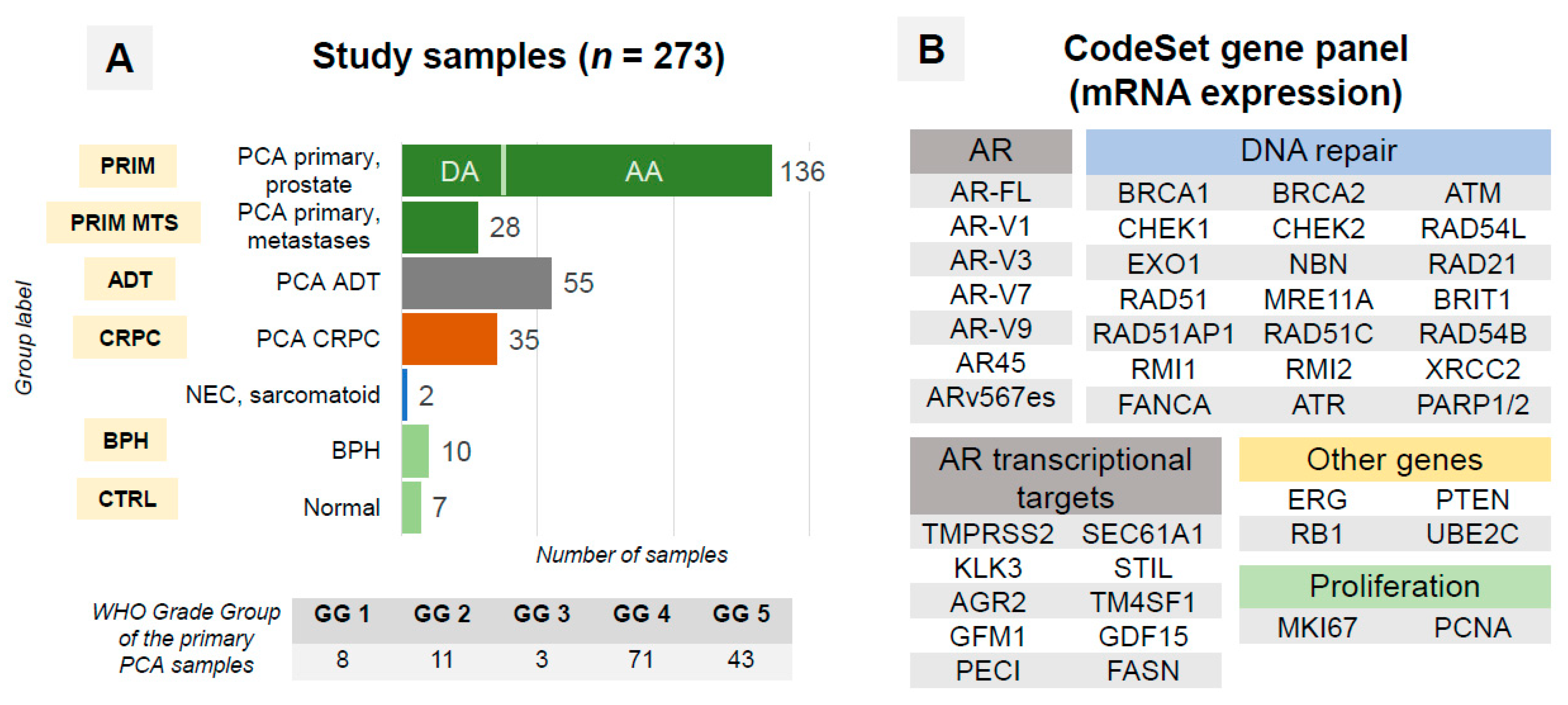
2.1. Patient Cohort
2.2. Samples
2.3. RNA Extraction
2.4. RNA Expression Analysis
2.5. Statistical Analysis
2.6. Linear Regression Analysis
2.7. Cell Culture and Irradiation
2.8. γH2A.X Assay
2.9. Immunoblot
2.10. Quantitative qRT-PCR
2.11. Ethical Considerations
3. Results
3.1. Quality Control (QC)
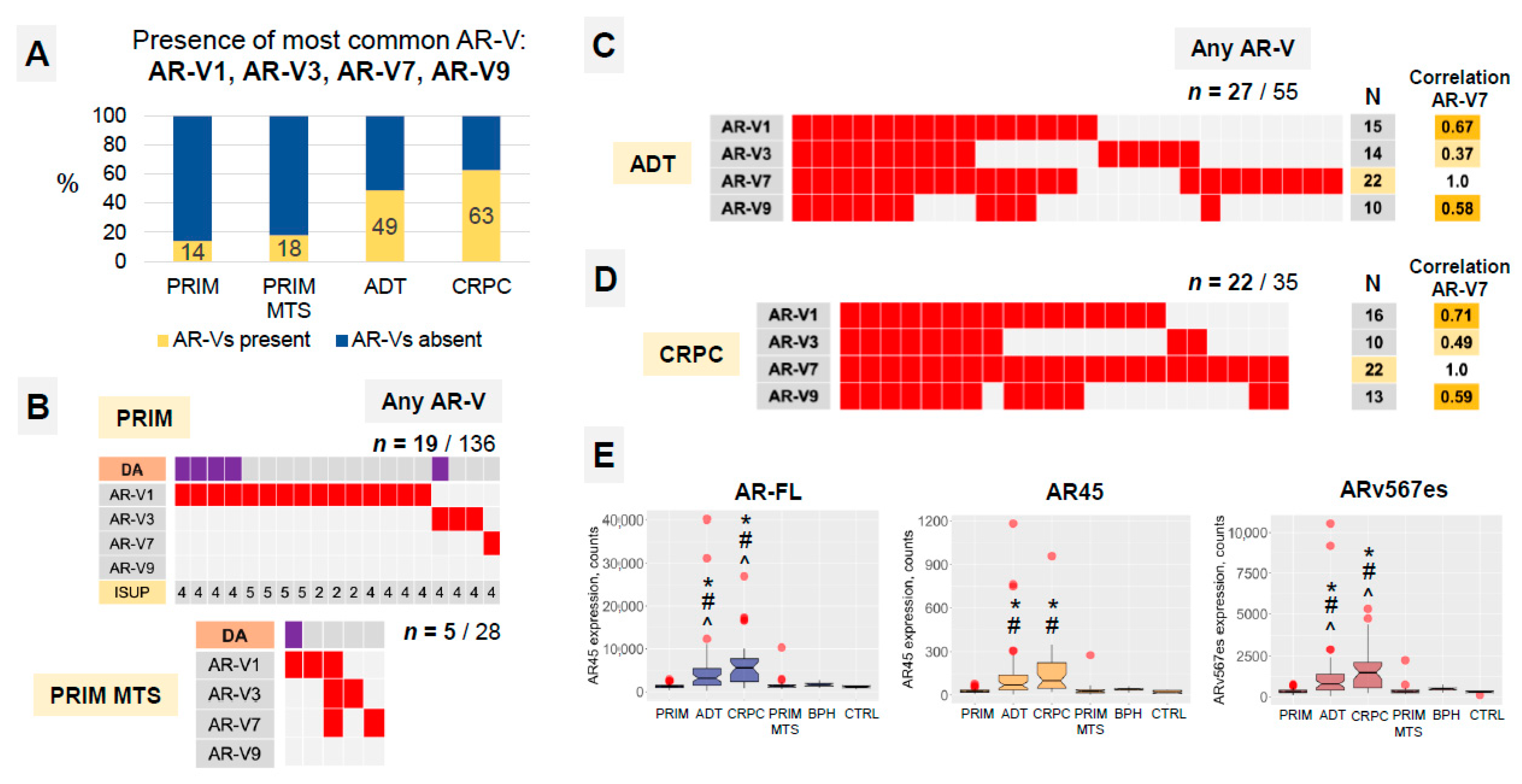
3.2. AR-Vs Appear Mostly as a Response to ADT
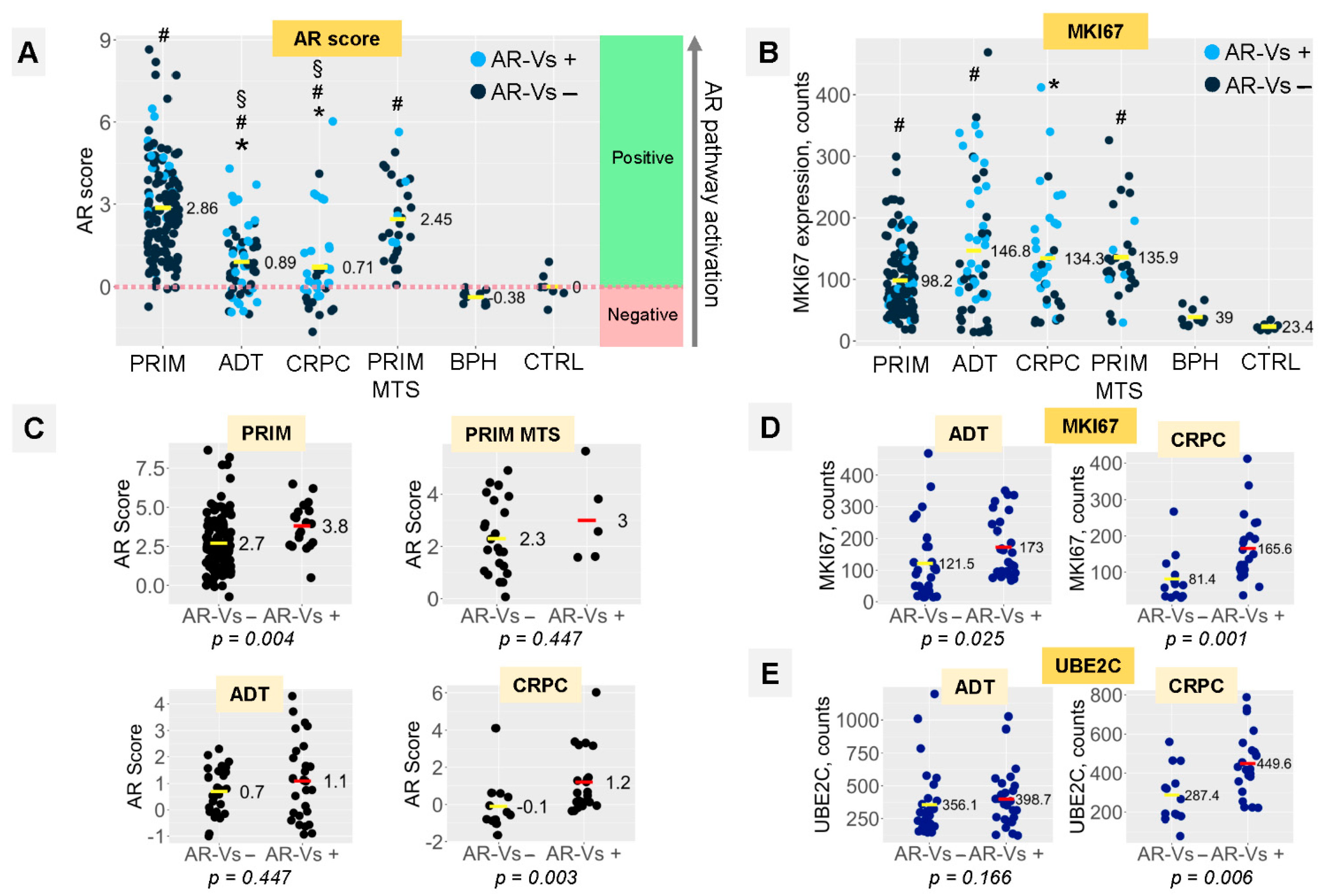
3.3. AR Signaling and Proliferation Depend on the Presence of AR-Vs in ADT and CRPC Tumors
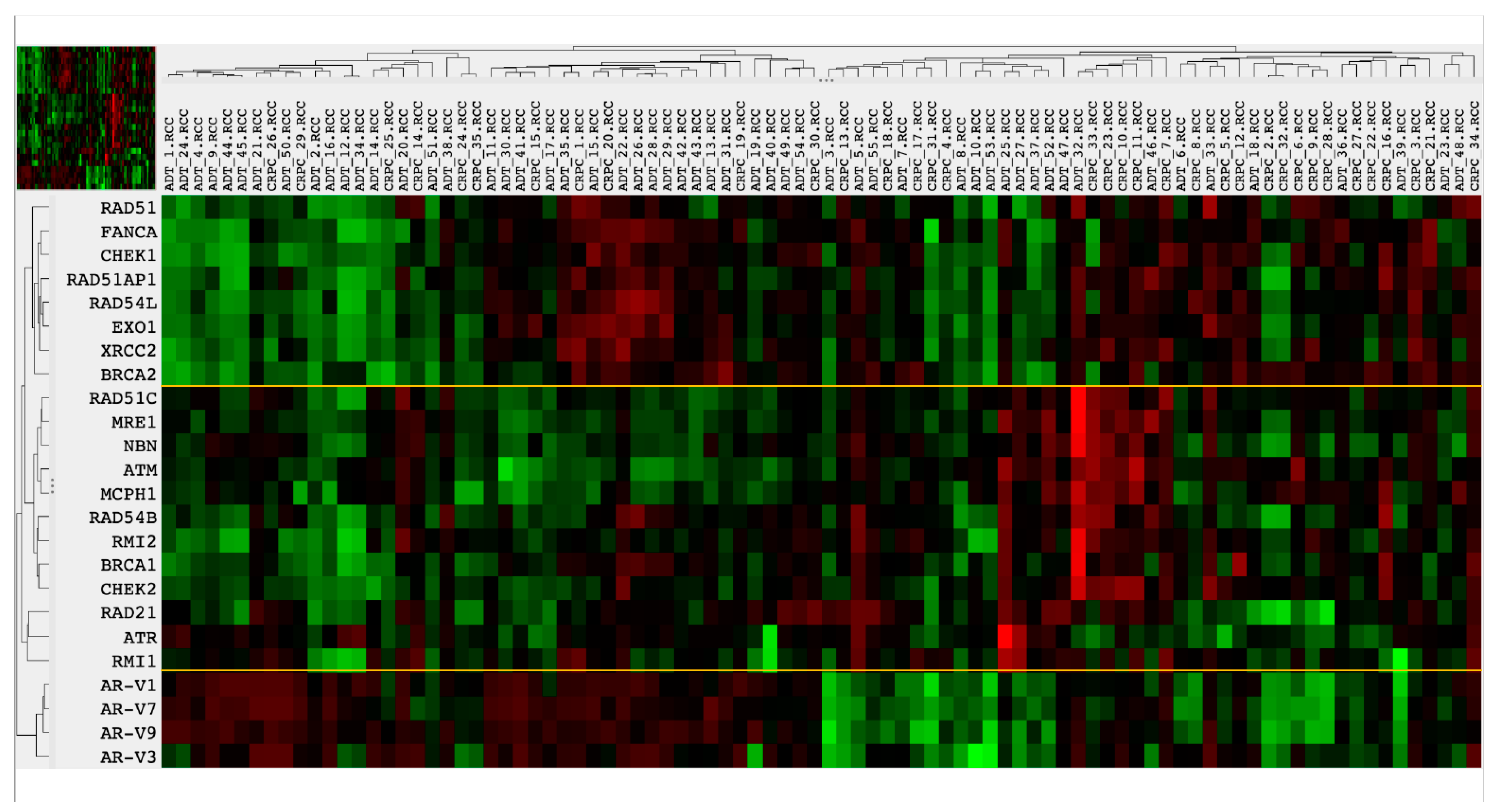
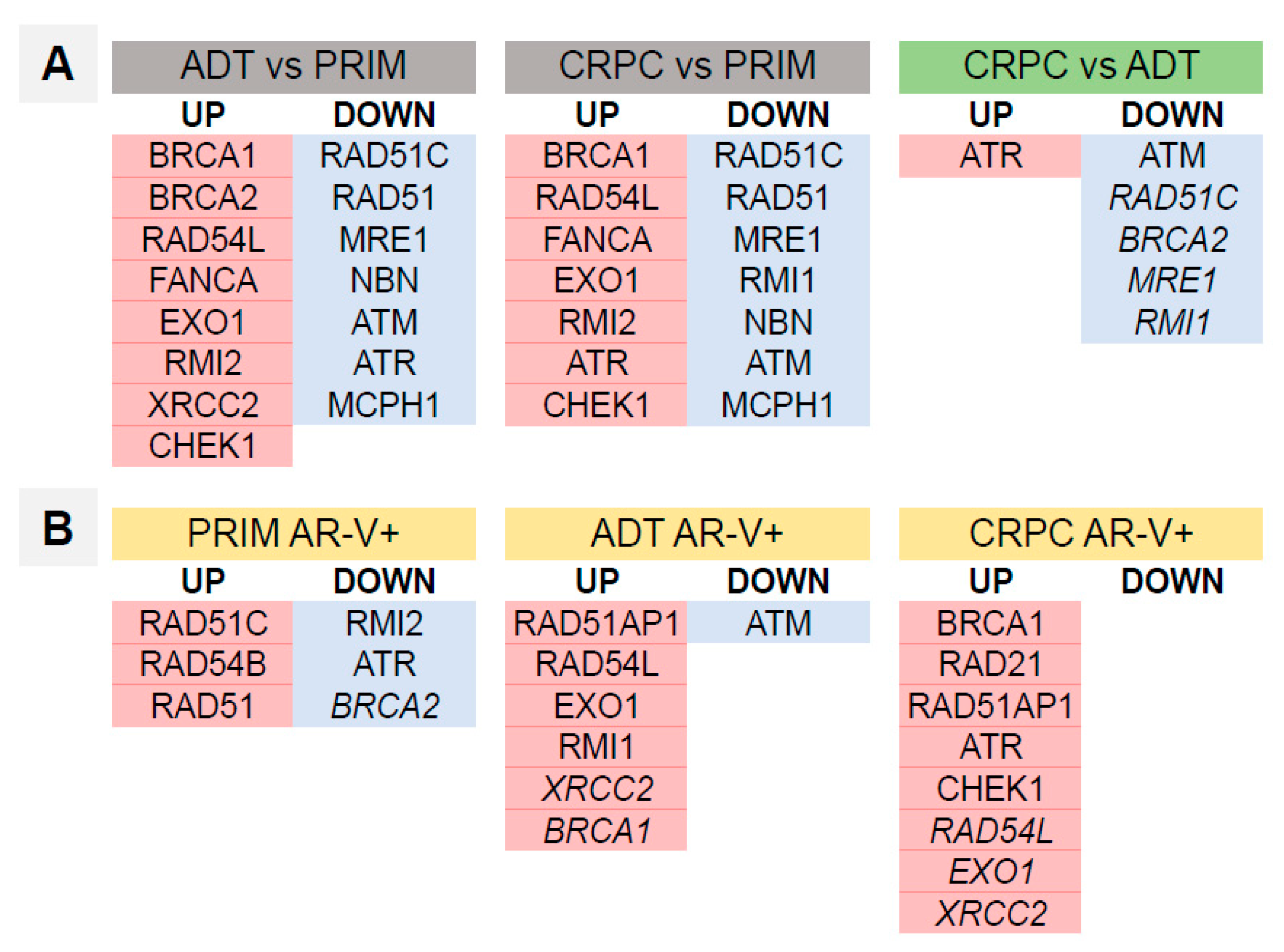
3.4. Homologous Recombination DNA Repair Activity Depends on the Presence of AR-Vs and AR Pathway Activation
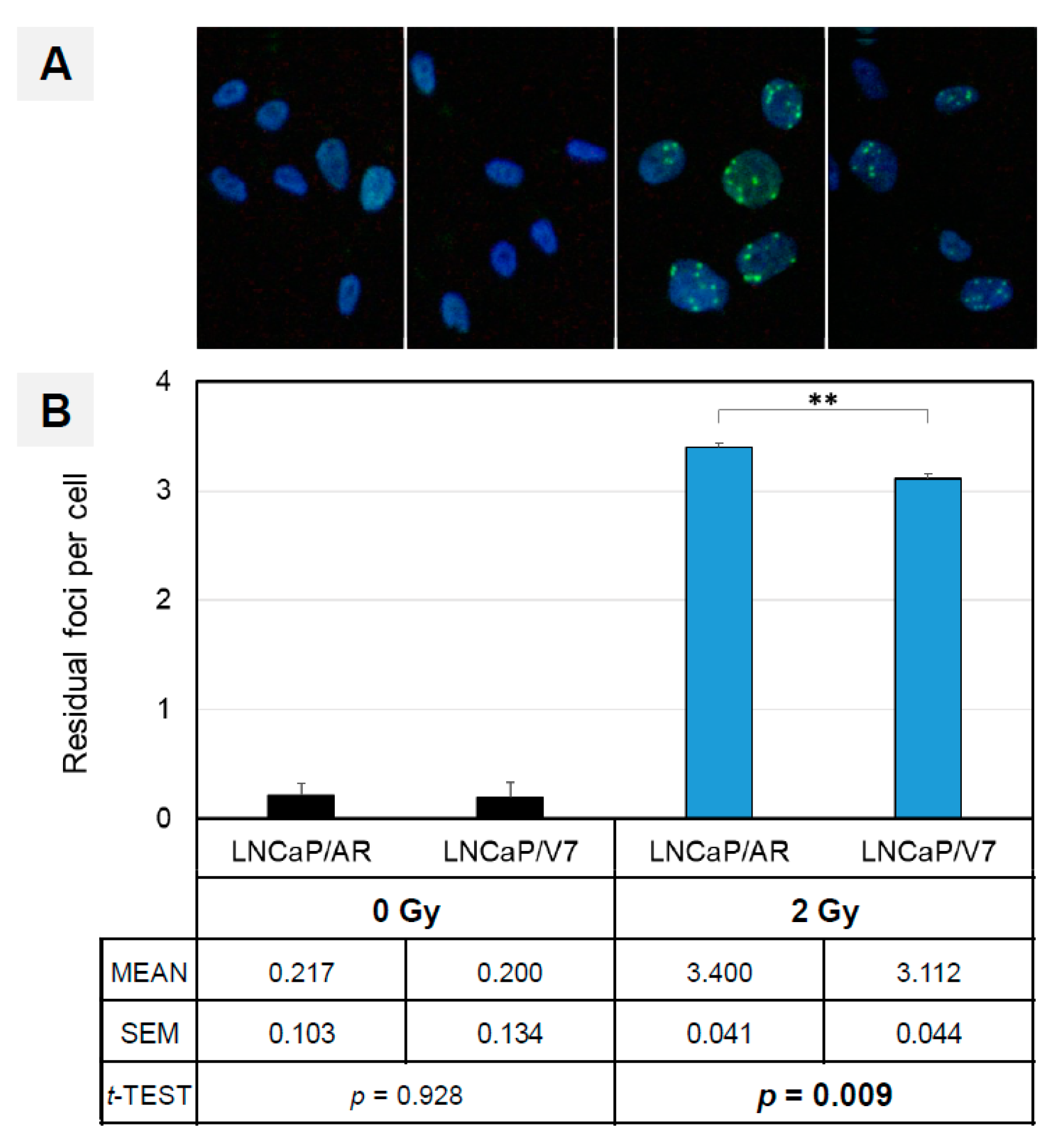
3.5. AR-V7 Enhances DNA Double-Strand Break Repair in an In Vitro PCA Model
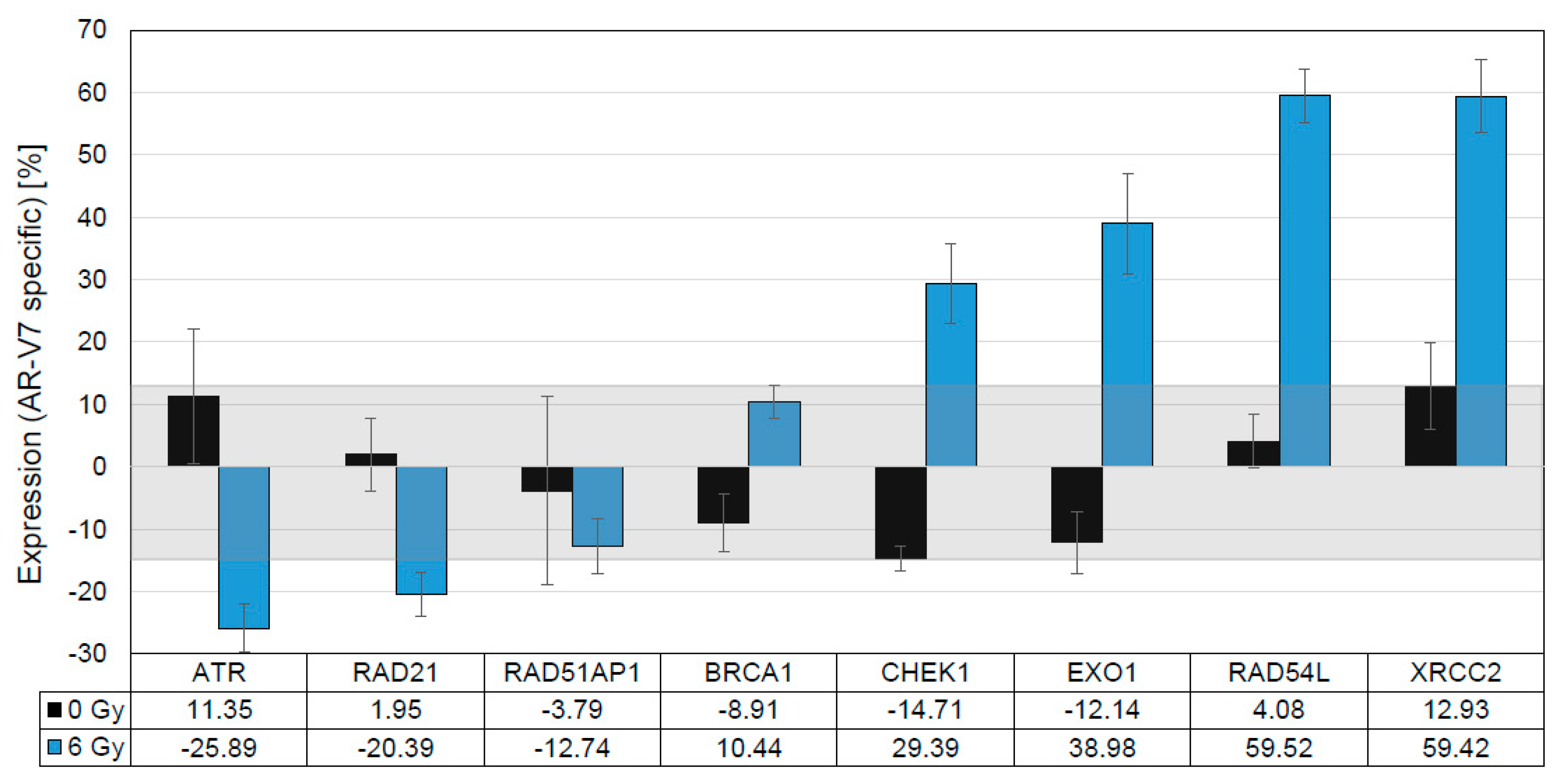
3.6. In Vitro Validation of Findings in Clinical Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Polkinghorn, W.R.; Parker, J.S.; Lee, M.X.; Kass, E.M.; Spratt, D.E.; Iaquinta, P.J.; Arora, V.K.; Yen, W.F.; Cai, L.; Zheng, D.; et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov. 2013, 3, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.F.; Schiewer, M.J.; Dean, J.L.; Schrecengost, R.S.; de Leeuw, R.; Han, S.; Ma, T.; Den, R.B.; Dicker, A.P.; Feng, F.Y.; et al. A hormone-DNA repair circuit governs the response to genotoxic insult. Cancer Discov. 2013, 3, 1254–1271. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Li, R.; Xu, K.; Ding, S.; Li, J.; Baek, G.; Ramanand, S.G.; Ding, S.; Liu, Z.; Gao, Y.; et al. Androgen Receptor Variants Mediate DNA Repair after Prostate Cancer Irradiation. Cancer Res. 2017, 77, 4745–4754. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liao, C.Y.; Chtatou, H.; Incrocci, L.; van Gent, D.C.; van Weerden, W.M.; Nonnekens, J. Apalutamide Sensitizes Prostate Cancer to Ionizing Radiation via Inhibition of Non-Homologous End-Joining DNA Repair. Cancers 2019, 11, 1593. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Karanika, S.; Yang, G.; Wang, J.; Park, S.; Broom, B.M.; Manyam, G.C.; Wu, W.; Luo, Y.; Basourakos, S.; et al. Androgen receptor inhibitor-induced ‘BRCAness’ and PARP inhibition are synthetically lethal for castration-resistant prostate cancer. Sci. Signal. 2017, 10, eaam7479. [Google Scholar] [CrossRef]
- Asim, M.; Tarish, F.; Zecchini, H.I.; Sanjiv, K.; Gelali, E.; Massie, C.E.; Baridi, A.; Warren, A.Y.; Zhao, W.; Ogris, C.; et al. Synthetic lethality between androgen receptor signalling and the PARP pathway in prostate cancer. Nat. Commun. 2017, 8, 374. [Google Scholar] [CrossRef]
- Schiewer, M.J.; Goodwin, J.F.; Han, S.; Brenner, J.C.; Augello, M.A.; Dean, J.L.; Liu, F.; Planck, J.L.; Ravindranathan, P.; Chinnaiyan, A.M.; et al. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov. 2012, 2, 1134–1149. [Google Scholar] [CrossRef]
- Kounatidou, E.; Nakjang, S.; McCracken, S.; Dehm, S.M.; Robson, C.N.; Jones, D.; Gaughan, L. A novel CRISPR-engineered prostate cancer cell line defines the AR-V transcriptome and identifies PARP inhibitor sensitivities. Nucleic Acids Res. 2019, 47, 5634–5647. [Google Scholar] [CrossRef]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef]
- Armenia, J.; Wankowicz, S.; Liu, D.; Gao, J.; Kundra, R.; Reznik, E.; Chatila, W.K.; Chakravarty, D.; Han, G.C.; Coleman, I.; et al. The long tail of oncogenic drivers in prostate cancer. Nat. Genet. 2018, 50, 645–651. [Google Scholar] [CrossRef]
- Clarke, N.; Wiechno, P.; Alekseev, B.; Sala, N.; Jones, R.; Kocak, I.; Chiuri, V.E.; Jassem, J.; Fléchon, A.; Redfern, C.; et al. Olaparib combined with abiraterone in patients with metastatic castration-resistant prostate cancer: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2018, 19, 975–986. [Google Scholar] [CrossRef]
- Byrum, A.K.; Vindigni, A.; Mosammaparast, N. Defining and Modulating ‘BRCAness’. Trends Cell Biol. 2019, 29, 740–751. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, D.; Qiu, Y. Role of PARP-1 in prostate cancer. Am. J. Clin. Exp. Urol. 2015, 3, 1–12. [Google Scholar] [PubMed]
- Schiewer, M.J.; Knudsen, K.E. Transcriptional roles of PARP1 in cancer. Mol. Cancer Res. 2014, 12, 1069–1080. [Google Scholar] [CrossRef]
- Gui, B.; Gui, F.; Takai, T.; Feng, C.; Bai, X.; Fazli, L.; Dong, X.; Liu, S.; Zhang, X.; Zhang, W.; et al. Selective targeting of PARP-2 inhibits androgen receptor signaling and prostate cancer growth through disruption of FOXA1 function. Proc. Natl. Acad. Sci. USA 2019, 116, 14573–14582. [Google Scholar] [CrossRef]
- Jividen, K.; Kedzierska, K.Z.; Yang, C.S.; Szlachta, K.; Ratan, A.; Paschal, B.M. Genomic analysis of DNA repair genes and androgen signaling in prostate cancer. BMC Cancer 2018, 18, 960. [Google Scholar] [CrossRef]
- Li, Y.; Yang, R.; Henzler, C.M.; Ho, Y.; Passow, C.; Auch, B.; Carreira, S.; Nava Rodrigues, D.; Bertan, C.; Hwang, T.H.; et al. Diverse AR Gene Rearrangements Mediate Resistance to Androgen Receptor Inhibitors in Metastatic Prostate Cancer. Clin. Cancer Res. 2020, 26, 1965–1976. [Google Scholar] [CrossRef]
- Wang, Q.; Li, W.; Zhang, Y.; Yuan, X.; Xu, K.; Yu, J.; Chen, Z.; Beroukhim, R.; Wang, H.; Lupien, M.; et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell 2009, 138, 245–256. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef]
- Kallio, H.M.L.; Hieta, R.; Latonen, L.; Brofeldt, A.; Annala, M.; Kivinummi, K.; Tammela, T.L.; Nykter, M.; Isaacs, W.B.; Lilja, H.G.; et al. Constitutively active androgen receptor splice variants AR-V3, AR-V7 and AR-V9 are co-expressed in castration-resistant prostate cancer metastases. Br. J. Cancer 2018, 119, 347–356. [Google Scholar] [CrossRef]
- Sharp, A.; Coleman, I.; Yuan, W.; Sprenger, C.; Dolling, D.; Rodrigues, D.N.; Russo, J.W.; Figueiredo, I.; Bertan, C.; Seed, G.; et al. Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J. Clin. Investig. 2019, 129, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Culig, Z. The way towards understanding possible multiple functions of AR V7 in prostate cancer. BJU Int. 2018, 122, 169. [Google Scholar] [CrossRef]
- Luo, H.; Liu, Y.; Li, Y.; Zhang, C.; Yu, B.; Shao, C. Androgen receptor splicing variant 7 promotes DNA damage response in prostate cancer cells. FASEB J. 2022, 36, e22495. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.R.; Azeem, W.; Hellem, M.R.; Marvyin, K.; Hua, Y.; Qu, Y.; Li, L.; Lin, B.; Ke, X.; Øyan, A.M.; et al. Context dependent regulatory patterns of the androgen receptor and androgen receptor target genes. BMC Cancer 2016, 16, 377. [Google Scholar] [CrossRef] [PubMed]
- Martz, C.A.; Ottina, K.A.; Singleton, K.R.; Jasper, J.S.; Wardell, S.E.; Peraza-Penton, A.; Anderson, G.R.; Winter, P.S.; Wang, T.; Alley, H.M.; et al. Systematic identification of signaling pathways with potential to confer anticancer drug resistance. Sci. Signal. 2014, 7, ra121. [Google Scholar] [CrossRef]
- Deville, S.S.; Luft, S.; Kaufmann, M.; Cordes, N. Keap1 inhibition sensitizes head and neck squamous cell carcinoma cells to ionizing radiation via impaired non-homologous end joining and induced autophagy. Cell Death Dis. 2020, 11, 887. [Google Scholar] [CrossRef]
- Becker, Y.; Förster, S.; Gielen, G.H.; Loke, I.; Thaysen-Andersen, M.; Laurini, C.; Wehrand, K.; Pietsch, T.; Diestel, S. Paucimannosidic glycoepitopes inhibit tumorigenic processes in glioblastoma multiforme. Oncotarget 2019, 10, 4449–4465. [Google Scholar] [CrossRef][Green Version]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef]
- Mathy, C.S.; Mayr, T.; Kürpig, S.; Meisenheimer, M.; Dolscheid-Pommerich, R.C.; Stoffel-Wagner, B.; Kristiansen, G.; Essler, M.; Muders, M.H.; Bundschuh, R.A. Antihormone treatment differentially regulates PSA secretion, PSMA expression and 68 Ga-PSMA uptake in LNCaP cells. J. Cancer Res. Clin. Oncol. 2021, 147, 1733–1743. [Google Scholar] [CrossRef]
- Wright, M.E.; Tsai, M.J.; Aebersold, R. Androgen receptor represses the neuroendocrine transdifferentiation process in prostate cancer cells. Mol. Endocrinol. 2003, 17, 1726–1737. [Google Scholar] [CrossRef]
- Hu, R.; Lu, C.; Mostaghel, E.A.; Yegnasubramanian, S.; Gurel, M.; Tannahill, C.; Edwards, J.; Isaacs, W.B.; Nelson, P.S.; Bluemn, E.; et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012, 72, 3457–3462. [Google Scholar] [CrossRef] [PubMed]
- Roggero, C.M.; Jin, L.; Cao, S.; Sonavane, R.; Kopplin, N.G.; Ta, H.Q.; Ekoue, D.N.; Witwer, M.; Ma, S.; Liu, H.; et al. A detailed characterization of stepwise activation of the androgen receptor variant 7 in prostate cancer cells. Oncogene 2021, 40, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.D.; Welsbie, D.S.; Tran, C.; Baek, S.H.; Chen, R.; Vessella, R.; Rosenfeld, M.G.; Sawyers, C.L. Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 2004, 10, 33–39. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Lu, J.; Ye, Z.; Hao, S.; Wang, L.; Kohli, M.; Tindall, D.J.; Li, B.; Zhu, R.; Wang, L.; et al. Androgen receptor splice variants bind to constitutively open chromatin and promote abiraterone-resistant growth of prostate cancer. Nucleic Acids Res. 2018, 46, 1895–1911. [Google Scholar] [CrossRef]
- Jones, D.; Wade, M.; Nakjang, S.; Chaytor, L.; Grey, J.; Robson, C.N.; Gaughan, L. FOXA1 regulates androgen receptor variant activity in models of castrate-resistant prostate cancer. Oncotarget 2015, 6, 29782–29794. [Google Scholar] [CrossRef]
- Yap, T.A.; Plummer, R.; Azad, N.S.; Helleday, T. The DNA Damaging Revolution: PARP Inhibitors and Beyond. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 185–195. [Google Scholar] [CrossRef]
- Spratt, D.E.; Evans, M.J.; Davis, B.J.; Doran, M.G.; Lee, M.X.; Shah, N.; Wongvipat, J.; Carnazza, K.E.; Klee, G.G.; Polkinghorn, W.; et al. Androgen Receptor Upregulation Mediates Radioresistance after Ionizing Radiation. Cancer Res. 2015, 75, 4688–4696. [Google Scholar] [CrossRef]
- Hussain, M.; Daignault-Newton, S.; Twardowski, P.W.; Albany, C.; Stein, M.N.; Kunju, L.P.; Siddiqui, J.; Wu, Y.M.; Robinson, D.; Lonigro, R.J.; et al. Targeting Androgen Receptor and DNA Repair in Metastatic Castration-Resistant Prostate Cancer: Results from NCI 9012. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 991–999. [Google Scholar] [CrossRef]
- Clarke, N.W.; Armstrong, A.J.; Thiery-Vuillemin, A.; Oya, M.; Shore, N.; Loredo, E.; Procopio, G.; de Menezes, J.; Girotto, G.; Arslan, C.; et al. Abiraterone and Olaparib for Metastatic Castration-Resistant Prostate Cancer. NEJM Evid. 2022, 1, 1–16. [Google Scholar] [CrossRef]
- Cato, L.; de Tribolet-Hardy, J.; Lee, I.; Rottenberg, J.T.; Coleman, I.; Melchers, D.; Houtman, R.; Xiao, T.; Li, W.; Uo, T.; et al. ARv7 Represses Tumor-Suppressor Genes in Castration-Resistant Prostate Cancer. Cancer Cell 2019, 35, 401–413.e6. [Google Scholar] [CrossRef]
- Veldscholte, J.; Berrevoets, C.A.; Ris-Stalpers, C.; Kuiper, G.G.; Jenster, G.; Trapman, J.; Brinkmann, A.O.; Mulder, E. The androgen receptor in LNCaP cells contains a mutation in the ligand binding domain which affects steroid binding characteristics and response to antiandrogens. J. Steroid Biochem. Mol. Biol. 1992, 41, 665–669. [Google Scholar] [CrossRef]
- Sfanos, K.S.; Aloia, A.L.; Hicks, J.L.; Esopi, D.M.; Steranka, J.P.; Shao, W.; Sanchez-Martinez, S.; Yegnasubramanian, S.; Burns, K.H.; Rein, A.; et al. Identification of replication competent murine gammaretroviruses in commonly used prostate cancer cell lines. PLoS ONE 2011, 6, e20874. [Google Scholar] [CrossRef] [PubMed]
- Daniel, R.; Ramcharan, J.; Rogakou, E.; Taganov, K.D.; Greger, J.G.; Bonner, W.; Nussenzweig, A.; Katz, R.A.; Skalka, A.M. Histone H2AX is phosphorylated at sites of retroviral DNA integration but is dispensable for postintegration repair. J. Biol. Chem. 2004, 279, 45810–45814. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative Genomic Profiling of Human Prostate Cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef]
- Chen, X.; Bernemann, C.; Tolkach, Y.; Heller, M.; Nientiedt, C.; Falkenstein, M.; Herpel, E.; Jenzer, M.; Grüllich, C.; Jäger, D.; et al. Overexpression of nuclear AR-V7 protein in primary prostate cancer is an independent negative prognostic marker in men with high-risk disease receiving adjuvant therapy. Urol. Oncol. 2018, 36, 161.e19–161.e30. [Google Scholar] [CrossRef]
- Xu, L.; Ma, E.; Zeng, T.; Zhao, R.; Tao, Y.; Chen, X.; Groth, J.; Liang, C.; Hu, H.; Huang, J. ATM deficiency promotes progression of CRPC by enhancing Warburg effect. Endocr.-Relat. Cancer 2019, 26, 59–71. [Google Scholar] [CrossRef]
- Wokołorczyk, D.; Kluźniak, W.; Huzarski, T.; Gronwald, J.; Szymiczek, A.; Rusak, B.; Stempa, K.; Gliniewicz, K.; Kashyap, A.; Morawska, S.; et al. Mutations in ATM, NBN and BRCA2 predispose to aggressive prostate cancer in Poland. Int. J. Cancer 2020, 147, 2793–2800. [Google Scholar] [CrossRef]
- Rai, R.; Dai, H.; Multani, A.S.; Li, K.; Chin, K.; Gray, J.; Lahad, J.P.; Liang, J.; Mills, G.B.; Meric-Bernstam, F.; et al. BRIT1 regulates early DNA damage response, chromosomal integrity, and cancer. Cancer Cell 2006, 10, 145–157. [Google Scholar] [CrossRef]
- de Bono, J.S.; Mehra, N.; Scagliotti, G.V.; Castro, E.; Dorff, T.; Stirling, A.; Stenzl, A.; Fleming, M.T.; Higano, C.S.; Saad, F.; et al. Talazoparib monotherapy in metastatic castration-resistant prostate cancer with DNA repair alterations (TALAPRO-1): An open-label, phase 2 trial. Lancet. Oncol. 2021, 22, 1250–1264. [Google Scholar] [CrossRef]
- Abida, W.; Armenia, J.; Gopalan, A.; Brennan, R.; Walsh, M.; Barron, D.; Danila, D.; Rathkopf, D.; Morris, M.; Slovin, S.; et al. Prospective Genomic Profiling of Prostate Cancer Across Disease States Reveals Germline and Somatic Alterations That May Affect Clinical Decision Making. JCO Precis. Oncol. 2017, 1, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Dai, Z.; Wang, L.; Gao, X.; Yang, L.; Wang, Z.; Wang, Q.; Liu, Z. MET inhibition enhances PARP inhibitor efficacy in castration-resistant prostate cancer by suppressing the ATM/ATR and PI3K/AKT pathways. J. Cell. Mol. Med. 2021, 25, 11157–11169. [Google Scholar] [CrossRef] [PubMed]
- Al-Ubaidi, F.L.T.; Schultz, N.; Loseva, O.; Egevad, L.; Granfors, T.; Helleday, T. Castration therapy results in decreased Ku70 levels in prostate cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 1547–1556. [Google Scholar] [CrossRef] [PubMed]
- Dylgjeri, E.; McNair, C.; Goodwin, J.F.; Raymon, H.K.; McCue, P.A.; Shafi, A.A.; Leiby, B.E.; de Leeuw, R.; Kothari, V.; McCann, J.J.; et al. Pleiotropic Impact of DNA-PK in Cancer and Implications for Therapeutic Strategies. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 5623–5638. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
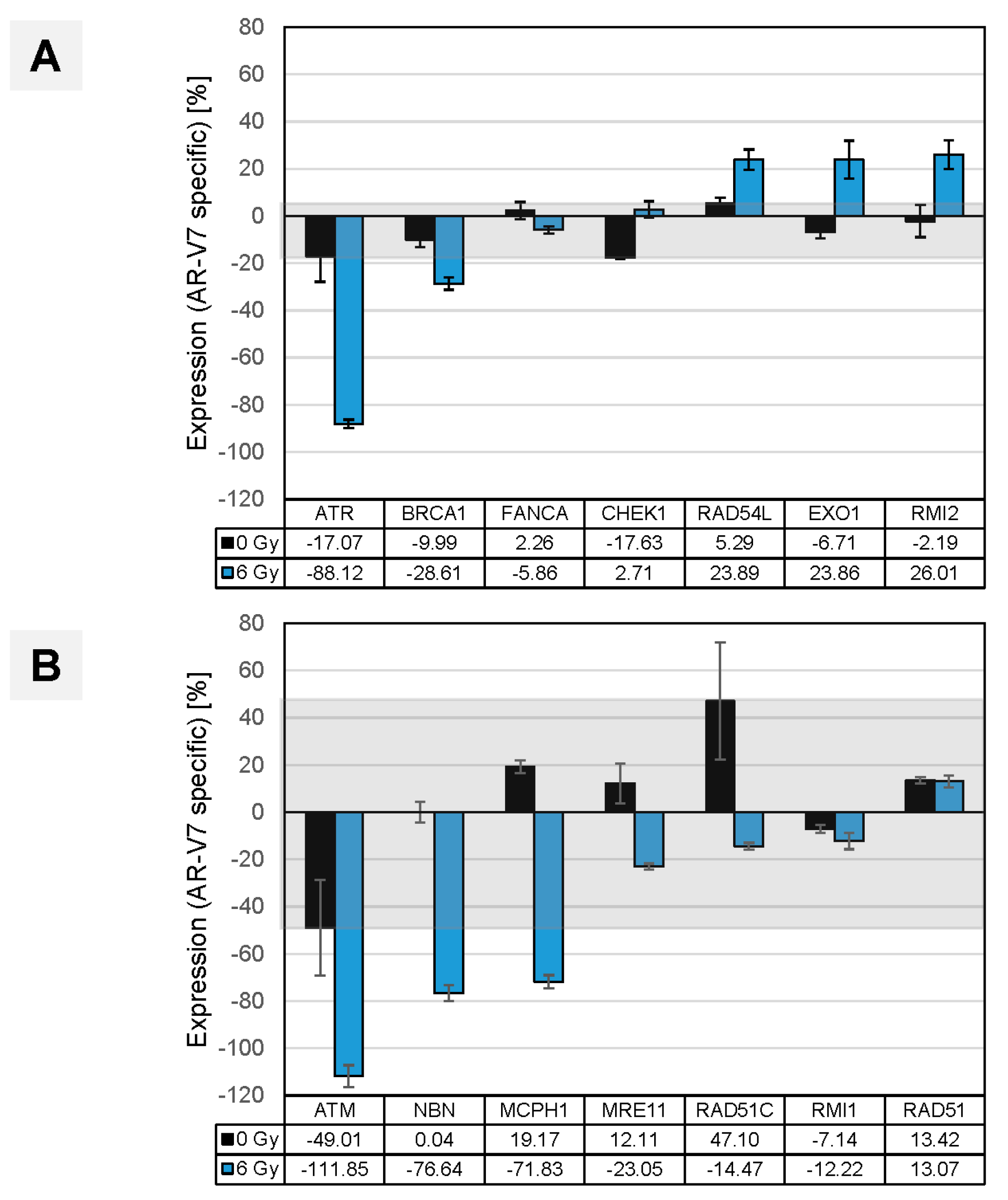{kind=link}
| Number of Patients | Number of Samples | |
|---|---|---|
| PCA, primary tumor, hormone-naïve | 77 | 136 |
| pT-stage | ||
| pT1b (TURP) | 20 | |
| pT2 | 14 | |
| pT3a | 9 | |
| pT3b | 21 | |
| pT4 | 3 | |
| unknown | 10 | |
| pN stage | ||
| pN0 | 32 | |
| pN1 | 12 | |
| pNx | 33 | |
| Prostatectomy ISUP/WHO grade group | ||
| ISUP 1 | 3 | |
| ISUP 2 | 6 | |
| ISUP 3 | 1 | |
| ISUP 4 | 37 | |
| ISUP 5 | 30 | |
| Morphology | ||
| Acinar adenocarcinoma | 52 | |
| Ductal/mixed adenocarcinoma § | 25 | |
| PCA, metastases, hormone-naïve | 23 | 28 |
| PCA ADT * | 42 | 55 |
| PCA CRPC *,# | 32 | 35 |
| BPH | 10 | 10 |
| Benign prostate tissue without hyperplasia | 7 | 7 |
| Neuroendocrine carcinoma of the prostate | 1 | 1 |
| Sarcomatoid carcinoma $ | 1 | 1 |
| OVERALL | 184 | 273 |
| AR splice variants vs. DNA repair. | ||||
| Patients | Coefficient Estimate | SD | p-Value | |
| ADT | 88 | 0.008013 | 0.003624 | 0.0297 * |
| ADT nonCRPC | 53 | 0.007468 | 0.004472 | 0.101 |
| CRPC | 35 | 0.012147 | 0.006103 | 0.0551 |
| Prim | 136 | 0.080467 | 0.038329 | 0.0377 * |
| AR splice variants vs. proliferation. | ||||
| Patients | Coefficient Estimate | SD | p-Value | |
| ADT | 88 | 0.028671 | 0.002070 | <2 × 10−16 *** |
| ADT nonCRPC | 53 | 0.030869 | 0.002317 | <2 × 10−16 *** |
| CRPC | 35 | 0.023398 | 0.003974 | 1.5 × 10−6 *** |
| Prim | 136 | 0.030435 | 0.002304 | <2 × 10−16 *** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tolkach, Y.; Kremer, A.; Lotz, G.; Schmid, M.; Mayr, T.; Förster, S.; Garbe, S.; Hosni, S.; Cronauer, M.V.; Kocsmár, I.; et al. Androgen Receptor Splice Variants Contribute to the Upregulation of DNA Repair in Prostate Cancer. Cancers 2022, 14, 4441. https://doi.org/10.3390/cancers14184441
Tolkach Y, Kremer A, Lotz G, Schmid M, Mayr T, Förster S, Garbe S, Hosni S, Cronauer MV, Kocsmár I, et al. Androgen Receptor Splice Variants Contribute to the Upregulation of DNA Repair in Prostate Cancer. Cancers. 2022; 14(18):4441. https://doi.org/10.3390/cancers14184441
Chicago/Turabian StyleTolkach, Yuri, Anika Kremer, Gábor Lotz, Matthias Schmid, Thomas Mayr, Sarah Förster, Stephan Garbe, Sana Hosni, Marcus V. Cronauer, Ildikó Kocsmár, and et al. 2022. "Androgen Receptor Splice Variants Contribute to the Upregulation of DNA Repair in Prostate Cancer" Cancers 14, no. 18: 4441. https://doi.org/10.3390/cancers14184441
APA StyleTolkach, Y., Kremer, A., Lotz, G., Schmid, M., Mayr, T., Förster, S., Garbe, S., Hosni, S., Cronauer, M. V., Kocsmár, I., Kocsmár, É., Riesz, P., Alajati, A., Ritter, M., Ellinger, J., Ohlmann, C.-H., & Kristiansen, G. (2022). Androgen Receptor Splice Variants Contribute to the Upregulation of DNA Repair in Prostate Cancer. Cancers, 14(18), 4441. https://doi.org/10.3390/cancers14184441