
Insights into Epigenetic Changes Related to Genetic Variants and Cells-of-Origin of Pancreatic Neuroendocrine Tumors: An Algorithm for Practical Workup


Abstract
:Simple Summary
Abstract
1. Introduction
2. Aim and Strategy
3. Origin of PanNETs
4. Pathology and Molecular Subtypes
5. Molecular Landscape
5.1. Genetic Basis: A-D-M Mutations
5.2. Epigenetic Basis: A-D-M Status in DNA Methylation
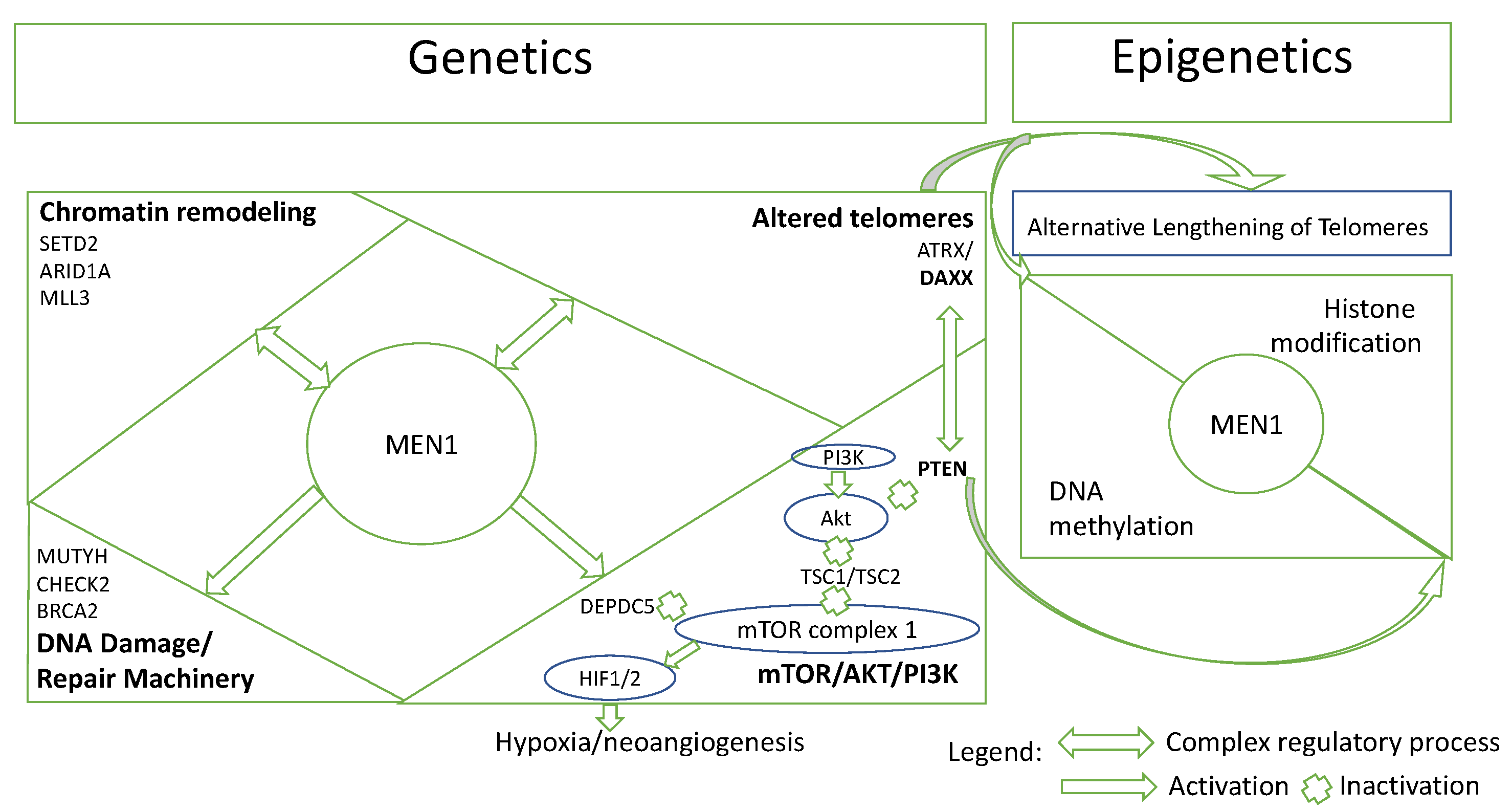
5.3. Epigenetics Basis: A-D-M and ALT Status in Chromatin Remodeling, Histone Changes and Telomere Alteration
5.4. Pathway-Based Interaction of PanNETs
6. Discussion
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pipinikas, C.P.; Berner, A.M.; Sposito, T.; Thirlwell, C. The Evolving (Epi)Genetic Landscape of Pancreatic Neuroendocrine Tumours. Endocr. Relat. Cancer 2019, 26, R519–R544. [Google Scholar] [CrossRef]
- Dasari, A.; Shen, C.; Halperin, D.; Zhao, B.; Zhou, S.; Xu, Y.; Shih, T.; Yao, J.C. Trends in the Incidence, Prevalence, and Survival Outcomes in Patients with Neuroendocrine Tumors in the United States. JAMA Oncol. 2017, 3, 1335–1342. [Google Scholar] [CrossRef]
- Das, S.; Dasari, A. Epidemiology, Incidence, and Prevalence of Neuroendocrine Neoplasms: Are There Global Differences? Curr. Oncol. Rep. 2021, 23, 43. [Google Scholar] [CrossRef]
- Buicko, J.L.; Finnerty, B.M.; Zhang, T.; Kim, B.J.; Fahey, T.J.; Nancy Du, Y.C. Insights into the Biology and Treatment Strategies of Pancreatic Neuroendocrine Tumors. Ann. Pancreat. Cancer 2019, 2, 12. [Google Scholar] [CrossRef]
- Singh, S.; Dey, C.; Kennecke, H.; Kocha, W.; Maroun, J.; Metrakos, P.; Mukhtar, T.; Pasieka, J.; Rayson, D.; Rowsell, C.; et al. Consensus Recommendations for the Diagnosis and Management of Pancreatic Neuroendocrine Tumors: Guidelines from a Canadian National Expert Group. Ann. Surg. Oncol. 2014, 22, 2685–2699. [Google Scholar] [CrossRef]
- Rindi, G.; Klimstra, D.S.; Abedi-Ardekani, B.; Asa, S.L.; Bosman, F.T.; Brambilla, E.; Busam, K.J.; de Krijger, R.R.; Dietel, M.; El-Naggar, A.K.; et al. A Common Classification Framework for Neuroendocrine Neoplasms: An International Agency for Research on Cancer (IARC) and World Health Organization (WHO) Expert Consensus Proposal. Mod. Pathol. 2018, 31, 1770–1786. [Google Scholar] [CrossRef]
- IARC Publications Website—WHO Classification of Tumours of Endocrine Organs. Available online: https://https://publications.iarc.fr/Book-And-Report-Series/Who-Classification-Of-Tumours/WHO-Classification-Of-Tumours-Of-Endocrine-Organs-2017 (accessed on 22 April 2022).
- Rindi, G.; Inzani, F. Neuroendocrine Neoplasm Update: Toward Universal Nomenclature. Endocr. Relat. Cancer 2020, 27, R211–R218. [Google Scholar] [CrossRef]
- Rindi, G.; Mete, O.; Uccella, S.; Basturk, O.; la Rosa, S.; Brosens, L.A.A.; Ezzat, S.; de Herder, W.W.; Klimstra, D.S.; Papotti, M.; et al. Overview of the 2022 WHO Classification of Neuroendocrine Neoplasms. Endocr. Pathol. 2022, 33, 115–154. [Google Scholar] [CrossRef]
- di Domenico, A.; Wiedmer, T.; Marinoni, I.; Perren, A. Genetic and Epigenetic Drivers of Neuroendocrine Tumours (NET). Endocr. Relat. Cancer 2017, 24, R315–R334. [Google Scholar] [CrossRef]
- Scarpa, A.; Chang, D.K.; Nones, K.; Corbo, V.; Patch, A.-M.; Bailey, P.; Lawlor, R.T.; Johns, A.L.; Miller, D.K.; Mafficini, A.; et al. Whole-Genome Landscape of Pancreatic Neuroendocrine Tumours. Nature 2017, 543, 65–71. [Google Scholar] [CrossRef] [Green Version]
- Conemans, E.B.; Lodewijk, L.; Moelans, C.B.; Offerhaus, G.J.A.; Pieterman, C.R.C.; Morsink, F.H.; Dekkers, O.M.; de Herder, W.W.; Hermus, A.R.; van der Horst-Schrivers, A.N.; et al. DNA Methylation Profiling in MEN1-Related Pancreatic Neuroendocrine Tumors Reveals a Potential Epigenetic Target for Treatment. Eur. J. Endocrinol. 2018, 179, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Park, J.K.; Paik, W.H.; Lee, K.; Ryu, J.K.; Lee, S.H.; Kim, Y.-T.; Park, J.K.; Paik, W.H.; Lee, K.; Ryu, J.K.; et al. DAXX/ATRX and MEN1 Genes Are Strong Prognostic Markers in Pancreatic Neuroendocrine Tumors. Oncotarget 2017, 8, 49796–49806. [Google Scholar] [CrossRef]
- Hackeng, W.M.; Brosens, L.A.A.; Poruk, K.E.; Noë, M.; Hosoda, W.; Poling, J.S.; Rizzo, A.; Campbell-Thompson, M.; Atkinson, M.A.; Konukiewitz, B.; et al. Aberrant Menin Expression Is an Early Event in Pancreatic Neuroendocrine Tumorigenesis. Hum. Pathol. 2016, 56, 93–100. [Google Scholar] [CrossRef]
- Heaphy, C.M.; de Wilde, R.F.; Jiao, Y.; Klein, A.P.; Edil, B.H.; Shi, C.; Bettegowda, C.; Rodriguez, F.J.; Eberhart, C.G.; Hebbar, S.; et al. Altered Telomeres in Tumors with ATRX and DAXX Mutations. Science 2011, 333, 425. [Google Scholar] [CrossRef]
- Cejas, P.; Drier, Y.; Dreijerink, K.M.A.; Brosens, L.A.A.; Deshpande, V.; Epstein, C.B.; Conemans, E.B.; Morsink, F.H.M.; Graham, M.K.; Valk, G.D.; et al. Enhancer Signatures Stratify and Predict Outcomes of Non-Functional Pancreatic Neuroendocrine Tumors. Nat. Med. 2019, 25, 1264–1269. [Google Scholar] [CrossRef] [PubMed]
- Consortium, R.E.; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative Analysis of 111 Reference Human Epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef]
- Tanaka, H.; Hijioka, S.; Hosoda, W.; Ueno, M.; Kobayashi, N.; Ikeda, M.; Ito, T.; Kodama, Y.; Morizane, C.; Notohara, K.; et al. Pancreatic Neuroendocrine Carcinoma G3 May Be Heterogeneous and Could Be Classified into Two Distinct Groups. Pancreatology 2020, 20, 1421–1427. [Google Scholar] [CrossRef]
- di Domenico, A.; Pipinikas, C.P.; Maire, R.S.; Bräutigam, K.; Simillion, C.; Dettmer, M.S.; Vassella, E.; Thirlwell, C.; Perren, A.; Marinoni, I. Epigenetic Landscape of Pancreatic Neuroendocrine Tumours Reveals Distinct Cells of Origin and Means of Tumour Progression. Commun. Biol. 2020, 3, 740. [Google Scholar] [CrossRef]
- Schimmack, S.; Svejda, B.; Lawrence, B.; Kidd, M.; Modlin, I.M. The Diversity and Commonalities of Gastroenteropancreatic Neuroendocrine Tumors. Langenbecks Arch. Surg. 2011, 396, 273–298. [Google Scholar] [CrossRef]
- Laidlaw, G.F. Nesidioblastoma, the Islet Tumor of the Pancreas. Am. J. Pathol. 1938, 14, 125. [Google Scholar] [PubMed]
- Vortmeyer, A.O.; Huang, S.; Lubensky, I.; Zhuang, Z. Non-Islet Origin of Pancreatic Islet Cell Tumors. J. Clin. Endocrinol. Metab. 2004, 89, 1934–1938. [Google Scholar] [CrossRef] [PubMed]
- Klöppel, G.; Anlauf, M.; Perren, A. Endocrine Precursor Lesions of Gastroenteropancreatic Neuroendocrine Tumors. Endocr. Pathol. 2007, 18, 150–155. [Google Scholar] [CrossRef]
- Jiao, Y.; Shi, C.; Edil, B.H.; de Wilde, R.F.; Klimstra, D.S.; Maitra, A.; Schulick, R.D.; Tang, L.H.; Wolfgang, C.L.; Choti, M.A.; et al. DAXX/ATRX, MEN1, and MTOR Pathway Genes Are Frequently Altered in Pancreatic Neuroendocrine Tumors. Science 2011, 331, 1199–1203. [Google Scholar] [CrossRef]
- Waldum, H.L.; Öberg, K.; Sørdal, Ø.F.; Sandvik, A.K.; Gustafsson, B.I.; Mjønes, P.; Fossmark, R. Not Only Stem Cells, but Also Mature Cells, Particularly Neuroendocrine Cells, May Develop into Tumours: Time for a Paradigm Shift. Ther. Adv. Gastroenterol. 2018, 11, 1756284818775054. [Google Scholar] [CrossRef]
- Suissa, Y.; Magenheim, J.; Stolovich-Rain, M.; Hija, A.; Collombat, P.; Mansouri, A.; Sussel, L.; Sosa-Pineda, B.; McCracken, K.; Wells, J.M.; et al. Gastrin: A Distinct Fate of Neurogenin3 Positive Progenitor Cells in the Embryonic Pancreas. PLoS ONE 2013, 8, e70397. [Google Scholar] [CrossRef]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-André, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-Enhancers in the Control of Cell Identity and Disease. Cell 2013, 155, 934. [Google Scholar] [CrossRef]
- Napolitano, T.; Avolio, F.; Courtney, M.; Vieira, A.; Druelle, N.; Ben-Othman, N.; Hadzic, B.; Navarro, S.; Collombat, P. Pax4 Acts as a Key Player in Pancreas Development and Plasticity. Semin. Cell Dev. Biol. 2015, 44, 107–114. [Google Scholar] [CrossRef]
- Collombat, P.; Hecksher-Sørensen, J.; Broccoli, V.; Krull, J.; Ponte, I.; Mundiger, T.; Smith, J.; Gruss, P.; Serup, P.; Mansouri, A. The Simultaneous Loss of Arx and Pax4 Genes Promotes a Somatostatin-Producing Cell Fate Specification at the Expense of the Alpha- and Beta-Cell Lineages in the Mouse Endocrine Pancreas. Development 2005, 132, 2969–2980. [Google Scholar] [CrossRef]
- Gannon, M.; Tweedie Ables, E.; Crawford, L.; Lowe, D.; Offield, M.F.; Magnuson, M.A.; Wright, C.V.E. Pdx-1 Function Is Specifically Required in Embryonic Beta Cells to Generate Appropriate Numbers of Endocrine Cell Types and Maintain Glucose Homeostasis. Dev. Biol. 2008, 314, 406–417. [Google Scholar] [CrossRef]
- Yang, Y.P.; Thorel, F.; Boyer, D.F.; Herrera, P.L.; Wright, C.V.E. Context-Specific α- to-β-Cell Reprogramming by Forced Pdx1 Expression. Genes. Dev. 2011, 25, 1680–1685. [Google Scholar] [CrossRef] [Green Version]
- Shih, H.P.; Wang, A.; Sander, M. Pancreas Organogenesis: From Lineage Determination to Morphogenesis. Annu. Rev. Cell Dev. Biol. 2013, 29, 81–105. [Google Scholar] [CrossRef]
- Larsen, H.L.; Grapin-Botton, A. The Molecular and Morphogenetic Basis of Pancreas Organogenesis. Semin. Cell Dev. Biol. 2017, 66, 51–68. [Google Scholar] [CrossRef]
- Hermann, G.; Konukiewitz, B.; Schmitt, A.; Perren, A.; Klöppel, G. Hormonally Defined Pancreatic and Duodenal Neuroendocrine Tumors Differ in Their Transcription Factor Signatures: Expression of ISL1, PDX1, NGN3, and CDX2. Virchows Arch. 2011, 459, 147–154. [Google Scholar] [CrossRef]
- Guz, Y.; Montminy, M.R.; Stein, R.; Leonard, J.; Gamer, L.W.; Wright, C.V.E.; Teitelman, G. Expression of Murine STF-1, a Putative Insulin Gene Transcription Factor, in Beta Cells of Pancreas, Duodenal Epithelium and Pancreatic Exocrine and Endocrine Progenitors during Ontogeny. Development 1995, 121, 11–18. [Google Scholar] [CrossRef]
- Chan, C.S.; Laddha, S.V.; Lewis, P.W.; Koletsky, M.S.; Robzyk, K.; da Silva, E.; Torres, P.J.; Untch, B.R.; Li, J.; Bose, P.; et al. ATRX, DAXX or MEN1 Mutant Pancreatic Neuroendocrine Tumors Are a Distinct Alpha-Cell Signature Subgroup. Nat. Commun. 2018, 9, 4158. [Google Scholar] [CrossRef]
- Boons, G.; Vandamme, T.; Ibrahim, J.; Roeyen, G.; Driessen, A.; Peeters, D.; Lawrence, B.; Print, C.; Peeters, M.; van Camp, G.; et al. PDX1 DNA Methylation Distinguishes Two Subtypes of Pancreatic Neuroendocrine Neoplasms with a Different Prognosis. Cancers 2020, 12, 1461. [Google Scholar] [CrossRef]
- Hackeng, W.M.; Morsink, F.H.M.; Moons, L.M.G.; Heaphy, C.M.; Offerhaus, G.J.A.; Dreijerink, K.M.A.; Brosens, L.A.A. Assessment of ARX Expression, a Novel Biomarker for Metastatic Risk in Pancreatic Neuroendocrine Tumors, in Endoscopic Ultrasound Fine-Needle Aspiration. Diagn. Cytopathol. 2020, 48, 308–315. [Google Scholar] [CrossRef]
- Brocks, D.; Assenov, Y.; Minner, S.; Bogatyrova, O.; Simon, R.; Koop, C.; Oakes, C.; Zucknick, M.; Lipka, D.B.; Weischenfeldt, J.; et al. Intratumor DNA Methylation Heterogeneity Reflects Clonal Evolution in Aggressive Prostate Cancer. Cell Rep. 2014, 8, 798–806. [Google Scholar] [CrossRef]
- Pommier, R.M.; Sanlaville, A.; Tonon, L.; Kielbassa, J.; Thomas, E.; Ferrari, A.; Sertier, A.S.; Hollande, F.; Martinez, P.; Tissier, A.; et al. Comprehensive Characterization of Claudin-Low Breast Tumors Reflects the Impact of the Cell-of-Origin on Cancer Evolution. Nat. Commun. 2020, 11, 3431. [Google Scholar] [CrossRef] [PubMed]
- Hackeng, W.M.; Schelhaas, W.; Morsink, F.H.M.; Heidsma, C.M.; van Eeden, S.; Valk, G.D.; Vriens, M.R.; Heaphy, C.M.; Nieveen van Dijkum, E.J.M.; Offerhaus, G.J.A.; et al. Alternative Lengthening of Telomeres and Differential Expression of Endocrine Transcription Factors Distinguish Metastatic and Non-Metastatic Insulinomas. Endocr. Pathol. 2020, 31, 108–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreijerink, K.M.A.; Hackeng, W.M.; Singhi, A.D.; Heaphy, C.M.; Brosens, L.A.A. Clinical Implications of Cell-of-Origin Epigenetic Characteristics in Non-Functional Pancreatic Neuroendocrine Tumors. J. Pathol. 2022, 256, 143–148. [Google Scholar] [CrossRef]
- Neiman, D.; Moss, J.; Hecht, M.; Magenheim, J.; Piyanzin, S.; Shapiro, A.M.J.; de Koning, E.J.P.; Razin, A.; Cedar, H.; Shemer, R.; et al. Islet Cells Share Promoter Hypomethylation Independently of Expression, but Exhibit Cell-Type-Specific Methylation in Enhancers. Proc. Natl. Acad. Sci. USA 2017, 114, 13525–13530. [Google Scholar] [CrossRef]
- Yang, K.C.; Kalloger, S.E.; Aird, J.J.; Lee, M.K.C.; Rushton, C.; Mungall, K.L.; Mungall, A.J.; Gao, D.; Chow, C.; Xu, J.; et al. Proteotranscriptomic Classification and Characterization of Pancreatic Neuroendocrine Neoplasms. Cell Rep. 2021, 37, 109817. [Google Scholar] [CrossRef]
- Raj, N.; Valentino, E.; Capanu, M.; Tang, L.H.; Basturk, O.; Untch, B.R.; Allen, P.J.; Klimstra, D.S.; Reidy-Lagunes, D. Treatment Response and Outcomes of Grade 3 Pancreatic Neuroendocrine Neoplasms Based on Morphology: Well Differentiated Versus Poorly Differentiated. Pancreas 2017, 46, 296–301. [Google Scholar] [CrossRef]
- Basturk, O.; Tang, L.; Hruban, R.H.; Adsay, V.; Yang, Z.; Krasinskas, A.M.; Vakiani, E.; la Rosa, S.; Jang, K.T.; Frankel, W.L.; et al. Poorly differentiated neuroendocrine carcinomas of the pancreas: A clinicopathologic analysis of 44 cases. Am. J. Surg. Pathol. 2014, 38, 437. [Google Scholar] [CrossRef]
- Sorbye, H.; Baudin, E.; Borbath, I.; Caplin, M.; Chen, J.; Cwikla, J.B.; Frilling, A.; Grossman, A.; Kaltsas, G.; Scarpa, A.; et al. Unmet Needs in High-Grade Gastroenteropancreatic Neuroendocrine Neoplasms (WHO G3). Neuroendocrinology 2019, 108, 54–62. [Google Scholar] [CrossRef]
- Khan, M.S.; Luong, T.V.; Watkins, J.; Toumpanakis, C.; Caplin, M.E.; Meyer, T. A Comparison of Ki-67 and Mitotic Count as Prognostic Markers for Metastatic Pancreatic and Midgut Neuroendocrine Neoplasms. Br. J. Cancer 2013, 108, 1838–1845. [Google Scholar] [CrossRef]
- Yang, Z.; Tang, L.H.; Klimstra, D.S. Effect of Tumor Heterogeneity on the Assessment of Ki67 Labeling Index in Well-Differentiated Neuroendocrine Tumors Metastatic to the Liver: Implications for Prognostic Stratification. Am. J. Surg. Pathol. 2011, 35, 853–860. [Google Scholar] [CrossRef]
- Luchini, C.; Pantanowitz, L.; Adsay, V.; Asa, S.L.; Antonini, P.; Girolami, I.; Veronese, N.; Nottegar, A.; Cingarlini, S.; Landoni, L.; et al. Ki-67 Assessment of Pancreatic Neuroendocrine Neoplasms: Systematic Review and Meta-Analysis of Manual vs. Digital Pathology Scoring. Mod. Pathol. 2022, 35, 712–720. [Google Scholar] [CrossRef]
- Partelli, S.; Bartsch, D.K.; Capdevila, J.; Chen, J.; Knigge, U.; Niederle, B.; Nieveen van Dijkum, E.J.M.; Pape, U.-F.; Pascher, A.; Ramage, J.; et al. ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Tumours: Surgery for Small Intestinal and Pancreatic Neuroendocrine Tumours. Neuroendocrinology 2017, 105, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Marinoni, I.; Kurrer, A.S.; Vassella, E.; Dettmer, M.; Rudolph, T.; Banz, V.; Hunger, F.; Pasquinelli, S.; Speel, E.J.; Perren, A. Loss of DAXX and ATRX Are Associated with Chromosome Instability and Reduced Survival of Patients with Pancreatic Neuroendocrine Tumors. Gastroenterology 2014, 146, 453–460. [Google Scholar] [CrossRef]
- Ali, A.S.; Grönberg, M.; Federspiel, B.; Scoazec, J.Y.; Hjortland, G.O.; Grønbæk, H.; Ladekarl, M.; Langer, S.W.; Welin, S.; Vestermark, L.W.; et al. Expression of P53 Protein in High-Grade Gastroenteropancreatic Neuroendocrine Carcinoma. PLoS ONE 2017, 12, e0187667. [Google Scholar] [CrossRef] [PubMed]
- Mafficini, A.; Scarpa, A. Genetics and Epigenetics of Gastroenteropancreatic Neuroendocrine Neoplasms. Endocr. Rev. 2019, 40, 506–536. [Google Scholar] [CrossRef] [PubMed]
- Yachida, S.; Vakiani, E.; White, C.M.; Zhong, Y.; Saunders, T.; Morgan, R.; de Wilde, R.F.; Maitra, A.; Hicks, J.; DeMarzo, A.M.; et al. Small Cell and Large Cell Neuroendocrine Carcinomas of the Pancreas Are Genetically Similar and Distinct from Well-Differentiated Pancreatic Neuroendocrine Tumors. Am. J. Surg. Pathol. 2012, 36, 173–184. [Google Scholar] [CrossRef]
- Scoazec, J.Y. Lung and Digestive Neuroendocrine Neoplasms. From WHO Classification to Biomarker Screening: Which Perspectives? Ann. Endocrinol. 2019, 80, 163–165. [Google Scholar] [CrossRef]
- Pavel, M.; Öberg, K.; Falconi, M.; Krenning, E.P.; Sundin, A.; Perren, A.; Berruti, A.; ESMO Guidelines Committee. Electronic address: [email protected] Gastroenteropancreatic Neuroendocrine Neoplasms: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2020, 31, 844–860. [Google Scholar] [CrossRef]
- Simon, T.; Riemer, P.; Jarosch, A.; Detjen, K.; di Domenico, A.; Bormann, F.; Menne, A.; Khouja, S.; Monjé, N.; Childs, L.H.; et al. DNA Methylation Reveals Distinct Cells of Origin for Pancreatic Neuroendocrine Carcinomas and Pancreatic Neuroendocrine Tumors. Genome Med. 2022, 14, 24. [Google Scholar] [CrossRef]
- Metz, D.C.; Jensen, R.T. Gastrointestinal Neuroendocrine Tumors: Pancreatic Endocrine Tumors. Gastroenterology 2008, 135, 1469. [Google Scholar] [CrossRef]
- Pea, A.; Hruban, R.H.; Wood, L.D. Genetics of Pancreatic Neuroendocrine Tumors: Implications for the Clinic. Expert Rev. Gastroenterol. Hepatol. 2015, 9, 1407. [Google Scholar] [CrossRef]
- Larsson, C.; Skogseid, B.; Öberg, K.; Nakamura, Y.; Nordenskjöld, M. Multiple Endocrine Neoplasia Type 1 Gene Maps to Chromosome 11 and Is Lost in Insulinoma. Nature 1988, 332, 85–87. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G. Mutation and Cancer: Statistical Study of Retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Cives, M.; Simone, V.; Rizzo, F.M.; Silvestris, F. NETs: Organ-Related Epigenetic Derangements and Potential Clinical Applications. Oncotarget 2016, 7, 57414. [Google Scholar] [CrossRef] [PubMed]
- Singhi, A.D.; Liu, T.C.; Roncaioli, J.L.; Cao, D.; Zeh, H.J.; Zureikat, A.H.; Tsung, A.; Marsh, J.W.; Lee, K.K.; Hogg, M.E.; et al. Alternative Lengthening of Telomeres and Loss of DAXX/ATRX Expression Predicts Metastatic Disease and Poor Survival in Patients with Pancreatic Neuroendocrine Tumors. Clin. Cancer Res. 2017, 23, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, A.; Kebebew, E. Genetic and Epigenetic Alterations in Pancreatic Neuroendocrine Tumors. J. Gastrointest. Oncol. 2020, 11, 567–577. [Google Scholar] [CrossRef]
- Lewis, P.W.; Elsaesser, S.J.; Noh, K.M.; Stadler, S.C.; Allis, C.D. Daxx Is an H3.3-Specific Histone Chaperone and Cooperates with ATRX in Replication-Independent Chromatin Assembly at Telomeres. Proc. Natl. Acad. Sci. USA 2010, 107, 14075–14080. [Google Scholar] [CrossRef]
- Zhang, H.; He, J.; Li, J.; Tian, D.; Gu, L.; Zhou, M. Methylation of RASSF1A Gene Promoter Is Regulated by P53 and DAXX. FASEB J. 2013, 27, 232. [Google Scholar] [CrossRef]
- Pipinikas, C.P.; Dibra, H.; Karpathakis, A.; Feber, A.; Novelli, M.; Oukrif, D.; Fusai, G.; Valente, R.; Caplin, M.; Meyer, T.; et al. Epigenetic Dysregulation and Poorer Prognosis in DAXX-Deficient Pancreatic Neuroendocrine Tumours. Endocr. Relat. Cancer 2015, 22, L13. [Google Scholar] [CrossRef]
- Kurihara, S.; Hiyama, E.; Onitake, Y.; Yamaoka, E.; Hiyama, K. Clinical Features of ATRX or DAXX Mutated Neuroblastoma. J. Pediatr. Surg. 2014, 49, 1835–1838. [Google Scholar] [CrossRef]
- Tirosh, A.; Mukherjee, S.; Lack, J.; Gara, S.K.; Wang, S.; Quezado, M.M.; Keutgen, X.M.; Wu, X.; Cam, M.; Kumar, S.; et al. Distinct Genome-Wide Methylation Patterns in Sporadic and Hereditary Nonfunctioning Pancreatic Neuroendocrine Tumors. Cancer 2019, 125, 1247–1257. [Google Scholar] [CrossRef]
- Lakis, V.; Lawlor, R.T.; Newell, F.; Patch, A.M.; Mafficini, A.; Sadanandam, A.; Koufariotis, L.T.; Johnston, R.L.; Leonard, C.; Wood, S.; et al. DNA Methylation Patterns Identify Subgroups of Pancreatic Neuroendocrine Tumors with Clinical Association. Commun. Biol. 2021, 4, 155. [Google Scholar] [CrossRef] [PubMed]
- Muraro, M.J.; Dharmadhikari, G.; Grün, D.; Groen, N.; Dielen, T.; Jansen, E.; van Gurp, L.; Engelse, M.A.; Carlotti, F.; de Koning, E.J.P.; et al. A Single-Cell Transcriptome Atlas of the Human Pancreas. Cell Syst. 2016, 3, 385–394.e3. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, J.S. Epigenetic Regulation in Gastroenteropancreatic Neuroendocrine Tumors. Front Oncol. 2022, 12, 901435. [Google Scholar] [CrossRef] [PubMed]
- Pea, A.; Yu, J.; Marchionni, L.; Noe, M.; Luchini, C.; Pulvirenti, A.; Wilde, R.F.d.; Brosens, L.A.; Rezaee, N.; Javed, A.; et al. Genetic Analysis of Small Well-Differentiated Pancreatic Neuroendocrine Tumors Identifies Subgroups with Differing Risks of Liver Metastases. Ann. Surg. 2020, 271, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Koneru, B.; Lopez, G.; Farooqi, A.; Conkrite, K.L.; Nguyen, T.H.; Macha, S.J.; Modi, A.; Rokita, J.L.; Urias, E.; Hindle, A.; et al. Telomere Maintenance Mechanisms Define Clinical Outcome in High-Risk Neuroblastoma. Cancer Res. 2020, 80, 2663–2675. [Google Scholar] [CrossRef]
- Ohba, S.; Kuwahara, K.; Yamada, S.; Abe, M.; Hirose, Y. Correlation between IDH, ATRX, and TERT Promoter Mutations in Glioma. Brain. Tumor. Pathol. 2020, 37, 33–40. [Google Scholar] [CrossRef]
- Heaphy, C.M.; Bi, W.L.; Coy, S.; Davis, C.; Gallia, G.L.; Santagata, S.; Rodriguez, F.J. Telomere Length Alterations and ATRX/DAXX Loss in Pituitary Adenomas. Mod. Pathol. 2020, 33, 1475–1481. [Google Scholar] [CrossRef]
- Ahvenainen, T.V.; Mäkinen, N.M.; von Nandelstadh, P.; Vahteristo, M.E.A.; Pasanen, A.M.; Bützow, R.C.; Vahteristo, P.M. Loss of ATRX/DAXX Expression and Alternative Lengthening of Telomeres in Uterine Leiomyomas. Cancer 2018, 124, 4650–4656. [Google Scholar] [CrossRef]
- de Wilde, R.F.; Heaphy, C.M.; Maitra, A.; Meeker, A.K.; Edil, B.H.; Wolfgang, C.L.; Ellison, T.A.; Schulick, R.D.; Molenaar, I.Q.; Valk, G.D.; et al. Loss of ATRX or DAXX Expression and Concomitant Acquisition of the Alternative Lengthening of Telomeres Phenotype Are Late Events in a Small Subset of MEN-1 Syndrome Pancreatic Neuroendocrine Tumors. Mod. Pathol. 2012, 25, 1033–1039. [Google Scholar] [CrossRef]
- Dogeas, E.; Karagkounis, G.; Heaphy, C.M.; Hirose, K.; Pawlik, T.M.; Wolfgang, C.L.; Meeker, A.; Hruban, R.H.; Cameron, J.L.; Choti, M.A. Alternative Lengthening of Telomeres Predicts Site of Origin in Neuroendocrine Tumor Liver Metastases. J. Am. Coll. Surg. 2014, 218, 628–635. [Google Scholar] [CrossRef]
- Luchini, C.; Lawlor, R.T.; Bersani, S.; Vicentini, C.; Paolino, G.; Mattiolo, P.; Pea, A.; Cingarlini, S.; Milella, M.; Scarpa, A. Alternative Lengthening of Telomeres (ALT) in Pancreatic Neuroendocrine Tumors: Ready for Prime-Time in Clinical Practice? Curr Oncol. Rep. 2021, 23, 106. [Google Scholar] [CrossRef] [PubMed]
- Hackeng, W.M.; Brosens, L.A.; Kim, J.Y.; O’Sullivan, R.; Sung, Y.N.; Liu, T.C.; Cao, D.; Heayn, M.; Cashman, J.B.; An, S.; et al. Non-Functional Pancreatic Neuroendocrine Tumours: ATRX/DAXX and Alternative Lengthening of Telomeres (ALT) Are Prognostically Independent from ARX/PDX1 Expression and Tumour Size. Gut 2022, 71, 961–973. [Google Scholar] [CrossRef]
- VandenBussche, C.J.; Allison, D.B.; Graham, M.K.; Charu, V.; Lennon, A.M.; Wolfgang, C.L.; Hruban, R.H.; Heaphy, C.M. Alternative Lengthening of Telomeres and ATRX/DAXX Loss Can Be Reliably Detected in FNAs of Pancreatic Neuroendocrine Tumors. Cancer Cytopathol. 2017, 125, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Maharjan, C.K.; Ear, P.H.; Tran, C.G.; Howe, J.R.; Chandrasekharan, C.; Quelle, D.E. Pancreatic Neuroendocrine Tumors: Molecular Mechanisms and Therapeutic Targets. Cancers 2021, 13, 5117. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.; Tang, L.H.; Davidson, C.; Vosburgh, E.; Chen, W.; Foran, D.J.; Notterman, D.A.; Levine, A.J.; Xu, E.Y. Two Well-Differentiated Pancreatic Neuroendocrine Tumor Mouse Models. Cell Death Differ. 2020, 27, 269. [Google Scholar] [CrossRef]
- Modlin, I.M.; Oberg, K.; Taylor, A.; Drozdov, I.; Bodei, L.; Kidd, M. Neuroendocrine Tumor Biomarkers: Current Status and Perspectives. Neuroendocrinology 2014, 100, 265–277. [Google Scholar] [CrossRef]
- Boninsegna, L.; Panzuto, F.; Partelli, S.; Capelli, P.; Fave, G.D.; Bettini, R.; Pederzoli, P.; Scarpa, A.; Falconi, M. Malignant Pancreatic Neuroendocrine Tumour: Lymph Node Ratio and Ki67 Are Predictors of Recurrence after Curative Resections. Eur. J. Cancer 2012, 48, 1608–1615. [Google Scholar] [CrossRef]
- Pompili, L.; Leonetti, C.; Biroccio, A.; Salvati, E. Diagnosis and Treatment of ALT Tumors: Is Trabectedin a New Therapeutic Option? J. Exp. Clin. Cancer Res. 2017, 36, 189. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
| P53 | Rb1 | DAXX | ATRX | |
|---|---|---|---|---|
| PanNETs | − | + | − | − |
| PanNECs | + | − | + | + |
| PanNEN 1 | PanNEN 2 | PanNEN 3 | PanNEN 4 | PanNEN 5 | |
|---|---|---|---|---|---|
| Cell of origin epigenetic markers | α- cell-like (ARX+/PDX−) | α- cell-like > β- cell-like | β- cell-like (ARX−/PDX+) | α- cell-like > β- cell-like | Unknown |
| Clinical features | Non-functional | Non-functional | Functional-insulinoma | Functional-insulinoma | Non-functional |
| Genetic signature | MEN1 | MEN1/ATRX/DAXX | Other | ATRX/DAXX | Other |
| Epigenetic pattern | Well-differentiated | Intermediate differentiation | Well-differentiated | Intermediate differentiation | Well and poor differentiated |
| ALT activation | No | High susceptibility | No | High susceptibility | Unknown |
| Outcome | ↑ | ↓ | ↑ | ↓ | ↓ |
| Risk Stratification | Type of Cell | A-D-M | ARX | PDX1 | ALT |
|---|---|---|---|---|---|
| Aggressive ++ NF tumor | α like | A-D-M− | +/DN | − | + |
| Aggressive NF tumor | α like > β like | M− | +/DN | − | − |
| Aggressive-F tumor (insulinoma) | α like > β like | A-D− | +/DN | − | + |
| Indolent-F tumor (insulinoma) | β like | A-D-M+ | − | +/DP | − |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciobanu, O.A.; Martin, S.C.; Herlea, V.; Fica, S. Insights into Epigenetic Changes Related to Genetic Variants and Cells-of-Origin of Pancreatic Neuroendocrine Tumors: An Algorithm for Practical Workup. Cancers 2022, 14, 4444. https://doi.org/10.3390/cancers14184444
Ciobanu OA, Martin SC, Herlea V, Fica S. Insights into Epigenetic Changes Related to Genetic Variants and Cells-of-Origin of Pancreatic Neuroendocrine Tumors: An Algorithm for Practical Workup. Cancers. 2022; 14(18):4444. https://doi.org/10.3390/cancers14184444
Chicago/Turabian StyleCiobanu, Oana A., Sorina C. Martin, Vlad Herlea, and Simona Fica. 2022. "Insights into Epigenetic Changes Related to Genetic Variants and Cells-of-Origin of Pancreatic Neuroendocrine Tumors: An Algorithm for Practical Workup" Cancers 14, no. 18: 4444. https://doi.org/10.3390/cancers14184444