
Emerging Roles of TRIM Family Proteins in Gliomas Pathogenesis

 and
and 
Abstract
Simple Summary
Abstract
1. Introduction
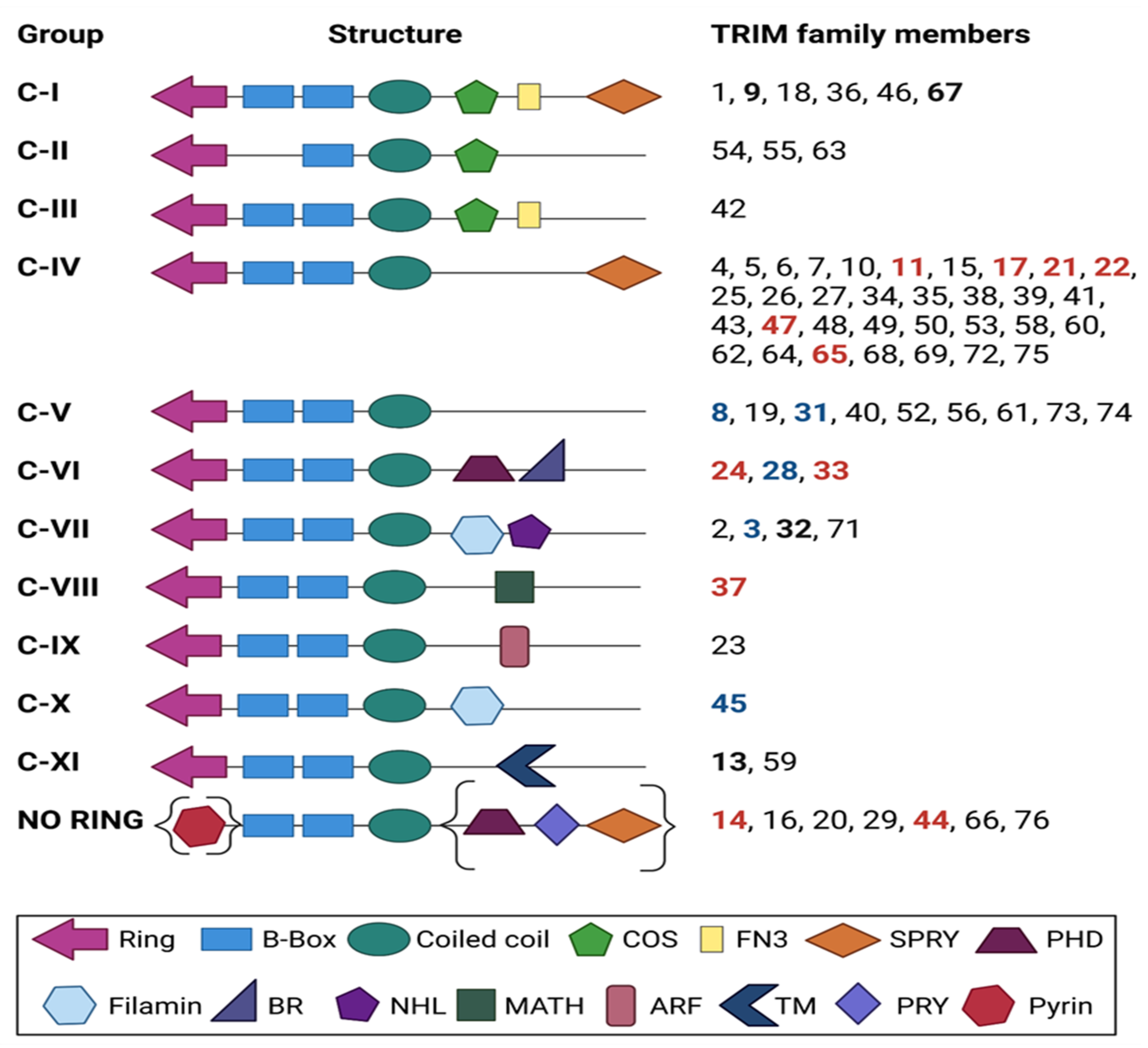
2. TRIM Proteins: An Overview of Structure and Function
3. TRIM Proteins and the Nervous System: Insights into Brain Physiology and Pathophysiology
3.1. Healthy Brain
3.2. Pathological Conditions-Injured Brain

{kind=link}
{kind=link}
| Group | TRIM | Role in Brain Physiology | Role in Brain Pathologies | References |
|---|---|---|---|---|
| C-I | TRIM67 |
| - | [66,67,68] |
| C-I | TRIM9 |
|
| [67,68] |
| C-IV | TRIM11 |
|
| [63,74] |
| C-V | TRIM8 |
|
| [75,76] |
| C-V | TRIM31 |
|
| [71] |
| C-VI | TRIM28 |
|
| [58,59,60,61,62] |
| C-VII | TRIM3 |
|
| [64,78] |
| C-VII | TRIM32 |
|
| [70] |
| C-X | TRIM45 |
|
| [65,72] |
| C-XI | TRIM13 |
|
| [72] |
4. TRIM Proteins in Brain Tumors
4.1. Family Group C-IV: TRIM11, TRIM17, TRIM21, TRIM22, TRIM47 and TRIM65
4.2. Family Group C-V: TRIM8 and TRIM31
4.3. Family Group C-VI: TRIM24, TRIM28, and TRIM33
4.4. Family Group C-VII: TRIM3 and TRIM32
4.5. Family Group C-VIII: TRIM37
4.6. Family Group X: TRIM45
4.7. No-Ring Group: TRIM14, TRIM44, and TRIM66
| Group | TRIM | Expression and Function | Role * | References |
|---|---|---|---|---|
| C-IV | TRIM11 |
| ONC | [86] |
| TRIM17 |
| TS | [87] | |
| TRIM21 |
| ONC | [88] | |
| TRIM22 |
| ONC | [89] | |
| TRIM47 |
| ONC | [90,91] | |
| TRIM65 |
| UNCLEAR | [92] | |
| C-V | TRIM8 |
| DUAL | [93,132] |
| TRIM31 |
| ONC | [94,95,133] | |
| C-VI | TRIM24 |
| ONC | [83,84,98] |
| TRIM28 |
| ONC | [35,82,103,108,112,134] | |
| TRIM33 |
| TS | [118,119,121] | |
| C-VII | TRIM3 |
| [124] | |
| TRIM32 |
| [126] | ||
| C-VIII | TRIM37 |
| ONC | [81] |
| C-X | TRIM45 |
| TS | [85] |
| no-Ring | TRIM14 |
| ONC | [127] |
| TRIM44 |
| ONC | [129,130] | |
| TRIM66 |
| ONC | [131] |
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ARF | ADP ribosylation factor-like |
| BioID | Biotin Identification |
| BRD | Bromodomain |
| CC | Coiled-Coil |
| CCND1 | Cyclin D1 |
| CL | Classical |
| CNS | Central Nervous System |
| COS | C-terminal subgroup one signature |
| DDR | DNA damage response |
| DEGs | Differentially Expressed Genes |
| DLGNT | Diffuse Leptomeningeal Glioneuronal Tumors |
| EE | Epileptic Encephalopathy |
| ESCs | Embryonic Stem Cells |
| EGFRvIII | EGF receptor vIII |
| EMT | Endothelial to Mesenchymal Transition |
| ERVs | Endogenous retroviruses |
| FBXO45 | F-Box protein 45 |
| FN3 | Fibronectin type III motif |
| GBM | Glioblastoma |
| GSCs | Glioma-derived stem cells |
| H3 | Histone 3 |
| H3K23ac | H3 lysine 23 acetylation |
| HB-EGF | Heparin-binding EGF-like growth factor |
| HFD | High Fat Diet |
| HGGs | High Grade Gliomas |
| ID1 | Inhibitor of DNA binding 1 |
| IRS1 | Insulin Receptor Substrate-1 |
| K195 | Lysine 195 |
| KAP1 | KRAB-associated protein-1 (TRIM28) |
| KEAP1 | Kelch-like ECH- Associated Protein 1 |
| LGGs | Low Grade Gliomas |
| LGMDR8 | Limb–Girdle Muscular Dystrophy R8 |
| lncRNAs | Long Non-Coding RNAs |
| LOH | Loss of Heterozygosity |
| MAPK | Mitogen-Activated Protein Kinase |
| MATH | Meprin And Tumor-necrosis factor receptor-associated factor Homology |
| MES | Mesenchymal |
| MG53 | Mitsugumin 53 |
| MGMT | O6-methylguanine-DNA methyltransferase |
| NPCs | Neural Progenitor Cells |
| NSCs | Neural Stem Cells |
| OGD/R | Oxygen-Glucose Deprivation/Reperfusion |
| OPCs | Oligodendrocyte Progenitor Cells |
| OPTN | Optineurin |
| OS | Overall Survival |
| OSKM | OCT4, SOX2, KLF4, and c-MYC |
| PFS | Progression Free Survival |
| PHD | Plant Homeodomain |
| PSD | Post Synaptic Density |
| R193 | arginine 193 |
| RBCC | RING-B-box-Coiled-Coil |
| RNAi | RNA interference |
| shRNAs | Short Hairpin RNAs |
| SLE | Systemic Lupus Erythematosus |
| TEs | Transposable Elements |
| TIF1 | Transcriptional Intermediary Factor 1 |
| TIF1γ | Transcriptional Intermediary Factor 1 γ |
| TMZ | Temozolomide |
| TRIM | Tripartite Motif-containing |
References
- Galbraith, K.; Snuderl, M. Molecular Pathology of Gliomas. Surg. Pathol. Clin. 2021, 14, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Llaguno, S.R.A.; Parada, L.F. Cell of Origin of Glioma: Biological and Clinical Implications. Br. J. Cancer 2016, 115, 1445–1450. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.L.; Wadhwani, N.; Horbinski, C. Major Features of the 2021 WHO Classification of CNS Tumors. Neurotherapeutics 2022. [Google Scholar] [CrossRef] [PubMed]
- Śledzińska, P.; Bebyn, M.G.; Furtak, J.; Kowalewski, J.; Lewandowska, M.A. Prognostic and Predictive Biomarkers in Gliomas. Int. J. Mol. Sci. 2021, 22, 10373. [Google Scholar] [CrossRef]
- Yang, K.; Wu, Z.; Zhang, H.; Zhang, N.; Wu, W.; Wang, Z.; Dai, Z.; Zhang, X.; Zhang, L.; Peng, Y.; et al. Glioma Targeted Therapy: Insight into Future of Molecular Approaches. Mol. Cancer 2022, 21, 39. [Google Scholar] [CrossRef]
- Mandell, M.A.; Saha, B.; Thompson, T.A. The Tripartite Nexus: Autophagy, Cancer, and Tripartite Motif-Containing Protein Family Members. Front. Pharmacol. 2020, 11, 308. [Google Scholar] [CrossRef]
- Meroni, G.; Diez-Roux, G. TRIM/RBCC, a Novel Class of “single Protein RING Finger” E3 Ubiquitin Ligases. BioEssays 2005, 27, 1147–1157. [Google Scholar] [CrossRef]
- Torok, M.; Etkin, L.D. Two B or Not Two B? Overview of the Rapidly Expanding B-Box Family of Proteins. Differentiation 2001, 67, 63–71. [Google Scholar] [CrossRef]
- Reymond, A.; Meroni, G.; Fantozzi, A.; Merla, G.; Cairo, S.; Luzi, L.; Riganelli, D.; Zanaria, E.; Messali, S.; Cainarca, S.; et al. The Tripartite Motif Family Identifies Cell Compartments. EMBO J. 2001, 20, 2140. [Google Scholar] [CrossRef]
- Komander, D.; Rape, M. The Ubiquitin Code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef]
- Napolitano, L.M.; Jaffray, E.G.; Hay, R.T.; Meroni, G. Functional Interactions between Ubiquitin E2 Enzymes and TRIM Proteins. Biochem. J. 2011, 434, 309–319. [Google Scholar] [CrossRef]
- Deshaies, R.J.; Joazeiro, C.A.P. RING Domain E3 Ubiquitin Ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef]
- Nenasheva, V.V.; Tarantul, V.Z. Many Faces of TRIM Proteins on the Road from Pluripotency to Neurogenesis. Stem Cells Dev. 2020, 29, 1–14. [Google Scholar] [CrossRef]
- Williams, F.P.; Haubrich, K.; Perez-Borrajero, C.; Hennig, J. Emerging RNA-Binding Roles in the TRIM Family of Ubiquitin Ligases. Biol. Chem. 2019, 400, 1443–1464. [Google Scholar] [CrossRef]
- Zhang, L.; Afolabi, L.O.; Wan, X.; Li, Y.; Chen, L. Emerging Roles of Tripartite Motif-Containing Family Proteins (TRIMs) in Eliminating Misfolded Proteins. Front. Cell Dev. Biol. 2020, 8, 802. [Google Scholar] [CrossRef]
- Short, K.M.; Cox, T.C. Subclassification of the RBCC/TRIM Superfamily Reveals a Novel Motif Necessary for Microtubule Binding. J. Biol. Chem. 2006, 281, 8970–8980. [Google Scholar] [CrossRef]
- McNab, F.W.; Rajsbaum, R.; Stoye, J.P.; O’Garra, A. Tripartite-Motif Proteins and Innate Immune Regulation. Curr. Opin. Immunol. 2011, 23, 46–56. [Google Scholar] [CrossRef]
- Ozato, K.; Shin, D.M.; Chang, T.H.; Morse, H.C. TRIM Family Proteins and Their Emerging Roles in Innate Immunity. Nat. Rev. Immunol. 2008, 8, 849–860. [Google Scholar] [CrossRef]
- Sardiello, M.; Cairo, S.; Fontanella, B.; Ballabio, A.; Meroni, G. Genomic Analysis of the TRIM Family Reveals Two Groups of Genes with Distinct Evolutionary Properties. BMC Evol. Biol. 2008, 8, 225. [Google Scholar] [CrossRef]
- Rhodes, D.A.; De Bono, B.; Trowsdale, J. Relationship between SPRY and B30.2 Protein Domains. Evolution of a Component of Immune Defence? Immunology 2005, 116, 411–417. [Google Scholar] [CrossRef]
- Tocchini, C.; Ciosk, R. TRIM-NHL Proteins in Development and Disease. Semin. Cell Dev. Biol. 2015, 47–48, 52–59. [Google Scholar] [CrossRef]
- Herquel, B.; Ouararhni, K.; Davidson, I. The TIF1α-Related TRIM Cofactors Couple Chromatin Modifications to Transcriptional Regulation, Signaling and Tumor Suppression. Transcription 2011, 2, 231. [Google Scholar] [CrossRef]
- Appikonda, S.; Thakkar, K.N.; Shah, P.K.; Dent, S.Y.R.; Andersen, J.N.; Barton, M.C. Cross-Talk between Chromatin Acetylation and SUMOylation of Tripartite Motif-Containing Protein 24 (TRIM24) Impacts Cell Adhesion. J. Biol. Chem. 2018, 293, 7476–7485. [Google Scholar] [CrossRef]
- Sparrer, K.M.J.; Gableske, S.; Zurenski, M.A.; Parker, Z.M.; Full, F.; Baumgart, G.J.; Kato, J.; Pacheco-Rodriguez, G.; Liang, C.; Pornillos, O.; et al. TRIM23 Mediates Virus-Induced Autophagy via Activation of TBK1. Nat. Microbiol. 2017, 2, 1543. [Google Scholar] [CrossRef]
- Skerra, A. Engineered Protein Scaffolds for Molecular Recognition. J. Mol. Recognit. 2000, 13, 167–187. [Google Scholar] [CrossRef]
- Meroni, G. Genomics and Evolution of the TRIM Gene Family. Adv. Exp. Med. Biol. 2012, 770, 1–9. [Google Scholar] [CrossRef]
- Nguyen, D.T.T.; Richter, D.; Michel, G.; Mitschka, S.; Kolanus, W.; Cuevas, E.; Gregory Wulczyn, F. The Ubiquitin Ligase LIN41/TRIM71 Targets P53 to Antagonize Cell Death and Differentiation Pathways during Stem Cell Differentiation. Cell Death Differ. 2017, 24, 1063–1078. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Fiskus, W.; Yong, B.; Atadja, P.; Takahashi, Y.; Pandita, T.K.; Wang, H.G.; Bhalla, K.N. Acetylated Hsp70 and KAP1-Mediated Vps34 SUMOylation Is Required for Autophagosome Creation in Autophagy. Proc. Natl. Acad. Sci. USA 2013, 110, 6841–6846. [Google Scholar] [CrossRef] [PubMed]
- Pineda, C.T.; Ramanathan, S.; Fon Tacer, K.; Weon, J.L.; Potts, M.B.; Ou, Y.H.; White, M.A.; Potts, P.R. Degradation of AMPK by a Cancer-Specific Ubiquitin Ligase. Cell 2015, 160, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Czerwińska, P.; Mazurek, S.; Wiznerowicz, M. The Complexity of TRIM28 Contribution to Cancer. J. Biomed. Sci. 2017, 24, 63. [Google Scholar] [CrossRef]
- Wong, X.; Cutler, J.A.; Hoskins, V.E.; Gordon, M.; Madugundu, A.K.; Pandey, A.; Reddy, K.L. Mapping the Micro-Proteome of the Nuclear Lamina and Lamina-Associated Domains. Life Sci. Alliance 2021, 4, e202000774. [Google Scholar] [CrossRef]
- Jin, X.; Pan, Y.; Wang, L.; Zhang, L.; Ravichandran, R.; Potts, P.R.; Jiang, J.; Wu, H.; Huang, H. MAGE-TRIM28 Complex Promotes the Warburg Effect and Hepatocellular Carcinoma Progression by Targeting FBP1 for Degradation. Oncogenesis 2017, 6, e312. [Google Scholar] [CrossRef]
- Randolph, K.; Hyder, U.; D’Orso, I. KAP1/TRIM28: Transcriptional Activator and/or Repressor of Viral and Cellular Programs? Front. Cell. Infect. Microbiol. 2022, 12, 834636. [Google Scholar] [CrossRef]
- Pineda, C.T.; Potts, P.R. Oncogenic MAGEA-TRIM28 Ubiquitin Ligase Downregulates Autophagy by Ubiquitinating and Degrading AMPK in Cancer. Autophagy 2015, 11, 844–846. [Google Scholar] [CrossRef]
- Peng, Y.; Zhang, M.; Jiang, Z.; Jiang, Y. TRIM28 Activates Autophagy and Promotes Cell Proliferation in Glioblastoma. OncoTargets Ther. 2019, 12, 397–404. [Google Scholar] [CrossRef]
- Hatakeyama, S. TRIM Family Proteins: Roles in Autophagy, Immunity, and Carcinogenesis. Trends Biochem. Sci. 2017, 42, 297–311. [Google Scholar] [CrossRef]
- Di Rienzo, M.; Romagnoli, A.; Antonioli, M.; Piacentini, M.; Fimia, G.M. TRIM Proteins in Autophagy: Selective Sensors in Cell Damage and Innate Immune Responses. Cell Death Differ. 2020, 27, 887–902. [Google Scholar] [CrossRef]
- Jena, K.K.; Kolapalli, S.P.; Mehto, S.; Nath, P.; Das, B.; Sahoo, P.K.; Ahad, A.; Syed, G.H.; Raghav, S.K.; Senapati, S.; et al. TRIM16 Controls Assembly and Degradation of Protein Aggregates by Modulating the P62-NRF2 Axis and Autophagy. EMBO J. 2018, 37, e98358. [Google Scholar] [CrossRef]
- Van Gent, M.; Sparrer, K.M.J.; Gack, M.U. TRIM Proteins and Their Roles in Antiviral Host Defenses. Annu. Rev. Virol. 2018, 5, 385–405. [Google Scholar] [CrossRef]
- Giraldo, M.I.; Hage, A.; van Tol, S.; Rajsbaum, R. TRIM Proteins in Host Defense and Viral Pathogenesis. Curr. Clin. Microbiol. Rep. 2020, 7, 101–114. [Google Scholar] [CrossRef]
- McAvera, R.M.; Crawford, L.J. TIF1 Proteins in Genome Stability and Cancer. Cancers 2020, 12, 2094. [Google Scholar] [CrossRef] [PubMed]
- Crawford, L.J.; Johnston, C.K.; Irvine, A.E. TRIM Proteins in Blood Cancers. J. Cell Commun. Signal. 2018, 12, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Marzano, F.; Caratozzolo, M.F.; Pesole, G.; Sbisà, E.; Tullo, A. TRIM Proteins in Colorectal Cancer: TRIM8 as a Promising Therapeutic Target in Chemo Resistance. Biomedicines 2021, 9, 241. [Google Scholar] [CrossRef] [PubMed]
- Gechijian, L.N.; Buckley, D.L.; Lawlor, M.A.; Reyes, J.M.; Paulk, J.; Ott, C.J.; Winter, G.E.; Erb, M.A.; Scott, T.G.; Xu, M.; et al. Functional TRIM24 Degrader via Conjugation of Ineffectual Bromodomain and VHL Ligands Article. Nat. Chem. Biol. 2018, 14, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wei, X.; Luo, Q.; Xia, Y.; Pan, T.; He, J.; Jahangir, A.; Jia, L.; Liu, W.; Zou, Y.; et al. Loss of TRIM67 Attenuates the Progress of Obesity-Induced Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 7475. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Xiao, R.P. MG53 and Disordered Metabolism in Striated Muscle. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1984–1990. [Google Scholar] [CrossRef]
- Dalgaard, K.; Landgraf, K.; Heyne, S.; Lempradl, A.; Longinotto, J.; Gossens, K.; Ruf, M.; Orthofer, M.; Strogantsev, R.; Selvaraj, M.; et al. Trim28 Haploinsufficiency Triggers Bi-Stable Epigenetic Obesity. Cell 2016, 164, 353–364. [Google Scholar] [CrossRef]
- Gao, W.; Li, Y.; Liu, X.; Wang, S.; Mei, P.; Chen, Z.; Liu, K.; Li, S.; Xu, X.W.; Gan, J.; et al. TRIM21 Regulates Pyroptotic Cell Death by Promoting Gasdermin D Oligomerization. Cell Death Differ. 2022, 29, 439–450. [Google Scholar] [CrossRef]
- Smith, S.; Nígabhann, J.; McCarthy, E.; Coffey, B.; Mahony, R.; Byrne, J.C.; Stacey, K.; Ball, E.; Bell, A.; Cunnane, G.; et al. Estrogen Receptor α Regulates Tripartite Motif-Containing Protein 21 Expression, Contributing to Dysregulated Cytokine Production in Systemic Lupus Erythematosus. Arthritis Rheumatol. 2014, 66, 163–172. [Google Scholar] [CrossRef]
- Liu, D.; Zhao, Z.; She, Y.; Zhang, L.; Chen, X.; Ma, L.; Cui, J. TRIM14 Inhibits OPTN-Mediated Autophagic Degradation of KDM4D to Epigenetically Regulate Inflammation. Proc. Natl. Acad. Sci. USA 2022, 119, e2113454119. [Google Scholar] [CrossRef]
- Bond, S.T.; King, E.J.; Henstridge, D.C.; Tran, A.; Moody, S.C.; Yang, C.; Liu, Y.; Mellett, N.A.; Nath, A.P.; Inouye, M.; et al. Deletion of Trim28 in Committed Adipocytes Promotes Obesity but Preserves Glucose Tolerance. Nat. Commun. 2021, 12, 74. [Google Scholar] [CrossRef]
- Watanabe, M.; Takahashi, H.; Saeki, Y.; Ozaki, T.; Itoh, S.; Suzuki, M.; Mizushima, W.; Tanaka, K.; Hatakeyama, S. The E3 Ubiquitin Ligase TRIM23 Regulates Adipocyte Differentiation via Stabilization of the Adipogenic Activator PPARγ. eLife 2015, 4, e05615. [Google Scholar] [CrossRef]
- Wan, T.; Li, X.; Li, Y. The Role of TRIM Family Proteins in Autophagy, Pyroptosis, and Diabetes Mellitus. Cell Biol. Int. 2021, 45, 913–926. [Google Scholar] [CrossRef]
- Zhang, J.R.; Li, X.X.; Hu, W.N.; Li, C.Y. Emerging Role of TRIM Family Proteins in Cardiovascular Disease. Cardiology 2020, 145, 390–400. [Google Scholar] [CrossRef]
- Song, R.; Peng, W.; Zhang, Y.; Lv, F.; Wu, H.K.; Guo, J.; Cao, Y.; Pi, Y.; Zhang, X.; Jin, L.; et al. Central Role of E3 Ubiquitin Ligase MG53 in Insulin Resistance and Metabolic Disorders. Nature 2013, 494, 375–379. [Google Scholar] [CrossRef]
- Jiang, W.; Liu, M.; Gu, C.; Ma, H. The Pivotal Role of Mitsugumin 53 in Cardiovascular Diseases. Cardiovasc. Toxicol. 2021, 21, 2–11. [Google Scholar] [CrossRef]
- Jaworska, A.M.; Wlodarczyk, N.A.; Mackiewicz, A.; Czerwinska, P. The Role of TRIM Family Proteins in the Regulation of Cancer Stem Cell Self-Renewal. Stem Cells 2020, 38, 165–173. [Google Scholar] [CrossRef]
- Miles, D.C.; de Vries, N.A.; Gisler, S.; Lieftink, C.; Akhtar, W.; Gogola, E.; Pawlitzky, I.; Hulsman, D.; Tanger, E.; Koppens, M.; et al. TRIM28 Is an Epigenetic Barrier to Induced Pluripotent Stem Cell Reprogramming. Stem Cells 2017, 35, 147–157. [Google Scholar] [CrossRef]
- Fasching, L.; Kapopoulou, A.; Sachdeva, R.; Petri, R.; Jönsson, M.E.; Männe, C.; Turelli, P.; Jern, P.; Cammas, F.; Trono, D.; et al. TRIM28 Represses Transcription of Endogenous Retroviruses in Neural Progenitor Cells. Cell Rep. 2015, 10, 20–28. [Google Scholar] [CrossRef]
- Brattås, P.L.; Jönsson, M.E.; Fasching, L.; Nelander Wahlestedt, J.; Shahsavani, M.; Falk, R.; Falk, A.; Jern, P.; Parmar, M.; Jakobsson, J. TRIM28 Controls a Gene Regulatory Network Based on Endogenous Retroviruses in Human Neural Progenitor Cells. Cell Rep. 2017, 18, 1–11. [Google Scholar] [CrossRef]
- Pavlaki, I.; Alammari, F.; Sun, B.; Clark, N.; Sirey, T.; Lee, S.; Woodcock, D.J.; Ponting, C.P.; Szele, F.G.; Vance, K.W. The Long Non-coding RNA Paupar Promotes KAP 1-dependent Chromatin Changes and Regulates Olfactory Bulb Neurogenesis. EMBO J. 2018, 37, e98219. [Google Scholar] [CrossRef] [PubMed]
- Grassi, D.A.; Jönsson, M.E.; Brattås, P.L.; Jakobsson, J. TRIM28 and the Control of Transposable Elements in the Brain. Brain Res. 2019, 1705, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Tuoc, T.C.; Stoykova, A. Trim11 Modulates the Function of Neurogenic Transcription Factor Pax6 through Ubiquitin-Proteosome System. Genes Dev. 2008, 22, 1972–1986. [Google Scholar] [CrossRef] [PubMed]
- Hung, A.Y.; Sung, C.C.; Brito, I.L.; Sheng, M. Degradation of Postsynaptic Scaffold GKAP and Regulation of Dendritic Spine Morphology by the TRIM3 Ubiquitin Ligase in Rat Hippocampal Neurons. PLoS ONE 2010, 5, e9842. [Google Scholar] [CrossRef]
- Choe, S.; Huh, T.L.; Rhee, M. Trim45 Is Essential to the Development of the Diencephalon and Eye in Zebrafish Embryos. Anim. Cells Syst. 2020, 24, 99–106. [Google Scholar] [CrossRef]
- Boyer, N.P.; Monkiewicz, C.; Menon, S.; Moy, S.S.; Gupton, S.L. Mammalian TRIM67 Functions in Brain Development and Behavior. eNeuro 2018, 5, 59. [Google Scholar] [CrossRef]
- Boyer, N.P.; McCormick, L.E.; Menon, S.; Urbina, F.L.; Gupton, S.L. A Pair of E3 Ubiquitin Ligases Compete to Regulate Filopodial Dynamics and Axon Guidance. J. Cell Biol. 2020, 219, e201902088. [Google Scholar]
- Menon, S.; Goldfarb, D.; Ho, C.T.; Cloer, E.W.; Boyer, N.P.; Hardie, C.; Bock, A.J.; Johnson, E.C.; Anil, J.; Major, M.B.; et al. The TRIM9/TRIM67 Neuronal Interactome Reveals Novel Activators of Morphogenesis. Mol. Biol. Cell 2021, 32, 314–330. [Google Scholar] [CrossRef]
- Zeng, J.; Wang, Y.; Luo, Z.; Chang, L.C.; Yoo, J.S.; Yan, H.; Choi, Y.; Xie, X.; Deverman, B.E.; Gradinaru, V.; et al. TRIM9-Mediated Resolution of Neuroinflammation Confers Neuroprotection upon Ischemic Stroke in Mice. Cell Rep. 2019, 27, 549–560.e6. [Google Scholar] [CrossRef]
- Wei, L.; Zhang, J.-S.; Ji, S.-F.; Xu, H.; Zhao, Z.-H.; Zhang, L.; Pang, L.; Zhang, J.-F.; Yang, P.-B.; Ma, H. Knockdown of TRIM32 Protects Hippocampal Neurons from Oxygen–Glucose Deprivation-Induced Injury. Neurochem. Res. 2019, 44, 2182–2189. [Google Scholar] [CrossRef]
- Zeng, S.; Zhao, Z.; Zheng, S.; Wu, M.; Song, X.; Li, Y.; Zheng, Y.; Liu, B.; Chen, L.; Gao, C.; et al. The E3 Ubiquitin Ligase TRIM31 Is Involved in Cerebral Ischemic Injury by Promoting Degradation of TIGAR. Redox Biol. 2021, 45, 102058. [Google Scholar] [CrossRef]
- Xia, Q.; Zhan, G.; Mao, M.; Zhao, Y.; Li, X. TRIM45 Causes Neuronal Damage by Aggravating Microglia-Mediated Neuroinflammation upon Cerebral Ischemia and Reperfusion Injury. Exp. Mol. Med. 2022, 54, 180–193. [Google Scholar] [CrossRef]
- Qian, Y.; Lei, G.; Wen, L. Brain-Specific Deletion of TRIM13 Promotes Metabolic Stress-Triggered Insulin Resistance, Glucose Intolerance, and Neuroinflammation. Biochem. Biophys. Res. Commun. 2020, 527, 138–145. [Google Scholar] [CrossRef]
- Niikura, T.; Hashimoto, Y.; Tajima, H.; Ishizaka, M.; Yamagishi, Y.; Kawasumi, M.; Nawa, M.; Terashita, K.; Also, S.; Nishimoto, I. A Tripartite Motif Protein TRIM11 Binds and Destabilizes Humanin, a Neuroprotective Peptide against Alzheimer’s Disease-Relevant Insults. Eur. J. Neurosci. 2003, 17, 1150–1158. [Google Scholar] [CrossRef]
- Ding, C.; Zhang, C.; Kopp, R.; Kuney, L.; Meng, Q.; Wang, L.; Xia, Y.; Jiang, Y.; Dai, R.; Min, S.; et al. Transcription Factor POU3F2 Regulates TRIM8 Expression Contributing to Cellular Functions Implicated in Schizophrenia. Mol. Psychiatry 2021, 26, 3444–3460. [Google Scholar] [CrossRef]
- Assoum, M.; Lines, M.A.; Elpeleg, O.; Darmency, V.; Whiting, S.; Edvardson, S.; Devinsky, O.; Heinzen, E.; Hernan, R.R.; Antignac, C.; et al. Further Delineation of the Clinical Spectrum of de Novo TRIM8 Truncating Mutations. Am. J. Med. Genet. Part A 2018, 176, 2470–2478. [Google Scholar] [CrossRef]
- Sakai, Y.; Fukai, R.; Matsushita, Y.; Miyake, N.; Saitsu, H.; Akamine, S.; Torio, M.; Sasazuki, M.; Ishizaki, Y.; Sanefuji, M.; et al. De Novo Truncating Mutation of TRIM8 Causes Early-Onset Epileptic Encephalopathy. Ann. Hum. Genet. 2016, 80, 235–240. [Google Scholar] [CrossRef]
- Dong, W.; Luo, B.; Qiu, C.; Jiang, X.; Shen, B.; Zhang, L.; Liu, W.; Zhang, W. TRIM3 Attenuates Apoptosis in Parkinson’s Disease via Activating PI3K/AKT Signal Pathway. Aging 2020, 13, 735–749. [Google Scholar] [CrossRef]
- Lazzari, E.; Meroni, G. TRIM32 Ubiquitin E3 Ligase, One Enzyme for Several Pathologies: From Muscular Dystrophy to Tumours. Int. J. Biochem. Cell Biol. 2016, 79, 469–477. [Google Scholar] [CrossRef]
- Kumarasinghe, L.; Xiong, L.; Garcia-gimeno, M.A.; Lazzari, E.; Sanz, P.; Meroni, G. TRIM32 and Malin in Neurological and Neuromuscular Rare Diseases. Cells 2021, 10, 820. [Google Scholar] [CrossRef]
- Tang, S.-L.; Gao, Y.-L.; Wen-Zhong, H. Knockdown of TRIM37 Suppresses the Proliferation, Migration and Invasion of Glioma Cells through the Inactivation of PI3K/Akt Signaling Pathway. Biomed. Pharmacother. 2018, 99, 59–64. [Google Scholar] [CrossRef]
- Porčnik, A.; Novak, M.; Breznik, B.; Majc, B.; Hrastar, B.; Šamec, N.; Zottel, A.; Jovčevska, I.; Vittori, M.; Rotter, A.; et al. Trim28 Selective Nanobody Reduces Glioblastoma Stem Cell Invasion. Molecules 2021, 26, 5141. [Google Scholar] [CrossRef]
- Lv, D.; Li, Y.; Zhang, W.; Alvarez, A.A.; Song, L.; Tang, J.; Gao, W.Q.; Hu, B.; Cheng, S.Y.; Feng, H. TRIM24 Is an Oncogenic Transcriptional Co-Activator of STAT3 in Glioblastoma. Nat. Commun. 2017, 8, 1454. [Google Scholar] [CrossRef]
- Zhang, L.H.; Yin, A.A.; Cheng, J.X.; Huang, H.Y.; Li, X.M.; Zhang, Y.Q.; Han, N.; Zhang, X. TRIM24 Promotes Glioma Progression and Enhances Chemoresistance through Activation of the PI3K/Akt Signaling Pathway. Oncogene 2015, 34, 600–610. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, C.; Cui, J.; Ou, J.; Han, J.; Qin, Y.; Zhi, F.; Wang, R.F. Trim45 Functions as a Tumor Suppressor in the Brain via Its E3 Ligase Activity by Stabilizing P53 through K63-Linked Ubiquitination. Cell Death Dis. 2017, 8, e2831. [Google Scholar] [CrossRef]
- Di, K.; Linskey, M.E.; Bota, D.A. TRIM11 Is Overexpressed in High-Grade Gliomas and Promotes Proliferation, Invasion, Migration and Glial Tumor Growth. Oncogene 2013, 32, 5038–5047. [Google Scholar] [CrossRef]
- Xiao, S.; Yu, J.; Yuan, X.; Chen, Q. Identification of a Tripartite Motif Family Gene Signature for Predicting the Prognosis of Patients with Glioma. Am. J. Transl. Res. 2022, 14, 1535–1550. [Google Scholar]
- Zhao, Z.; Wang, Y.; Yun, D.; Huang, Q.; Meng, D.; Li, Q.; Zhang, P.; Wang, C.; Chen, H.; Lu, D. Original Article TRIM21 Overexpression Promotes Tumor Progression by Regulating Cell Proliferation, Cell Migration and Cell Senescence in Human Glioma. Am. J. Cancer Res. 2020, 10, 114–130. [Google Scholar]
- Ji, J.; Ding, K.; Luo, T.; Zhang, X.; Chen, A.; Zhang, D.; Li, G.; Thorsen, F.; Huang, B.; Li, X.; et al. TRIM22 Activates NF-ΚB Signaling in Glioblastoma by Accelerating the Degradation of IκBα. Cell Death Differ. 2021, 28, 367–381. [Google Scholar] [CrossRef]
- Chen, L.; Li, M.; Li, Q.; Xu, M.; Zhong, W. Knockdown of TRIM47 Inhibits Glioma Cell Proliferation, Migration and Invasion through the Inactivation of Wnt/β-Catenin Pathway. Mol. Cell. Probes 2020, 53, 101623. [Google Scholar] [CrossRef]
- Ji, B.; Liu, L.; Guo, Y.; Ming, F.; Jiang, J.; Li, F.; Zhao, G.; Wen, J.; Li, N. Upregulated Tripartite Motif 47 Could Facilitate Glioma Cell Proliferation and Metastasis as a Tumorigenesis Promoter. Comput. Math. Methods Med. 2021, 2021, 5594973. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Liu, N.; Wang, H.; Wang, Y.; Guo, Z. LncRNA LINC01857 Promotes Growth, Migration, and Invasion of Glioma by Modulating MiR-1281/TRIM65 Axis. J. Cell. Physiol. 2019, 234, 22009–22016. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Mukherjee, S.; Tucker-Burden, C.; Ross, J.L.; Chau, M.J.; Kong, J.; Brat, D.J. TRIM8 Regulates Stemness in Glioblastoma through PIAS3-STAT3. Mol. Oncol. 2017, 11, 280–294. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Deng, Z.Z.; Li, H.Y.; Jiang, N.; Wei, Z.S.; Hong, M.F.; Chen, X.D.; Wang, J.H.; Zhang, M.X.; Shi, Y.H.; et al. TRIM31 Promotes Glioma Proliferation and Invasion through Activating NF-ΚB Pathway. Onco. Targets. Ther. 2019, 12, 2289–2297. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Lv, C.; Yang, Z.; Qin, T.; Sun, L.; Pan, P.; Wang, D. TRIM31 Promotes Proliferation, Invasion and Migration of Glioma Cells through Akt Signaling Pathway. Neoplasma 2019, 66, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.H.; Yin, Y.H.; Chen, H.Z.; Feng, S.Y.; Liu, J.L.; Chen, L.; Fu, W.L.; Sun, G.C.; Yu, X.G.; Xu, D.G. TRIM24 Promotes Stemness and Invasiveness of Glioblastoma Cells via Activating Sox2 Expression. Neuro. Oncol. 2020, 22, 1797–1808. [Google Scholar] [CrossRef]
- Berghoff, A.S.; Kiesel, B.; Widhalm, G.; Rajky, O.; Ricken, G.; Wohrer, A.; Dieckmann, K.; Filipits, M.; Brandstetter, A.; Weller, M.; et al. Programmed Death Ligand 1 Expression and Tumor-Infiltrating Lymphocytes in Glioblastoma. Neuro Oncology 2015, 17, 1064–1075. [Google Scholar] [CrossRef]
- Han, M.; Sun, Y. Pharmacological Targeting of Tripartite Motif Containing 24 for the Treatment of Glioblastoma. J. Transl. Med. 2021, 19, 505. [Google Scholar] [CrossRef]
- Golding, S.E.; Rosenberg, E.; Adams, B.R.; Wignarajah, S.; Beckta, J.M.; O’Connor, M.J.; Valerie, K. Dynamic Inhibition of ATM Kinase Provides a Strategy for Glioblastoma Multiforme Radiosensitization and Growth Control. Cell Cycle 2012, 11, 1167–1173. [Google Scholar] [CrossRef]
- Goodarzi, A.A.; Kurka, T.; Jeggo, P.A. KAP-1 Phosphorylation Regulates CHD3 Nucleosome Remodeling during the DNA Double-Strand Break Response. Nat. Struct. Mol. Biol. 2011, 18, 831–839. [Google Scholar] [CrossRef]
- Gupta, S.K.; Kizilbash, S.H.; Carlson, B.L.; Mladek, A.C.; Boakye-Agyeman, F.; Bakken, K.K.; Pokorny, J.L.; Schroeder, M.A.; Decker, P.A.; Cen, L.; et al. Delineation of MGMT Hypermethylation as a Biomarker for Veliparib-Mediated Temozolomide-Sensitizing Therapy of Glioblastoma. J. Natl. Cancer Inst. 2015, 108, djv369. [Google Scholar] [CrossRef]
- Jovčevska, I.; Zupanec, N.; Urlep, Ž.; Vranic, A.; Matos, B.; Stokin, C.L.; Muyldermans, S.; Myers, M.P.; Buzdin, A.A.; Petrov, I.; et al. Differentially Expressed Proteins in Glioblastoma Multiforme Identified with a Nanobody-Based Anti-Proteome Approach and Confirmed by OncoFinder as Possible Tumor-Class Predictive Biomarker Candidates. Oncotarget 2017, 8, 44141–44158. [Google Scholar] [CrossRef]
- Qi, Z.X.; Cai, J.J.; Chen, L.C.; Yue, Q.; Gong, Y.; Yao, Y.; Mao, Y. TRIM28 as an Independent Prognostic Marker Plays Critical Roles in Glioma Progression. J. Neurooncol. 2016, 126, 19–26. [Google Scholar] [CrossRef]
- Gursoy-Yuzugullu, O.; Carman, C.; Serafim, R.B.; Myronakis, M.; Valente, V.; Price, B.D.; Gursoy-Yuzugullu, O.; Carman, C.; Serafim, R.B.; Myronakis, M.; et al. Epigenetic Therapy with Inhibitors of Histone Methylation Suppresses DNA Damage Signaling and Increases Glioma Cell Radiosensitivity. Oncotarget 2017, 8, 24518–24532. [Google Scholar] [CrossRef]
- Spyropoulou, A.; Gargalionis, A.; Dalagiorgou, G.; Adamopoulos, C.; Papavassiliou, K.A.; Lea, R.W.; Piperi, C.; Papavassiliou, A.G. Role of Histone Lysine Methyltransferases SUV39H1 and SETDB1 in Gliomagenesis: Modulation of Cell Proliferation, Migration, and Colony Formation. Neuromol. Med. 2014, 16, 70–82. [Google Scholar] [CrossRef]
- Sepsa, A.; Levidou, G.; Gargalionis, A.; Adamopoulos, C.; Spyropoulou, A.; Dalagiorgou, G.; Thymara, I.; Boviatsis, E.; Themistocleous, M.S.; Petraki, K.; et al. Emerging Role of Linker Histone Variant H1x as a Biomarker with Prognostic Value in Astrocytic Gliomas. A Multivariate Analysis Including Trimethylation of H3K9 and H4K20. PLoS ONE 2015, 10, e0115101. [Google Scholar] [CrossRef]
- Wang, C.; Songyang, Z.; Huang, Y. TRIM28 Inhibits Alternative Lengthening of Telomere Phenotypes by Protecting SETDB1 from Degradation. Cell Biosci. 2021, 11, 149. [Google Scholar] [CrossRef]
- Yu, Z.; Feng, J.; Wang, W.; Deng, Z.; Zhang, Y.; Xiao, L.; Wang, Z.; Liu, C.; Liu, Q.; Chen, S.; et al. The EGFR-ZNF263 Signaling Axis Silences SIX3 in Glioblastoma Epigenetically. Oncogene 2020, 39, 3163–3178. [Google Scholar] [CrossRef]
- Zhang, B.; Shen, C.; Ge, F.; Ma, T.; Zhang, Z. Epigenetically Controlled Six3 Expression Regulates Glioblastoma Cell Proliferation and Invasion alongside Modulating the Activation Levels of WNT Pathway Members. J. Neurooncol. 2017, 133, 509–518. [Google Scholar] [CrossRef]
- Bunina, D.; Abazova, N.; Diaz, N.; Noh, K.M.; Krijgsveld, J.; Zaugg, J.B. Genomic Rewiring of SOX2 Chromatin Interaction Network during Differentiation of ESCs to Postmitotic Neurons. Cell Syst. 2020, 10, 480. [Google Scholar] [CrossRef]
- Ma, T.; Hu, C.; Lal, B.; Zhou, W.; Ma, Y.; Ying, M.; Prinos, P.; Quiñones-Hinojosa, A.; Lim, M.; Laterra, J.; et al. Reprogramming Transcription Factors Oct4 and Sox2 Induces a BRD-Dependent Immunosuppressive Transcriptome in GBM-Propagating Cells. Cancer Res. 2021, 81, 2457. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.J.; Ren, J.; Jiang, H.; Liu, W.; Hu, L.Y.; Pan, Y.X.; Sun, B.; Sun, Q.F.; Bian, L.G. MAGEA6 Promotes Human Glioma Cell Survival via Targeting AMPKα1. Cancer Lett. 2018, 412, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Zhong, S.; Sun, B.M.; Sun, Q.F.; Hu, L.Y.; Pan, S.J. Lnc-THOR Silencing Inhibits Human Glioma Cell Survival by Activating MAGEA6-AMPK Signaling. Cell Death Dis. 2019, 10, 866. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.J.; Li, J.W.; Bao, B.H.; Wu, H.C.; Du, Z.H.; Su, J.L.; Zhang, M.H.; Liang, H.Q. MicroRNA-873 (MiRNA-873) Inhibits Glioblastoma Tumorigenesis and Metastasis by Suppressing the Expression of IGF2BP1. J. Biol. Chem. 2015, 290, 8938–8948. [Google Scholar] [CrossRef] [PubMed]
- Zhan, W.L.; Gao, N.; Tu, G.L.; Tang, H.; Gao, L.; Xia, Y. LncRNA LINC00689 Promotes the Tumorigenesis of Glioma via Mediation of MiR-526b-3p/IGF2BP1 Axis. NeuroMolecular Med. 2021, 23, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Scarcella, D.L.; Chow, C.W.; Gonzales, M.F.; Economou, C.; Brasseur, F.; Ashley, D.M. Expression of MAGE and GAGE in High-Grade Brain Tumors: A Potential Target for Specific Immunotherapy and Diagnostic Markers. Clin. Cancer Res. 1999, 5, 335–341. [Google Scholar] [PubMed]
- Shi, Y.; Lim, S.K.; Liang, Q.; Iyer, S.V.; Wang, H.Y.; Wang, Z.; Xie, X.; Sun, D.; Chen, Y.J.; Tabar, V.; et al. Gboxin Is an Oxidative Phosphorylation Inhibitor That Targets Glioblastoma. Nature 2019, 567, 341. [Google Scholar] [CrossRef]
- Xue, J.; Chen, Y.; Wu, Y.; Wang, Z.; Zhou, A.; Zhang, S.; Lin, K.; Aldape, K.; Majumder, S.; Lu, Z.; et al. Tumour Suppressor TRIM33 Targets Nuclear β-Catenin Degradation. Nat. Commun. 2015, 6, 6156. [Google Scholar] [CrossRef]
- Verma, B.K.; Kondaiah, P. Regulation of β-Catenin by IGFBP2 and Its Cytoplasmic Actions in Glioma. J. Neurooncol. 2020, 149, 209–217. [Google Scholar] [CrossRef]
- Peluffo, G.; Subedee, A.; Harper, N.W.; Kingston, N.; Jovanovic, B.; Flores, F.; Stevens, L.E.; Beca, F.; Trinh, A.; Chilamakuri, C.S.R.; et al. EN1 Is a Transcriptional Dependency in Triple-Negative Breast Cancer Associated with Brain Metastasis. Cancer Res. 2019, 79, 4173–4183. [Google Scholar] [CrossRef]
- Deng, M.Y.; Sill, M.; Chiang, J.; Schittenhelm, J.; Ebinger, M.; Schuhmann, M.U.; Monoranu, C.M.; Milde, T.; Wittmann, A.; Hartmann, C.; et al. Molecularly Defined Diffuse Leptomeningeal Glioneuronal Tumor (DLGNT) Comprises Two Subgroups with Distinct Clinical and Genetic Features. Acta Neuropathol. 2018, 136, 239–253. [Google Scholar] [CrossRef]
- Mukherjee, S.; Tucker-Burden, C.; Zhang, C.; Moberg, K.; Read, R.; Hadjipanayis, C.; Brat, D.J. Drosophila Brat and Human Ortholog TRIM3 Maintain Stem Cell Equilibrium and Suppress Brain Tumorigenesis by Attenuating Notch Nuclear Transport. Cancer Res. 2016, 76, 2443–2452. [Google Scholar] [CrossRef]
- Chen, G.; Kong, J.; Tucker-Burden, C.; Anand, M.; Rong, Y.; Rahman, F.; Moreno, C.S.; Van Meir, E.G.; Hadjipanayis, C.G.; Brat, D.J. Human Brat Ortholog TRIM3 Is a Tumor Suppressor That Regulates Asymmetric Cell Division in Glioblastoma. Cancer Res. 2014, 74, 4536–4548. [Google Scholar] [CrossRef]
- Boulay, J.L.; Stiefel, U.; Taylor, E.; Dolder, B.; Merlo, A.; Hirth, F. Loss of Heterozygosity of TRIM3 in Malignant Gliomas. BMC Cancer 2009, 9, 71. [Google Scholar] [CrossRef]
- Altinoz, M.A.; Elmaci, I.; Ince, B.; Ozpinar, A.; Sav, A.M. Hemoglobins, Hemorphins, and 11p15.5 Chromosomal Region in Cancer Biology and İmmunity with Special Emphasis for Brain Tumors. J. Neurol. Surg. A Cent. Eur. Neurosurg. 2016, 77, 247–257. [Google Scholar] [CrossRef]
- Cai, Y.; Gu, W.T.; Cheng, K.; Jia, P.F.; Li, F.; Wang, M.; Zhang, W.F.; Qiu, J.T.; Wu, Z.B.; Zhao, W.G. Knockdown of TRIM32 Inhibits Tumor Growth and Increases the Therapeutic Sensitivity to Temozolomide in Glioma in a P53-Dependent and -Independent Manner. Biochem. Biophys. Res. Commun. 2021, 550, 134–141. [Google Scholar] [CrossRef]
- Feng, S.; Cai, X.; Li, Y.; Jian, X.; Zhang, L.; Li, B. Tripartite Motif-Containing 14 (TRIM14) Promotes Epithelial-Mesenchymal Transition via ZEB2 in Glioblastoma Cells. J. Exp. Clin. Cancer Res. 2019, 38, 57. [Google Scholar] [CrossRef]
- Deng, Y.; Zhu, H.; Xiao, L.; Liu, C.; Meng, X. Circ_0005198 Enhances Temozolomide Resistance of Glioma Cells through MiR-198/TRIM14 Axis. Aging 2020, 13, 2198–2211. [Google Scholar] [CrossRef]
- Li, L.; Shao, M.Y.; Zou, S.C.; Xiao, Z.F.; Chen, Z.C. MiR-101-3p Inhibits EMT to Attenuate Proliferation and Metastasis in Glioblastoma by Targeting TRIM44. J. Neurooncol. 2019, 141, 19–30. [Google Scholar] [CrossRef]
- Zhou, X.; Yang, Y.; Ma, P.; Wang, N.; Yang, D.; Tu, Q.; Sun, B.; Xiang, T.; Zhao, X.; Hou, Z.; et al. TRIM44 Is Indispensable for Glioma Cell Proliferation and Cell Cycle Progression through AKT/P21/P27 Signaling Pathway. J. Neurooncol. 2019, 145, 211–222. [Google Scholar] [CrossRef]
- Song, Y.; Meng, L.; Yu, J.; Cao, Z.; Sun, J.; Zhao, H. TRIM66 Overexpression Promotes Glioma Progression and Regulates Glucose Uptake Through CMyc/GLUT3 Signaling. Cancer Manag. Res. 2021, 13, 5187–5201. [Google Scholar] [CrossRef]
- Micale, L.; Fusco, C.; Fontana, A.; Barbano, R.; Augello, B.; De Nittis, P.; Copetti, M.; Pellico, M.T.; Mandriani, B.; Cocciadiferro, D.; et al. TRIM8 Downregulation in Glioma Affects Cell Proliferation and It Is Associated with Patients Survival. BMC Cancer 2015, 15, 470. [Google Scholar] [CrossRef]
- Fan, M.; Zhao, X.; Qi, J.; Jiang, Y.; Liu, B.; Dun, Z.; Zhang, R.; Wang, C.; Pang, Q. TRIM31 Enhances Chemoresistance in Glioblastoma through Activation of the PI3K/Akt Signaling Pathway. Exp. Ther. Med. 2020, 20, 802–809. [Google Scholar] [CrossRef]
- Qi, Z.; Cai, S.; Cai, J.; Chen, L.; Yao, Y.; Chen, L.; Mao, Y. MiR-491 Regulates Glioma Cells Proliferation by Targeting TRIM28 in Vitro. BMC Neurol. 2016, 16, 248. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giannopoulou, A.-I.; Xanthopoulos, C.; Piperi, C.; Kostareli, E. Emerging Roles of TRIM Family Proteins in Gliomas Pathogenesis. Cancers 2022, 14, 4536. https://doi.org/10.3390/cancers14184536
Giannopoulou A-I, Xanthopoulos C, Piperi C, Kostareli E. Emerging Roles of TRIM Family Proteins in Gliomas Pathogenesis. Cancers. 2022; 14(18):4536. https://doi.org/10.3390/cancers14184536
Chicago/Turabian StyleGiannopoulou, Angeliki-Ioanna, Charalampos Xanthopoulos, Christina Piperi, and Efterpi Kostareli. 2022. "Emerging Roles of TRIM Family Proteins in Gliomas Pathogenesis" Cancers 14, no. 18: 4536. https://doi.org/10.3390/cancers14184536
APA StyleGiannopoulou, A.-I., Xanthopoulos, C., Piperi, C., & Kostareli, E. (2022). Emerging Roles of TRIM Family Proteins in Gliomas Pathogenesis. Cancers, 14(18), 4536. https://doi.org/10.3390/cancers14184536