
EZH2 Inhibition and Cisplatin as a Combination Anticancer Therapy: An Overview of Preclinical Studies



Abstract
:Simple Summary
Abstract
1. Introduction
2. Cisplatin in Anticancer Therapies
Resistance to Cisplatin
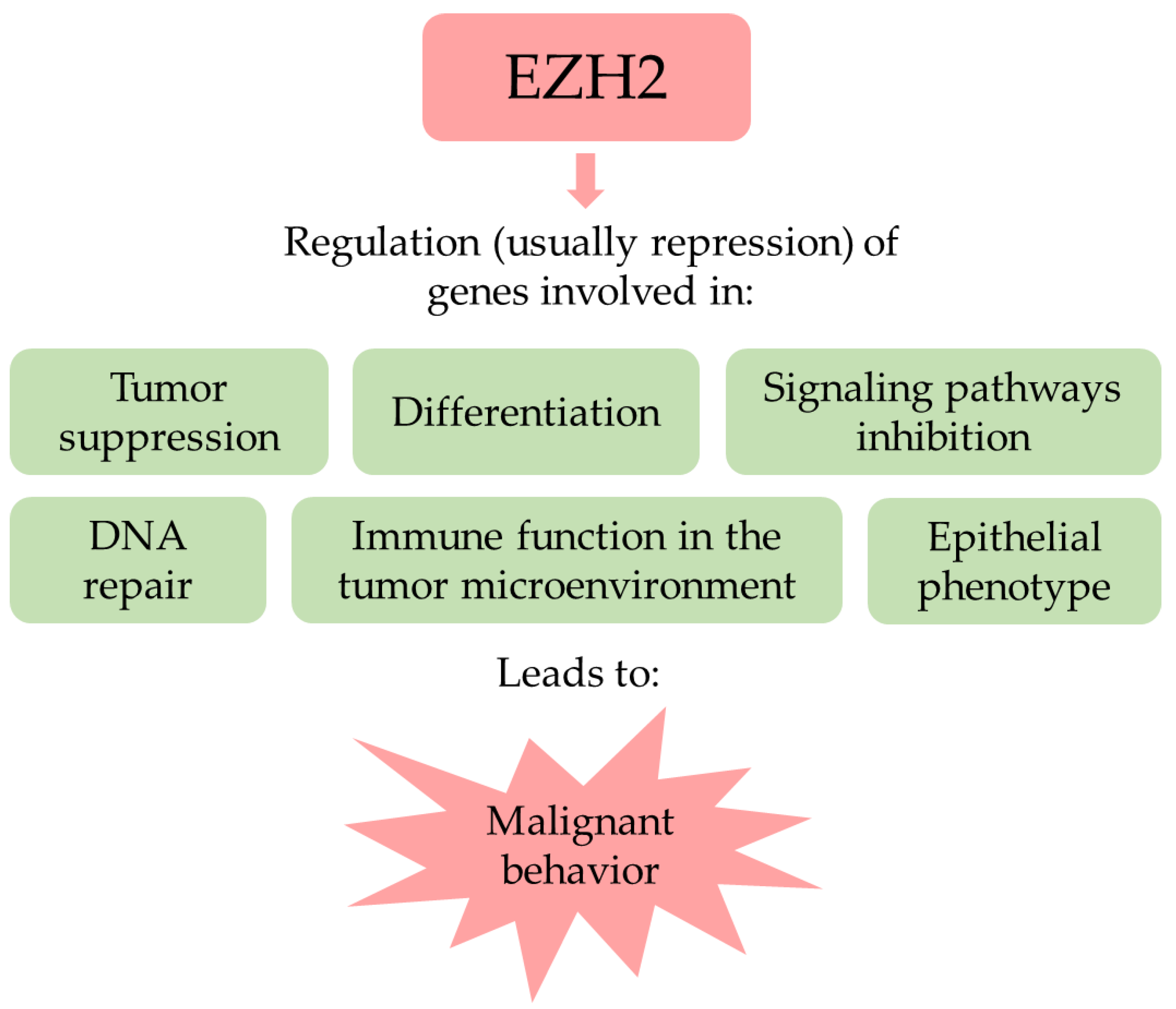
3. The Polycomb Repressive Complex 2 Related and Unrelated Roles of EZH2
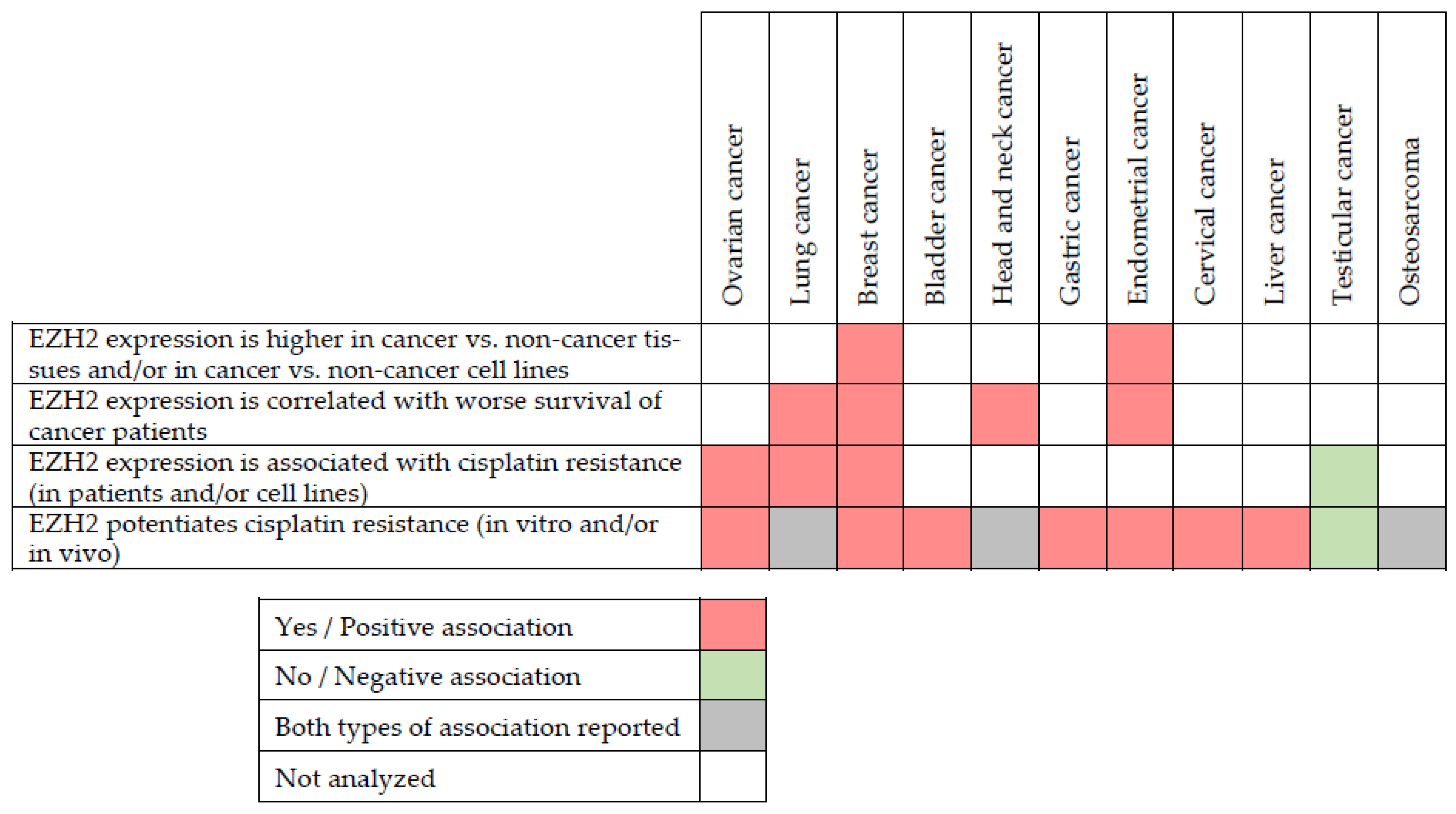
4. EZH2 in Cancer
5. EZH2 Inhibitors
6. Combined Effects of EZH2 Inhibition and Cisplatin in Anticancer Therapies

{kind=link}
{kind=link}
| Cancer Type | In Vitro Model | In Vivo Model | Cisplatin Concentration | EZH2 Silencing/ Inhibition | Possible Mechanism of Action | Effect of Combined Treatment | Ref. |
|---|---|---|---|---|---|---|---|
| Bladder cancer | Bladder cancer cell lines: HT1376, T24, and UM-UC-3 | HT1376 cell line xenograft in nude mice | In vitro: 0.83 µM. In vivo: 3 mg/kg, intraperitoneally (i.p.), once per week | In vitro: EPZ011989 (1 µM). In vivo: EPZ011989 (500 mg/kg, oral gavage, every 12 h) | Inhibition of EZH2 induces natural killer cell-mediated differentiation and death in HT1376-derived xenografts. | Additive/synergistic: In vitro: Combined application of EPZ011989 and cisplatin caused G2/M arrest and reduced clonogenicity of T24 and UM-UC-3 cell lines. In vivo: Combined application reduced xenograft growth. | [74] |
| Bladder cancer cell line: T24 | / | 2 μM | EZH2 siRNA | / | Additive/synergistic: EZH2 knockdown increased cisplatin cytotoxicity, while EZH2 overexpression reduced it. | [75] | |
| Breast cancer | Breast cancer cell lines: MCF-7 and MDA-MB-231 | MCF-7 xenograft in BALB/c nude mice | In vitro: 0.1–100 µM, mostly 10 µM. In vivo: i.p., 5 mg/kg, weekly | EZH2 siRNA | EZH2 knockdown increased expression of miR-381. | Additive/synergistic: In vitro: EZH2 knockdown sensitizes cells to cisplatin. In vivo: EZH2 knockdown sensitizes cells to cisplatin. | [76] |
| Breast cancer cell lines: from BRCA1-deficient and BRCA1-proficient mice | BRCA1-deficient tumors in FvB/Ola mice | In vitro: 0.5 µM. In vivo: 3 mg/kg. | In vitro: GSK126 (8 µM). In vivo: GSK126 150 mg/kg daily | / | Additive/synergistic: In vitro: GSK126 increased cisplatin-induced growth inhibition only in BRCA1-deficient cells. In vivo: Combined application of GSK126 and cisplatin increased overall survival. | [77] | |
| Cervical cancer | Cervical cancer cell line: HeLa | / | Range of concentrations (0–3333 µM) | EZH2 shRNA | / | Additive/synergistic: EZH2 knockdown reduced resistance to cisplatin. | [78] |
| Endometrial cancer | Endometrial cancer cell lines: Ishikawa, HEC1A, and KLE | / | Range of concentrations (0.1–100 µM) | EZH2 siRNA | EZH2 knockdown decreased the level of Peroxiredoxin 6 (PRDX6) protein. | Additive/synergistic: EZH2 knockdown sensitizes cell lines with higher EZH2 levels (Ishikawa and HEC1A) to cisplatin, but not cell line KLE which has lower levels of EZH2. | [79] |
| Endometrial Cancer cell line: HEC1B | / | 1 μM | GSK126 (7.5 µM) | / | Additive/synergistic: GSK126 increased cytotoxic effect of cisplatin. | [80] | |
| Gastric cancer | Gastric cancer cell lines: MKN45 and MGC803 | / | Range of concentrations (5–25 µM) | EZH2 siRNA | EZH2 inhibition reduced the activity of PI3K/AKT pathway. | Additive/synergistic: EZH2 knockdown enhances cisplatin-induced apoptosis. | [81] |
| Head and neck cancer | Head and neck squamous cell carcinoma cell lines: FaDu andSNU1041 | / | Range of concentrations (5–50 µM) | EZH2 siRNA | EZH2 knockdown reduced N-cadherin and vimentin and increased E-cadherin expression. | Additive/synergistic: EZH2 knockdown increased cytotoxic effect of cisplatin. | [82] |
| Head and neck cancer cell line: SCC-11 | / | 33.3 µM | EZH2 siRNA | / | Additive/synergistic: EZH2 knockdown increased cancer cells’ sensitivity to cisplatin. | [83] | |
| Head and neck cancer cell lines: CNE and 8F cells | / | Range of concentrations (0–64 µM) | GSK126 (1 µM) | EZH2 suppresses the nucleotide excision repair by silencing XPA. | Antagonistic: GSK126 doubled the resistance to cisplatin. | [84] | |
| Liver cancer | Liver cancer cell lines: HepG2 and SNU449 | / | Range of concentrations (0–20 µM) | EZH2 siRNA | miR138 targets the EZH2/EMT axis. | Additive/synergistic: EZH2 knockdown enhanced sensitivity to cisplatin. | [85] |
| Lung cancer | Lung cancer cell line: A549 | / | Range of concentrations (2–16 µM), mostly 4 µM. | EZH2 siRNA, Tazemetostat (40 µM) | Knockdown of EZH2 increased levels of cleaved caspase 3 and 9, E-cadherin and reduced expression of N-cadherin and vimentin. | Additive/synergistic: EZH2 knockdown or inhibition with tazemetostat increased cisplatin cytotoxicity. | [86] |
| / | Range of concentrations (20–140 µM) | shRNA | EZH2 knockdown reduced MRP1 mRNA levels. | Additive/synergistic: EZH2 knockdown increased cancer cells’ sensitivity to cisplatin. | [87] | ||
| Lung cancer cell lines: H128 and H146 | H128 cells xenograft in Nu/Nu mice | In vitro: Range of concentrations (5–25 µM). In vivo: 2.5 mg/kg; 2 times weekly, for 4 weeks. | In vitro: EZH2 siRNA, DZNep (2.5–5 µM), Tazemetostat (1 µM). In vivo: DZNep (2.5 mg/kg; 2 times weekly) | EZH2 interacts with and stabilizes components of nucleotide excision repair (DDB2). | Additive/synergistic: In vitro: siRNA and DZNep caused sensitization of cancer cells to cisplatin. No significant effect caused by combining cisplatin and tazemetostat. In vivo: Reduced tumor growth in mice treated with DZNep and cisplatin compared to individual agents. | [31] | |
| Lung cancer cell lines: H1299, H23, and H460 | / | 1–2 µM | EZH2 shRNA | EZH2 silencing upregulates PUMA, a proapoptotic protein. | Additive/synergistic: EZH2 knockdown increased cytotoxic effect of cisplatin. | [88] | |
| Lung cancer cell lines: A549, HCC4006, and H2073 | / | Range of concentrations (0–80 µM). | DZNep (2.5 µM) EZH2 siRNA | / | Additive/synergistic: DZNep and EZH2 knockdown sensitized cells to cisplatin. | [89] | |
| Lung cancer cell line: H1299 | / | 66.6 µM | DZNep (range 0–10 μM) | / | Additive/synergistic: DZNep increased cisplatin cytotoxicity in H1299 cell line. | [90] | |
| Osteosarcoma | Osteosarcoma cell lines: HOS and 143B | / | Range of concentrations (0.2–1.6 µM), but mostly 1 µM | Tazemetostat (10 µM) | Reductions in H3K27me3 levels induce PRKCA and MCL1 expression, BCL2 phosphorylation and activation of RAF/ERK/MAPK cascades. | Antagonistic: Tazemetostat increased resistance to cisplatin, leading to lower rates of apoptosis. | [91] |
| Ovarian cancer | Ovarian cancer cell line: HeyA8 | / | 1.25 µM | EZH2 siRNA | Reduced β-catenin levels after EZH2 silencing. | Additive/synergistic: EZH2 knockdown decreased proliferative capacity of cells exposed to cisplatin. | [92] |
| Ovarian cancer cell lines: IGROV1, PEO1, and PEO4 | / | Range of concentrations (0–80 µM) | miR-137, GSK343 (0–25 µM), EZH2 siRNA | miR-137 mediates the functional link between c-Myc and EZH2 that regulates cisplatin resistance. | Additive/synergistic: siRNA-EZH2 and GSK343 sensitized resistant cell lines to cisplatin. EZH2-depleted cells treated with cisplatin showed an increase in cell apoptosis (elevated level of cleaved PARP). | [93] | |
| Ovarian cancer cell line: SKOV3 | / | 25.53 µM | EZH2 shRNA | EZH2 silencing upregulated p14, p16, p53, and pRB. | Additive/synergistic: EZH2 knockdown increased cytotoxic effect of cisplatin. | [94] | |
| Ovarian cancer cell lines: A2780 and ES2 | / | 2, 4, or 8 µM | EZH2 shRNA | Reduced EZH2 levels increased cisplatin intake. High EZH2 levels promoted degradation of CTR1. | Additive/synergistic: shRNA sensitized cells to cisplatin. Overexpression of EZH2 leads to increased resistance to cisplatin. | [95] | |
| Ovarian cancer cell line: A2780 | A2780 xenograft in female BALB/c nude mice | In vitro: range of concentrations, mostly 10 µM. In vivo: 2 mg/kg tail vein injection | EZH2 siRNA (bound to Fe3O4 particles with cisplatin prodrug) | / | Additive/synergistic: In vitro: EZH2 knock-down increased sensitivity to cisplatin. In vivo: siRNA-EZH2 coated nanoparticles and cisplatin inhibited tumor growth more than nanoparticles with control siRNA and cisplatin. | [61] | |
| In vitro: Range of concentrations (10–120 µM). In vivo: 6 mg/kg, i.p., once, on day 0 | EZH2 shRNA | / | Additive/synergistic: In vitro: EZH2 knock-down increased sensitivity to cisplatin. In vivo: EZH2 knockdown combined with cisplatin led to greater reduction of tumor growth than cisplatin alone. | [96] | |||
| / | Range of concentrations (2–165 µM) | EZH2 shRNA | Depletion of EZH2 in cisplatin-resistant cells reduced BRCA1 expression. | Additive/synergistic: EZH2 knockdown increased cancer cells’ sensitivity to cisplatin. | [97] | ||
| Testicular cancer | Testicular cancer cell lines: NT2/D1, 2102EP, and 833K | / | Range of concentrations (0.1–10 µM) | GSK126 (1µM) | / | Antagonistic: Inhibition of EZH2 with GSK126 increased cancer cell resistance to cisplatin. | [98] |
6.1. EZH2 as a Potentiator of Cisplatin Resistance
6.1.1. Ovarian Cancer
6.1.2. Lung Cancer
6.1.3. Breast Cancer
6.1.4. Muscle-Invasive Bladder Cancer
6.1.5. Head and Neck Squamous Cell Carcinoma
6.1.6. Gastric Cancer
6.1.7. Endometrial Cancer
6.1.8. Other Cancer Types
6.2. EZH2 as a Negative Regulator of Cisplatin Resistance
7. EZH2 Inhibition and Cisplatin Toxicity
8. Combined Effects of EZH2 Inhibition and Other Platinum Compounds in Anticancer Therapies
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yip, H.Y.K.; Papa, A. Signaling Pathways in Cancer: Therapeutic Targets, Combinatorial Treatments, and New Developments. Cells 2021, 10, 659. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, D.M.; Gellhorn, A. Combinations of Chemical Compounds in Experimental Cancer Therapy. Cancer Res. 1951, 11, 35–41. [Google Scholar] [PubMed]
- Bhatia, K.; Bhumika; Das, A. Combinatorial Drug Therapy in Cancer—New Insights. Life Sci. 2020, 258, 118134. [Google Scholar] [CrossRef] [PubMed]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination Therapy in Combating Cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef]
- Al-Lazikani, B.; Banerji, U.; Workman, P. Combinatorial Drug Therapy for Cancer in the Post-Genomic Era. Nat. Biotechnol. 2012, 30, 679–692. [Google Scholar] [CrossRef]
- Sicklick, J.K.; Kato, S.; Okamura, R.; Schwaederle, M.; Hahn, M.E.; Williams, C.B.; De, P.; Krie, A.; Piccioni, D.E.; Miller, V.A.; et al. Molecular Profiling of Cancer Patients Enables Personalized Combination Therapy: The I-PREDICT Study. Nat. Med. 2019, 25, 744–750. [Google Scholar] [CrossRef]
- Klauschen, F.; Andreeff, M.; Keilholz, U.; Dietel, M.; Stenzinger, A. The Combinatorial Complexity of Cancer Precision Medicine. Oncoscience 2014, 1, 504–509. [Google Scholar] [CrossRef]
- Cerrato, A.; Mattheolabakis, G.; Spano, D. Editorial: Combinatorial Approaches for Cancer Treatment: From Basic to Translational Research. Front. Oncol. 2022, 12, 122. [Google Scholar] [CrossRef]
- Dasari, S.; Bernard Tchounwou, P. Cisplatin in Cancer Therapy: Molecular Mechanisms of Action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Brown, A.; Kumar, S.; Tchounwou, P.B. Cisplatin-Based Chemotherapy of Human Cancers. J. Cancer Sci. 2019, 11, 97. [Google Scholar]
- Amable, L. Cisplatin Resistance and Opportunities for Precision Medicine. Pharm. Res. 2016, 106, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular Mechanisms of Cisplatin Resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Kang, Y.; Chen, L.; Wang, H.; Liu, J.; Zeng, S.; Yu, L. The Drug-Resistance Mechanisms of Five Platinum-Based Antitumor Agents. Front. Pharm. 2020, 11, 343. [Google Scholar] [CrossRef] [PubMed]
- Rottenberg, S.; Disler, C.; Perego, P. The Rediscovery of Platinum-Based Cancer Therapy. Nat. Rev. Cancer 2021, 21, 37–50. [Google Scholar] [CrossRef]
- Chen, S.H.; Chang, J.Y. New Insights into Mechanisms of Cisplatin Resistance: From Tumor Cell to Microenvironment. Int. J. Mol. Sci. 2019, 20, 4136. [Google Scholar] [CrossRef]
- Shen, D.W.; Pouliot, L.M.; Hall, M.D.; Gottesman, M.M. Cisplatin Resistance: A Cellular Self-Defense Mechanism Resulting from Multiple Epigenetic and Genetic Changes. Pharm. Rev. 2012, 64, 706–721. [Google Scholar] [CrossRef]
- Gray, S.G. Epigenetics of Cisplatin Resistance. In Epigenetic Cancer Therapy; Academic Press: Cambridge, MA, USA, 2015; pp. 613–637. [Google Scholar]
- Zeller, C.; Dai, W.; Steele, N.L.; Siddiq, A.; Walley, A.J.; Wilhelm-Benartzi, C.S.M.; Rizzo, S.; van der Zee, A.; Plumb, J.A.; Brown, R. Candidate DNA Methylation Drivers of Acquired Cisplatin Resistance in Ovarian Cancer Identified by Methylome and Expression Profiling. Oncogene 2012, 31, 4567–4576. [Google Scholar] [CrossRef]
- Yu, W.; Jin, C.; Lou, X.; Han, X.; Li, L.; He, Y.; Zhang, H.; Ma, K.; Zhu, J.; Cheng, L.; et al. Global Analysis of DNA Methylation by Methyl-Capture Sequencing Reveals Epigenetic Control of Cisplatin Resistance in Ovarian Cancer Cell. PLoS ONE 2011, 6, e29450. [Google Scholar] [CrossRef]
- Gall Trošelj, K.; Novak Kujundzic, R.; Ugarkovic, D. Polycomb Repressive Complex’s Evolutionary Conserved Function: The Role of EZH2 Status and Cellular Background. Clin. Epigenetics 2016, 8, 55. [Google Scholar] [CrossRef]
- Margueron, R.; Reinberg, D. The Polycomb Complex PRC2 and Its Mark in Life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Hung, M.C. Regulation and Role of EZH2 in Cancer. Cancer Res. Treat. 2014, 46, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.Z.; Yan, Y.; Wang, X.X.; Jiang, Y.; Xu, H.E. EZH2: Biology, Disease, and Structure-Based Drug Discovery. Acta Pharm. Sin. 2014, 35, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Fong, K.W.; Mong, E.; Martin, M.C.; Schiltz, G.E.; Yu, J. Going beyond Polycomb: EZH2 Functions in Prostate Cancer. Oncogene 2021, 40, 5788–5798. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Kim, M.; Woo, D.H.; Shin, Y.; Shin, J.; Chang, N.; Oh, Y.T.; Kim, H.; Rheey, J.; Nakano, I.; et al. Phosphorylation of EZH2 Activates STAT3 Signaling via STAT3 Methylation and Promotes Tumorigenicity of Glioblastoma Stem-like Cells. Cancer Cell 2013, 23, 839–852. [Google Scholar] [CrossRef]
- Zhao, Y.; Hu, Z.; Li, J.; Hu, T. EZH2 Exacerbates Breast Cancer by Methylating and Activating STAT3 Directly. J. Cancer 2021, 12, 5220–5230. [Google Scholar] [CrossRef]
- Lee, J.M.; Lee, J.S.; Kim, H.; Kim, K.K.; Park, H.; Kim, J.Y.; Lee, S.H.; Kim, I.S.; Kim, J.; Lee, M.; et al. EZH2 Generates a Methyl Degron That Is Recognized by the DCAF1/DDB1/CUL4 E3 Ubiquitin Ligase Complex. Mol. Cell 2012, 48, 572–586. [Google Scholar] [CrossRef]
- Li, X.; Gonzalez, M.E.; Toy, K.; Filzen, T.; Merajver, S.D.; Kleer, C.G. Targeted Overexpression of EZH2 in the Mammary Gland Disrupts Ductal Morphogenesis and Causes Epithelial Hyperplasia. Am. J. Pathol. 2009, 175, 1246–1254. [Google Scholar] [CrossRef]
- Le, H.Q.; Hill, M.A.; Kollak, I.; Keck, M.; Schroeder, V.; Wirth, J.; Skronska-Wasek, W.; Schruf, E.; Strobel, B.; Stahl, H.; et al. An EZH2-dependent Transcriptional Complex Promotes Aberrant Epithelial Remodelling after Injury. EMBO Rep. 2021, 22, e52785. [Google Scholar] [CrossRef]
- Vasanthakumar, A.; Xu, D.; Lun, A.T.; Kueh, A.J.; Gisbergen, K.P.; Iannarella, N.; Li, X.; Yu, L.; Wang, D.; Williams, B.R.; et al. A Non-canonical Function of Ezh2 Preserves Immune Homeostasis. EMBO Rep. 2017, 18, 619–631. [Google Scholar] [CrossRef]
- Koyen, A.E.; Madden, M.Z.; Park, D.; Minten, E.V.; Kapoor-Vazirani, P.; Werner, E.; Pfister, N.T.; Haji-Seyed-Javadi, R.; Zhang, H.; Xu, J.; et al. EZH2 Has a Non-Catalytic and PRC2-Independent Role in Stabilizing DDB2 to Promote Nucleotide Excision Repair. Oncogene 2020, 39, 4798–4813. [Google Scholar] [CrossRef]
- Koubi, M.; Poplineau, M.; Vernerey, J.; N’Guyen, L.; Tiberi, G.; Garciaz, S.; El-Kaoutari, A.; Maqbool, M.A.; Andrau, J.C.; Guillouf, C.; et al. Regulation of the Positive Transcriptional Effect of PLZF through a Non-Canonical EZH2 Activity. Nucleic Acids Res. 2018, 46, 3339–3350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, L.; Yang, Y.; Li, Q.; Feng, Y.; Liu, T.; Guo, W. Epigenetic Regulation of Cancer Progression by EZH2: From Biological Insights to Therapeutic Potential. Biomark Res. 2018, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, M.; Wang, D.; Hou, P.; Chen, X.; Chu, S.; Chai, D.; Zheng, J.; Bai, J. Post-Translational Modifications of EZH2 in Cancer. Cell Biosci 2020, 10, 143. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Roberts, C.W.M. Targeting EZH2 in Cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Cai, J.; Hou, Y.; Huang, Z.; Wang, Z. Role of EZH2 in Cancer Stem Cells: From Biological Insight to a Therapeutic Target. Oncotarget 2017, 8, 37974–37990. [Google Scholar] [CrossRef]
- Chang, C.J.; Yang, J.Y.; Xia, W.; Chen, C.T.; Xie, X.; Chao, C.H.; Woodward, W.A.; Hsu, J.M.; Hortobagyi, G.N.; Hung, M.C. EZH2 Promotes Expansion of Breast Tumor Initiating Cells through Activation of RAF1-β-Catenin Signaling. Cancer Cell 2011, 19, 86–100. [Google Scholar] [CrossRef]
- Tsang, D.P.F.; Cheng, A.S.L. Epigenetic Regulation of Signaling Pathways in Cancer: Role of the Histone Methyltransferase EZH2. J. Gastroenterol. Hepatol. 2011, 26, 19–27. [Google Scholar] [CrossRef]
- Duan, R.; Du, W.; Guo, W. EZH2: A Novel Target for Cancer Treatment. J. Hematol. Oncol. 2020, 13, 104. [Google Scholar] [CrossRef]
- Yamagishi, M.; Uchimaru, K. Targeting EZH2 in Cancer Therapy. Curr. Opin. Oncol. 2017, 29, 375–381. [Google Scholar] [CrossRef]
- Zhang, L.; Qu, J.; Qi, Y.; Duan, Y.; Huang, Y.W.; Zhou, Z.; Li, P.; Yao, J.; Huang, B.; Zhang, S.; et al. EZH2 Engages TGFβ Signaling to Promote Breast Cancer Bone Metastasis via Integrin Β1-FAK Activation. Nat. Commun. 2022, 13, 2543. [Google Scholar] [CrossRef]
- Hirukawa, A.; Smith, H.W.; Zuo, D.; Dufour, C.R.; Savage, P.; Bertos, N.; Johnson, R.M.; Bui, T.; Bourque, G.; Basik, M.; et al. Targeting EZH2 Reactivates a Breast Cancer Subtype-Specific Anti-Metastatic Transcriptional Program. Nat. Commun. 2018, 9, 2547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Zhang, R. Role of EZH2 in Epithelial Ovarian Cancer: From Biological Insights to Therapeutic Target. Front. Oncol. 2013, 3, 47. [Google Scholar] [CrossRef] [PubMed]
- Serresi, M.; Gargiulo, G.; Proost, N.; Siteur, B.; Cesaroni, M.; Koppens, M.; Xie, H.; Sutherland, K.D.; Hulsman, D.; Citterio, E.; et al. Polycomb Repressive Complex 2 Is a Barrier to KRAS-Driven Inflammation and Epithelial-Mesenchymal Transition in Non-Small-Cell Lung Cancer. Cancer Cell 2016, 29, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, B.K.; Anderman, M.F.; Bhargava, D.; Boccuzzi, L.; Qian, X.; Wang, D.; Durkin, M.E.; Papageorge, A.G.; de Miguel, F.J.; Politi, K.; et al. Inhibition of Cytoplasmic EZH2 Induces Antitumor Activity through Stabilization of the DLC1 Tumor Suppressor Protein. Nat. Commun. 2021, 12, 6941. [Google Scholar] [CrossRef]
- Wang, L.; Chen, C.; Song, Z.; Wang, H.; Ye, M.; Wang, D.; Kang, W.; Liu, H.; Qing, G. EZH2 Depletion Potentiates MYC Degradation Inhibiting Neuroblastoma and Small Cell Carcinoma Tumor Formation. Nat. Commun. 2022, 13, 12. [Google Scholar] [CrossRef]
- Martínez-Fernández, M.; Rubio, C.; Segovia, C.; López-Calderón, F.F.; Dueñas, M.; Paramio, J.M. EZH2 in Bladder Cancer, a Promising Therapeutic Target. Int. J. Mol. Sci. 2015, 16, 27107–27132. [Google Scholar] [CrossRef]
- Simon, C.; Chagraoui, J.; Krosl, J.; Gendron, P.; Wilhelm, B.; Lemieux, S.; Boucher, G.; Chagnon, P.; Drouin, S.; Lambert, R.; et al. A Key Role for EZH2 and Associated Genes in Mouse and Human Adult T-Cell Acute Leukemia. Genes Dev. 2012, 26, 651–656. [Google Scholar] [CrossRef]
- Ntziachristos, P.; Tsirigos, A.; van Vlierberghe, P.; Nedjic, J.; Trimarchi, T.; Flaherty, M.S.; Ferres-Marco, D.; da Ros, V.; Tang, Z.; Siegle, J.; et al. Genetic Inactivation of the Polycomb Repressive Complex 2 in T Cell Acute Lymphoblastic Leukemia. Nat. Med. 2012, 18, 298–302. [Google Scholar] [CrossRef]
- Wang, X.; Brea, L.T.; Yu, J. Immune Modulatory Functions of EZH2 in the Tumor Microenvironment: Implications in Cancer Immunotherapy. Am. J. Clin. Exp. Urol. 2019, 7, 85–91. [Google Scholar]
- Sun, S.; Yu, F.; Xu, D.; Zheng, H.; Li, M. EZH2, a Prominent Orchestrator of Genetic and Epigenetic Regulation of Solid Tumor Microenvironment and Immunotherapy. Biochim. Biophys Acta Rev. Cancer 2022, 1877, 188700. [Google Scholar] [CrossRef]
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.; Wei, S.; Wang, W.; Sun, Y.; Zhao, E.; Vatan, L.; Szeliga, W.; et al. Epigenetic Silencing of TH1-Type Chemokines Shapes Tumour Immunity and Immunotherapy. Nature 2015, 527, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Kang, N.; Eccleston, M.; Clermont, P.L.; Latarani, M.; Male, D.K.; Wang, Y.; Crea, F. EZH2 Inhibition: A Promising Strategy to Prevent Cancer Immune Editing. Epigenomics 2020, 12, 1457–1476. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.S.; Park, K.; Garon, E.; Dubinett, S. Targeting EZH2 to Overcome the Resistance to Immunotherapy in Lung Cancer. Semin Oncol. 2022, in press. [CrossRef] [PubMed]
- Bray, M.; Driscoll, J.; Huggins, J.W. Treatment of Lethal Ebola Virus Infection in Mice with a Single Dose of an S-Adenosyl-L-Homocysteine Hydrolase Inhibitor. Antivir. Res. 2000, 45, 135–147. [Google Scholar] [CrossRef]
- Yap, T.A.; Winter, J.N.; Giulino-Roth, L.; Longley, J.; Lopez, J.; Michot, J.M.; Leonard, J.P.; Ribrag, V.; McCabe, M.T.; Creasy, C.L.; et al. Phase I Study of the Novel Enhancer of Zeste Homolog 2 (EZH2) Inhibitor GSK2816126 in Patients with Advanced Hematologic and Solid Tumors. Clin. Cancer Res. 2019, 25, 7331–7339. [Google Scholar] [CrossRef]
- Campbell, J.E.; Kuntz, K.W.; Knutson, S.K.; Warholic, N.M.; Keilhack, H.; Wigle, T.J.; Raimondi, A.; Klaus, C.R.; Rioux, N.; Yokoi, A.; et al. EPZ011989, A Potent, Orally-Available EZH2 Inhibitor with Robust in Vivo Activity. ACS Med. Chem. Lett. 2015, 6, 491–495. [Google Scholar] [CrossRef]
- Kurmasheva, R.T.; Erickson, S.W.; Earley, E.; Smith, M.A.; Houghton, P.J. In Vivo Evaluation of the EZH2 Inhibitor (EPZ011989) Alone or in Combination with Standard of Care Cytotoxic Agents against Pediatric Malignant Rhabdoid Tumor Preclinical Models—A Report from the Pediatric Preclinical Testing Consortium. Pediatr. Blood Cancer 2021, 68, e28772. [Google Scholar] [CrossRef]
- Eich, M.L.; Athar, M.; Ferguson, J.E.; Varambally, S. EZH2-Targeted Therapies in Cancer: Hype or a Reality. Cancer Res. 2020, 80, 5449–5458. [Google Scholar] [CrossRef]
- Batlevi, C.L. A Closer Look at Tazemetostat. Clin. Adv. Hematol. Oncol. 2020, 18, 820–822. [Google Scholar]
- Yu, C.; Ding, B.; Zhang, X.; Deng, X.; Deng, K.; Cheng, Z.; Xing, B.; Jin, D.; Ma, P.; Lin, J. Targeted Iron Nanoparticles with Platinum-(IV) Prodrugs and Anti-EZH2 SiRNA Show Great Synergy in Combating Drug Resistance in Vitro and in Vivo. Biomaterials 2018, 155, 112–123. [Google Scholar] [CrossRef]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus Gemcitabine versus Gemcitabine for Biliary Tract Cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Gardner, E.E.; Lok, B.H.; Schneeberger, V.E.; Desmeules, P.; Miles, L.A.; Arnold, P.K.; Ni, A.; Khodos, I.; de Stanchina, E.; Nguyen, T.; et al. Chemosensitive Relapse in Small Cell Lung Cancer Proceeds through an EZH2-SLFN11 Axis. Cancer Cell 2017, 31, 286–299. [Google Scholar] [CrossRef]
- Qiu, X.; Wang, W.; Li, B.; Cheng, B.; Lin, K.; Bai, J.; Li, H.; Yang, G. Targeting Ezh2 Could Overcome Docetaxel Resistance in Prostate Cancer Cells. BMC Cancer 2019, 19, 27. [Google Scholar] [CrossRef] [PubMed]
- Fillmore, C.M.; Xu, C.; Desai, P.T.; Berry, J.M.; Rowbotham, S.P.; Lin, Y.J.; Zhang, H.; Marquez, V.E.; Hammerman, P.S.; Wong, K.K.; et al. EZH2 Inhibition Sensitizes BRG1 and EGFR Mutant Lung Tumours to TopoII Inhibitors. Nature 2015, 520, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.Y.; Wang, H.; Xiang, P.; Liu, Y.W.; Li, H.Z.; Lei, B.X.; Yu, M.; Qi, S.T. Inhibition of EZH2 Reverses Chemotherapeutic Drug TMZ Chemosensitivity in Glioblastoma. Int. J. Clin. Exp. Pathol. 2014, 7, 6662–6670. [Google Scholar]
- Zhou, L.; Mudianto, T.; Ma, X.; Riley, R.; Uppaluri, R. Targeting EZH2 Enhances Antigen Presentation, Antitumor Immunity, and Circumvents Anti–PD-1 Resistance in Head and Neck Cancer. Clin. Cancer Res. 2020, 26, 290–300. [Google Scholar] [CrossRef]
- Bai, Y.; Zhang, Z.; Cheng, L.; Wang, R.; Chen, X.; Kong, Y.; Feng, F.; Ahmad, N.; Li, L.; Liu, X. Inhibition of Enhancer of Zeste Homolog 2 (EZH2) Overcomes Enzalutamide Resistance in Castration-Resistant Prostate Cancer. J. Biol. Chem. 2019, 294, 9911–9923. [Google Scholar] [CrossRef]
- Karakashev, S.; Fukumoto, T.; Zhao, B.; Lin, J.; Wu, S.; Fatkhutdinov, N.; Park, P.H.; Semenova, G.; Jean, S.; Cadungog, M.G.; et al. EZH2 Inhibition Sensitizes CARM1-High, Homologous Recombination Proficient Ovarian Cancers to PARP Inhibition. Cancer Cell 2020, 37, 157–167.e6. [Google Scholar] [CrossRef]
- Fiskus, W.; Wang, Y.; Sreekumar, A.; Buckley, K.M.; Shi, H.; Jillella, A.; Ustun, C.; Rao, R.; Fernandez, P.; Chen, J.; et al. Combined Epigenetic Therapy with the Histone Methyltransferase EZH2 Inhibitor 3-Deazaneplanocin A and the Histone Deacetylase Inhibitor Panobinostat against Human AML Cells. Blood 2009, 114, 2733–2743. [Google Scholar] [CrossRef]
- Li, C.; Wang, Y.; Gong, Y.; Zhang, T.; Huang, J.; Tan, Z.; Xue, L. Finding an Easy Way to Harmonize: A Review of Advances in Clinical Research and Combination Strategies of EZH2 Inhibitors. Clin. Epigenetics 2021, 13, 62. [Google Scholar] [CrossRef]
- Caron, P.; van der Linden, J.; van Attikum, H. Bon Voyage: A Transcriptional Journey around DNA Breaks. DNA Repair 2019, 82, 102686. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Chen, C.H.; Xiao, T.; de la Peña Avalos, B.; Dray, E.V.; Cai, C.; Gao, S.; Shah, N.; Zhang, Z.; Feit, A.; et al. Inhibition of EZH2 Transactivation Function Sensitizes Solid Tumors to Genotoxic Stress. Proc. Natl. Acad. Sci. USA 2022, 119, e2105898119. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, S.; Granger, V.; Rak, M.; Hu, Q.; Attwood, K.; Aquila, L.; Krishnan, N.; Osiecki, R.; Azabdaftari, G.; Guru, K.; et al. Inhibition of EZH2 Induces NK Cell-Mediated Differentiation and Death in Muscle-Invasive Bladder Cancer. Cell Death Differ. 2019, 26, 2100–2114. [Google Scholar] [CrossRef] [PubMed]
- Bi, H.; Zhang, Z.; Guo, L.; Fu, C. Effect of Wound Fluid on Chemotherapy Sensitivity of T24 Bladder Cancer Cells with Different Enhancer of Zeste Homolog 2 Status. Oncotarget 2017, 8, 63258–63264. [Google Scholar] [CrossRef]
- Dou, D.; Ge, X.; Wang, X.; Xu, X.; Zhang, Z.; Seng, J.; Cao, Z.; Gu, Y.; Han, M. Ezh2 Contributes to Cisplatin Resistance in Breast Cancer by Epigenetically Suppressing Mir-381 Expression. Onco Targets 2019, 12, 9627–9637. [Google Scholar] [CrossRef]
- Puppe, J.; Opdam, M.; Schouten, P.C.; Jozwiak, K.; Lips, E.; Severson, T.; van de Ven, M.; Brambillasca, C.; Bouwman, P.; van Tellingen, O.; et al. EZH2 Is Overexpressed in BRCA1-like Breast Tumors and Predictive for Sensitivity to High-Dose Platinum-Based Chemotherapy. Clin. Cancer Res. 2019, 25, 4351–4362. [Google Scholar] [CrossRef]
- Cai, L.; Wang, Z.; Liu, D. Interference with Endogenous EZH2 Reverses the Chemotherapy Drug Resistance in Cervical Cancer Cells Partly by Up-Regulating Dicer Expression. Tumor Biol. 2016, 37, 6359–6369. [Google Scholar] [CrossRef]
- Roh, J.W.; Choi, J.E.; Han, H.D.; Hu, W.; Matsuo, K.; Nishimura, M.; Lee, J.S.; Kwon, S.Y.; Cho, C.H.; Kim, J.; et al. Clinical and Biological Significance of EZH2 Expression in Endometrial Cancer. Cancer Biol. 2020, 21, 147–156. [Google Scholar] [CrossRef]
- Oki, S.; Sone, K.; Oda, K.; Hamamoto, R.; Ikemura, M.; Maeda, D.; Takeuchi, M.; Tanikawa, M.; Mori-Uchino, M.; Nagasaka, K.; et al. Oncogenic Histone Methyltransferase EZH2: A Novel Prognostic Marker with Therapeutic Potential in Endometrial Cancer. Oncotarget 2017, 8, 40402–40411. [Google Scholar] [CrossRef]
- Dai, Q.; Zhang, T.; Pan, J.; Li, C. LncRNA UCA1 Promotes Cisplatin Resistance in Gastric Cancer via Recruiting EZH2 and Activating PI3K/AKT Pathway. J. Cancer 2020, 11, 3882–3892. [Google Scholar] [CrossRef]
- Chang, J.W.; Gwak, S.Y.; Shim, G.A.; Liu, L.; Lim, Y.C.; Kim, J.M.; Jung, M.G.; Koo, B.S. EZH2 Is Associated with Poor Prognosis in Head-and-Neck Squamous Cell Carcinoma via Regulating the Epithelial-to-Mesenchymal Transition and Chemosensitivity. Oral Oncol. 2016, 52, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Kesselman, D.; Kizub, D.; Guerrero-Preston, R.; Ratovitski, E.A. Phospho-ΔNp63α/MicroRNA Feedback Regulation in Squamous Carcinoma Cells upon Cisplatin Exposure. Cell Cycle 2013, 12, 684–697. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wang, X.; Niu, X.; Wang, X.; Jiang, R.; Xu, T.; Liu, Y.; Liang, L.; Ou, X.; Xing, X.; et al. EZH2 Suppresses the Nucleotide Excision Repair in Nasopharyngeal Carcinoma by Silencing XPA Gene. Mol. Carcinog. 2017, 56, 447–463. [Google Scholar] [CrossRef] [PubMed]
- Zeng, T.; Luo, L.; Huang, Y.; Ye, X.; Lin, J. Upregulation of MiR-138 Increases Sensitivity to Cisplatin in Hepatocellular Carcinoma by Regulating EZH2. Biomed. Res. Int. 2021, 2021, 6665918. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Wu, W.; Wei, H.; Zhang, W.; Huang, Y.; Dong, Z. Downregulation of Histone-Lysine N-Methyltransferase EZH2 Inhibits Cell Viability and Enhances Chemosensitivity in Lung Cancer Cells. Oncol. Lett. 2021, 21, 26. [Google Scholar] [CrossRef]
- Lv, Y.; Yuan, C.; Xiao, X.; Wang, X.; Ji, X.; Yu, H.; Wu, Z.; Zhang, J. The Expression and Significance of the Enhancer of Zeste Homolog 2 in Lung Adenocarcinoma. Oncol. Rep. 2012, 28, 147–154. [Google Scholar] [CrossRef]
- Liu, H.; Li, W.; Yu, X.; Gao, F.; Duan, Z.; Ma, X.; Tan, S.; Yuan, Y.; Liu, L.; Wang, J.; et al. EZH2-Mediated Puma Gene Repression Regulates Non-Small Cell Lung Cancer Cell Proliferation and Cisplatin-Induced Apoptosis. Oncotarget 2016, 7, 56338–56354. [Google Scholar] [CrossRef]
- Riquelme, E.; Suraokar, M.; Behrens, C.; Lin, H.Y.; Girard, L.; Nilsson, M.B.; Simon, G.; Wang, J.; Coombes, K.R.; Lee, J.J.; et al. VEGF/VEGFR-2 Upregulates EZH2 Expression in Lung Adenocarcinoma Cells and EZH2 Depletion Enhances the Response to Platinum-Based and VEGFR-2-Targeted Therapy. Clin. Cancer Res. 2014, 20, 3849–3861. [Google Scholar] [CrossRef]
- Ni, J.; Hou, X.; Wang, X.; Shi, Y.; Xu, L.; Zheng, X.; Liu, N.; Qiu, A.; Zhuang, S. 3-Deazaneplanocin A Protects against Cisplatin-Induced Renal Tubular Cell Apoptosis and Acute Kidney Injury by Restoration of E-Cadherin Expression. Cell Death Dis. 2019, 10, 355. [Google Scholar] [CrossRef]
- He, C.; Sun, J.; Liu, C.; Jiang, Y.; Hao, Y. Elevated H3K27me3 Levels Sensitize Osteosarcoma to Cisplatin. Clin. Epigenetics 2019, 11, 8. [Google Scholar] [CrossRef]
- Sun, Y.; Wu, J.; Dong, X.; Zhang, J.; Meng, C.; Liu, G. MicroRNA-506-3p Increases the Response to PARP Inhibitors and Cisplatin by Targeting EZH2/β-Catenin in Serous Ovarian Cancers. Transl. Oncol. 2021, 14, 100987. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Cai, X.; Yung, M.M.; Zhou, W.; Li, J.; Zhang, Y.; Li, Z.; Liu, S.S.; Cheung, A.N.Y.; Ngan, H.Y.S.; et al. MiR-137 Mediates the Functional Link between c-Myc and EZH2 That Regulates Cisplatin Resistance in Ovarian Cancer. Oncogene 2019, 38, 564–580. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jin, L.; Liu, J.H.; Sui, Y.X.; Han, L.L.; Shen, X.L. Interfering EZH2 Expression Reverses the Cisplatin Resistance in Human Ovarian Cancer by Inhibiting Autophagy. Cancer Biother. Radiopharm. 2016, 31, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Zhao, S.; Yang, Q.; Wang, W.; Cai, E.; Wen, Y.; Yu, L.; Wang, Z.; Cai, J. Enhancer of Zeste Homolog 2 Promotes Cisplatin Resistance by Reducing Cellular Platinum Accumulation. Cancer Sci. 2018, 109, 1853–1864. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.; Yu, L.; Li, Z.; Shen, Y.; Wang, J.; Cai, J.; Xiao, L.; Wang, Z. Overexpression of EZH2 Contributes to Acquired Cisplatin Resistance in Ovarian Cancer Cells in Vitro and in Vivo. Cancer Biol. 2010, 10, 788–795. [Google Scholar] [CrossRef]
- Li, T.; Cai, J.; Ding, H.; Xu, L.; Yang, Q.; Wang, Z. EZH2 Participates in Malignant Biological Behavior of Epithelial Ovarian Cancer through Regulating the Expression of BRCA1. Cancer Biol. 2014, 15, 271–278. [Google Scholar] [CrossRef]
- Singh, R.; Fazal, Z.; Corbet, A.K.; Bikorimana, E.; Rodriguez, J.C.; Khan, E.M.; Shahid, K.; Freemantle, S.J.; Spinella, M.J. Epigenetic Remodeling through Downregulation of Polycomb Repressive Complex 2 Mediates Chemotherapy Resistance in Testicular Germ Cell Tumors. Cancers 2019, 11, 796. [Google Scholar] [CrossRef]
- Helm, C.W.; States, J.C. Enhancing the Efficacy of Cisplatin in Ovarian Cancer Treatment—Could Arsenic Have a Role. J. Ovarian Res. 2009, 2, 2–7. [Google Scholar] [CrossRef]
- Song, M.; Cui, M.; Liu, K. Therapeutic Strategies to Overcome Cisplatin Resistance in Ovarian Cancer. Eur. J. Med. Chem. 2022, 232, 114205. [Google Scholar] [CrossRef]
- Kuang, Y.; Cai, J.; Li, D.; Han, Q.; Cao, J.; Wang, Z. Repression of Dicer Is Associated with Invasive Phenotype and Chemoresistance in Ovarian Cancer. Oncol. Lett. 2013, 5, 1149–1154. [Google Scholar] [CrossRef]
- Neff, R.T.; Senter, L.; Salani, R. BRCA Mutation in Ovarian Cancer: Testing, Implications and Treatment Considerations. Adv. Med. Oncol. 2017, 9, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Husain, A.; He, G.; Venkatraman, E.S.; Spriggs, D.R. BRCA1 Up-Regulation Is Associated with Repair-Mediated Resistance to Cis-Diamminedichloroplatinum(II). Cancer Res. 1998, 58, 1120–1123. [Google Scholar]
- Kanakkanthara, A.; Kurmi, K.; Ekstrom, T.L.; Hou, X.; Purfeerst, E.R.; Heinzen, E.P.; Correia, C.; Huntoon, C.J.; O’Brien, D.; Wahner Hendrickson, A.E.; et al. BRCA1 Deficiency Upregulates NNMT, Which Reprograms Metabolism and Sensitizes Ovarian Cancer Cells to Mitochondrial Metabolic Targeting Agents. Cancer Res. 2019, 79, 5920–5929. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.M.; Kim, H.K.; Shim, W.; Anwar, M.A.; Kwon, J.W.; Kwon, H.K.; Kim, H.J.; Jeong, H.; Kim, H.M.; Hwang, D.; et al. Mechanism of Cisplatin-Induced Cytotoxicity Is Correlated to Impaired Metabolism Due to Mitochondrial ROS Generation. PLoS ONE 2015, 10, e0135083. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yu, Y.; Chen, C.; Wang, Q.; Huang, T.; Hong, F.; Zhu, L. Involvement of Enhancer of Zeste Homolog 2 in Cisplatin-Resistance in Ovarian Cancer Cells by Interacting with Several Genes. Mol. Med. Rep. 2015, 12, 2503–2510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.; Li, L.; Lu, Y.; Jiang, F.; Yang, X.A. SREBP2 Contributes to Cisplatin Resistance in Ovarian Cancer Cells. Exp. Biol. Med. 2018, 243, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Özeş, A.R.; Wang, Y.; Zong, X.; Fang, F.; Pilrose, J.; Nephew, K.P. Therapeutic Targeting Using Tumor Specific Peptides Inhibits Long Non-Coding RNA HOTAIR Activity in Ovarian and Breast Cancer. Sci. Rep. 2017, 7, 894. [Google Scholar] [CrossRef]
- Zhang, Y.; Ai, H.; Fan, X.; Chen, S.; Wang, Y.; Liu, L. Knockdown of Long Non-Coding RNA HOTAIR Reverses Cisplatin Resistance of Ovarian Cancer Cells through Inhibiting MiR-138-5p-Regulated EZH2 and SIRT1. Biol. Res. 2020, 53, 18. [Google Scholar] [CrossRef]
- Sajadpoor, Z.; Amini-Farsani, Z.; Teimori, H.; Shamsara, M.; Sangtarash, M.H.; Ghasemi-Dehkordi, P.; Yadollahi, F. Valproic Acid Promotes Apoptosis and Cisplatin Sensitivity Through Downregulation of H19 Noncoding RNA in Ovarian A2780 Cells. Appl Biochem. Biotechnol. 2018, 185, 1132–1144. [Google Scholar] [CrossRef]
- Liu, L.; Guo, J.; Yu, L.; Cai, J.; Gui, T.; Tang, H.; Song, L.; Wang, J.; Han, F.; Yang, C.; et al. MiR-101 Regulates Expression of EZH2 and Contributes to Progression of and Cisplatin Resistance in Epithelial Ovarian Cancer. Tumor Biol. 2014, 35, 12619–12626. [Google Scholar] [CrossRef]
- Cai, J.; Yang, C.; Yang, Q.; Ding, H.; Jia, J.; Guo, J.; Wang, J.; Wang, Z. Deregulation of Let-7e in Epithelial Ovarian Cancer Promotes the Development of Resistance to Cisplatin. Oncogenesis 2013, 2, e75. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Hao, K.; Hu, H.; Sheng, Z.; Yan, J.; Wang, Q.; Yu, L. Expression of the Enhancer of Zeste Homolog 2 in Biopsy Specimen Predicts Chemoresistance and Survival in Advanced Non-Small Cell Lung Cancer Receiving First-Line Platinum-Based Chemotherapy. Lung Cancer 2014, 86, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Wang, J.; Man, W.Y.; Zhang, Q.W.; Xu, W.G. SiRNA Silencing EZH2 Reverses Cisplatin-Resistance of Human Non-Small Cell Lung and Gastric Cancer Cells. Asian Pac. J. Cancer Prev. 2015, 16, 2425–2430. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Gu, X.; Chen, Y.; Zhou, M.; Jiang, F.; Zheng, S. Identification of Potential Prognostic and Predictive Biomarkers for Immune-Checkpoint Inhibitor Response in Small Cell Lung Cancer. Med. Sci. Monit. 2021, 27, 318. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Liu, J.; Li, K.; Yang, G.; Chen, S.; Wu, J.; Xie, X.; Ren, H.; Pang, Y. An SETD1A/Wnt/β-Catenin Feedback Loop Promotes NSCLC Development. J. Exp. Clin. Cancer Res. 2021, 40, 318. [Google Scholar] [CrossRef]
- Liu, Y.; Hu, Q.; Wang, X. AFAP1-AS1 Induces Cisplatin Resistance in Non-Small Cell Lung Cancer through PI3K/AKT Pathway. Oncol. Lett. 2020, 19, 1024–1030. [Google Scholar] [CrossRef] [PubMed]
- Zhan, J.; Wang, P.; Li, S.; Song, J.; He, H.; Wang, Y.; Liu, Z.; Wang, F.; Bai, H.; Fang, W.; et al. HOXB13 Networking with ABCG1/EZH2/Slug Mediates Metastasis and Confers Resistance to Cisplatin in Lung Adenocarcinoma Patients. Theranostics 2019, 9, 2084–2099. [Google Scholar] [CrossRef]
- Yang, D.; Feng, W.; Zhuang, Y.; Liu, J.; Feng, Z.; Xu, T.; Wang, W.; Zhu, Y.; Wang, Z. Long Non-Coding RNA Linc00665 Inhibits CDKN1C Expression by Binding to EZH2 and Affects Cisplatin Sensitivity of NSCLC Cells. Mol. Nucleic Acids 2021, 23, 1053–1065. [Google Scholar] [CrossRef]
- Xia, L.; Liu, Y.; Wang, Y. PD-1/PD-L1 Blockade Therapy in Advanced Non-Small-Cell Lung Cancer: Current Status and Future Directions. Oncologist 2019, 24, S31–S41. [Google Scholar] [CrossRef]
- Fournel, L.; Wu, Z.; Stadler, N.; Damotte, D.; Lococo, F.; Boulle, G.; Ségal-Bendirdjian, E.; Bobbio, A.; Icard, P.; Trédaniel, J.; et al. Cisplatin Increases PD-L1 Expression and Optimizes Immune Check-Point Blockade in Non-Small Cell Lung Cancer. Cancer Lett. 2019, 464, 5–14. [Google Scholar] [CrossRef]
- Lin, W.; Qian, J.; Wang, H.; Ren, L.; Yang, Y.; Chen, C.; Chen, X.; Huang, Y.; Liu, J.; Xu, N.; et al. Cisplatin plus Anti-PD-1 Enhanced Treatment Efficacy in Advanced Esophageal Squamous Cell Carcinoma. Am. J. Cancer Res. 2022, 12, 451–468. [Google Scholar] [PubMed]
- Zhang, M.; Yang, L.; Hou, L.; Tang, X. LncRNA SNHG1 Promotes Tumor Progression and Cisplatin Resistance through Epigenetically Silencing MiR-381 in Breast Cancer. Bioengineered 2021, 12, 9239–9250. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.; Yang, C.; Tang, Y.; Zheng, W.; Zhou, Y.; Zhang, S.; Song, M.; Cheng, P.; Wei, Z.; Zhong, C.; et al. Pharmacological Manipulation of Ezh2 with Salvianolic Acid B Results in Tumor Vascular Normalization and Synergizes with Cisplatin and T Cell-Mediated Immunotherapy. Pharm. Res. 2022, 182, 106333. [Google Scholar] [CrossRef]
- Yamamoto, M.; Jin, C.; Hata, T.; Yasumizu, Y.; Zhang, Y.; Hong, D.; Maeda, T.; Miyo, M.; Hiraki, M.; Suzuki, Y.; et al. MUC1-C Integrates Chromatin Remodeling and PARP1 Activity in the DNA Damage Response of Triple-Negative Breast Cancer Cells. Cancer Res. 2019, 79, 2031–2041. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tian, J.; Qu, C.; Peng, Y.; Lei, J.; Li, K.; Zong, B.; Sun, L.; Liu, S. Overexpression of SERPINA3 Promotes Tumor Invasion and Migration, Epithelial-Mesenchymal-Transition in Triple-Negative Breast Cancer Cells. Breast Cancer 2021, 28, 859–873. [Google Scholar] [CrossRef]
- Yu, G.; Zhou, H.; Yao, W.; Meng, L.; Lang, B. LncRNA TUG1 Promotes Cisplatin Resistance by Regulating CCND2 via Epigenetically Silencing MiR-194-5p in Bladder Cancer. Mol. Nucleic. Acids 2019, 16, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.D.; Grandis, J.R. The Molecular Pathogenesis of Head and Neck Cancer. Cancer Biol. Ther. 2010, 9, 1–7. [Google Scholar] [CrossRef]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and Neck Squamous Cell Carcinoma. Nat. Rev. Dis. Primers 2020, 6, 92. [Google Scholar] [CrossRef]
- Kanno, Y.; Chen, C.Y.; Lee, H.L.; Chiou, J.F.; Chen, Y.J. Molecular Mechanisms of Chemotherapy Resistance in Head and Neck Cancers. Front. Oncol 2021, 11, 640392. [Google Scholar] [CrossRef]
- Sun, S.; Wu, Y.; Guo, W.; Yu, F.; Kong, L.; Ren, Y.; Wang, Y.; Yao, X.; Jing, C.; Zhang, C.; et al. Stat3/Hotair Signaling Axis Regulates Hnscc Growth in an Ezh2-Dependent Manner. Clin. Cancer Res. 2018, 24, 2665–2677. [Google Scholar] [CrossRef]
- Huang, Y.; Chuang, A.; Hao, H.; Talbot, C.; Sen, T.; Trink, B.; Sidransky, D.; Ratovitski, E. Phospho-ΔNp63α Is a Key Regulator of the Cisplatin-Induced MicroRNAome in Cancer Cells. Cell Death Differ. 2011, 18, 1220–1230. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xu, Z.; Sun, J.; Lv, H.; Wang, Y.; Ni, Y.; Chen, S.; Hu, C.; Wang, L.; Chen, W.; et al. Cisplatin Resistance in Gastric Cancer Cells Is Involved with GPR30-Mediated Epithelial-Mesenchymal Transition. J. Cell Mol. Med. 2020, 24, 3625–3633. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ma, X.; Yang, D.; Suo, Z.; Dai, R.; Liu, C. PCAT-1 Contributes to Cisplatin Resistance in Gastric Cancer through Epigenetically Silencing PTEN via Recruiting EZH2. J. Cell Biochem. 2020, 121, 1353–1361. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Yang, S.; Han, Y.; Sun, J.; Xv, L.; Wu, L.; Ming, L. HOXD-AS1 Confers Cisplatin Resistance in Gastric Cancer through Epigenetically Silencing PDCD4 via Recruiting EZH2. Open Biol. 2019, 9, 190068. [Google Scholar] [CrossRef]
- Xu, C.; Guo, Y.; Liu, H.; Chen, G.; Yan, Y.; Liu, T. TUG1 Confers Cisplatin Resistance in Esophageal Squamous Cell Carcinoma by Epigenetically Suppressing PDCD4 Expression via EZH2. Cell Biosci. 2018, 8, 61. [Google Scholar] [CrossRef] [PubMed]
- Lobo, J.; Jerónimo, C.; Henrique, R. Cisplatin Resistance in Testicular Germ Cell Tumors: Current Challenges from Various Perspectives. Cancers 2020, 12, 1601. [Google Scholar] [CrossRef]
- Zhu, Z.; Tang, J.; Wang, J.; Duan, G.; Zhou, L.; Zhou, X. MIR-138 Acts as a Tumor Suppressor by Targeting EZH2 and Enhances Cisplatin-Induced Apoptosis in Osteosarcoma Cells. PLoS ONE 2016, 11, e0150026. [Google Scholar] [CrossRef]
- Liu, X.H.; Zhang, X.F.; Du, S.Z. Long Non-Coding RNA ACTA2-AS1 Inhibits the Cisplatin Resistance of Non-Small Cell Lung Cancer Cells through Inhibiting Autophagy by Suppressing TSC2. Cell Cycle 2022, 21, 368–378. [Google Scholar] [CrossRef]
- Chen, S.Q.; Li, J.Q.; Wang, X.Q.; Lei, W.J.; Li, H.; Wan, J.; Hu, Z.; Zou, Y.W.; Wu, X.Y.; Niu, H.X. EZH2-Inhibitor DZNep Enhances Apoptosis of Renal Tubular Epithelial Cells in Presence and Absence of Cisplatin. Cell Div. 2020, 15, 8. [Google Scholar] [CrossRef]
- Wen, L.; Tao, S.; Guo, F.; Li, L.; Yang, H.; Liang, Y.; Zhang, L.; Ma, L.; Fu, P. Selective EZH2 Inhibitor Zld1039 Alleviates Inflammation in Cisplatin-Induced Acute Kidney Injury Partially by Enhancing RKIP and Suppressing NF-ΚB P65 Pathway. Acta Pharm. Sin. 2021, 43, 2067–2080. [Google Scholar] [CrossRef]
- Yu, C.; Li, T.; Li, J.; Cui, B.; Liu, N.; Bayliss, G.; Zhuang, S. Inhibition of Polycomb Repressive Complex 2 by Targeting EED Protects against Cisplatin-induced Acute Kidney Injury. J. Cell Mol. Med. 2022, 26, 4061–4075. [Google Scholar] [CrossRef]
- Li, P.; Zhang, X.; Wang, H.; Wang, L.; Liu, T.; Du, L.; Yang, Y.; Wang, C. MALAT1 Is Associated with Poor Response to Oxaliplatin-Based Chemotherapy in Colorectal Cancer Patients and Promotes Chemoresistance through EZH2. Mol. Cancer 2017, 16, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Peng, J.; Xiao, L.; Zhou, C.; Fang, Y.; Ou, Q.; Qin, J.; Liu, M.; Pan, Z.; Hou, Z. TRIM25 Regulates Oxaliplatin Resistance in Colorectal Cancer by Promoting EZH2 Stability. Cell Death Dis. 2021, 12, 463. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Kong, F.; Li, S.; Jiang, H.; Dong, L.; Xu, X.; Zhang, X.; Yuan, H.; Xu, Y.; Chu, Y.; et al. A KLF4/PiHL/EZH2/HMGA2 Regulatory Axis and Its Function in Promoting Oxaliplatin-Resistance of Colorectal Cancer. Cell Death Dis. 2021, 12, 485. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gan, Y.; Liu, J.; Li, J.; Zhou, Z.; Tian, R.; Sun, R.; Liu, J.; Xiao, Q.; Li, Y.; et al. Downregulation of MEIS1 Mediated by ELFN1-AS1/EZH2/DNMT3a Axis Promotes Tumorigenesis and Oxaliplatin Resistance in Colorectal Cancer. Signal. Transduct Target. 2022, 7, 87. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, X.; Jiang, Y.; Liu, S.; Liu, H.; Sun, X.; Zhang, H.; Liu, Z.; Tao, Y.; Li, C.; et al. Elevating H3K27me3 Level Sensitizes Colorectal Cancer to Oxaliplatin. J. Mol. Cell Biol. 2020, 12, 125–137. [Google Scholar] [CrossRef]
- Naskou, J.; Beiter, Y.; van Rensburg, R.; Honisch, E.; Rudelius, M.; Schlensog, M.; Gottstein, J.; Walter, L.; Braicu, E.I.; Sehouli, J.; et al. EZH2 Loss Drives Resistance to Carboplatin and Paclitaxel in Serous Ovarian Cancers Expressing ATM. Mol. Cancer Res. 2020, 18, 278–286. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samaržija, I.; Tomljanović, M.; Novak Kujundžić, R.; Trošelj, K.G. EZH2 Inhibition and Cisplatin as a Combination Anticancer Therapy: An Overview of Preclinical Studies. Cancers 2022, 14, 4761. https://doi.org/10.3390/cancers14194761
Samaržija I, Tomljanović M, Novak Kujundžić R, Trošelj KG. EZH2 Inhibition and Cisplatin as a Combination Anticancer Therapy: An Overview of Preclinical Studies. Cancers. 2022; 14(19):4761. https://doi.org/10.3390/cancers14194761
Chicago/Turabian StyleSamaržija, Ivana, Marko Tomljanović, Renata Novak Kujundžić, and Koraljka Gall Trošelj. 2022. "EZH2 Inhibition and Cisplatin as a Combination Anticancer Therapy: An Overview of Preclinical Studies" Cancers 14, no. 19: 4761. https://doi.org/10.3390/cancers14194761
APA StyleSamaržija, I., Tomljanović, M., Novak Kujundžić, R., & Trošelj, K. G. (2022). EZH2 Inhibition and Cisplatin as a Combination Anticancer Therapy: An Overview of Preclinical Studies. Cancers, 14(19), 4761. https://doi.org/10.3390/cancers14194761