
Spatial Transcriptomic Analysis Reveals Associations between Genes and Cellular Topology in Breast and Prostate Cancers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Datasets
2.2. Statistical and Correlation Analyses
2.3. ITF Generation
2.4. Clustering Correlated Gene Expression and Topology to Identify Sets of TAGs
2.5. Functional Enrichment Analysis
2.6. Integrative ITF Analysis in FFPE Human Breast Cancer ST Data
3. Results
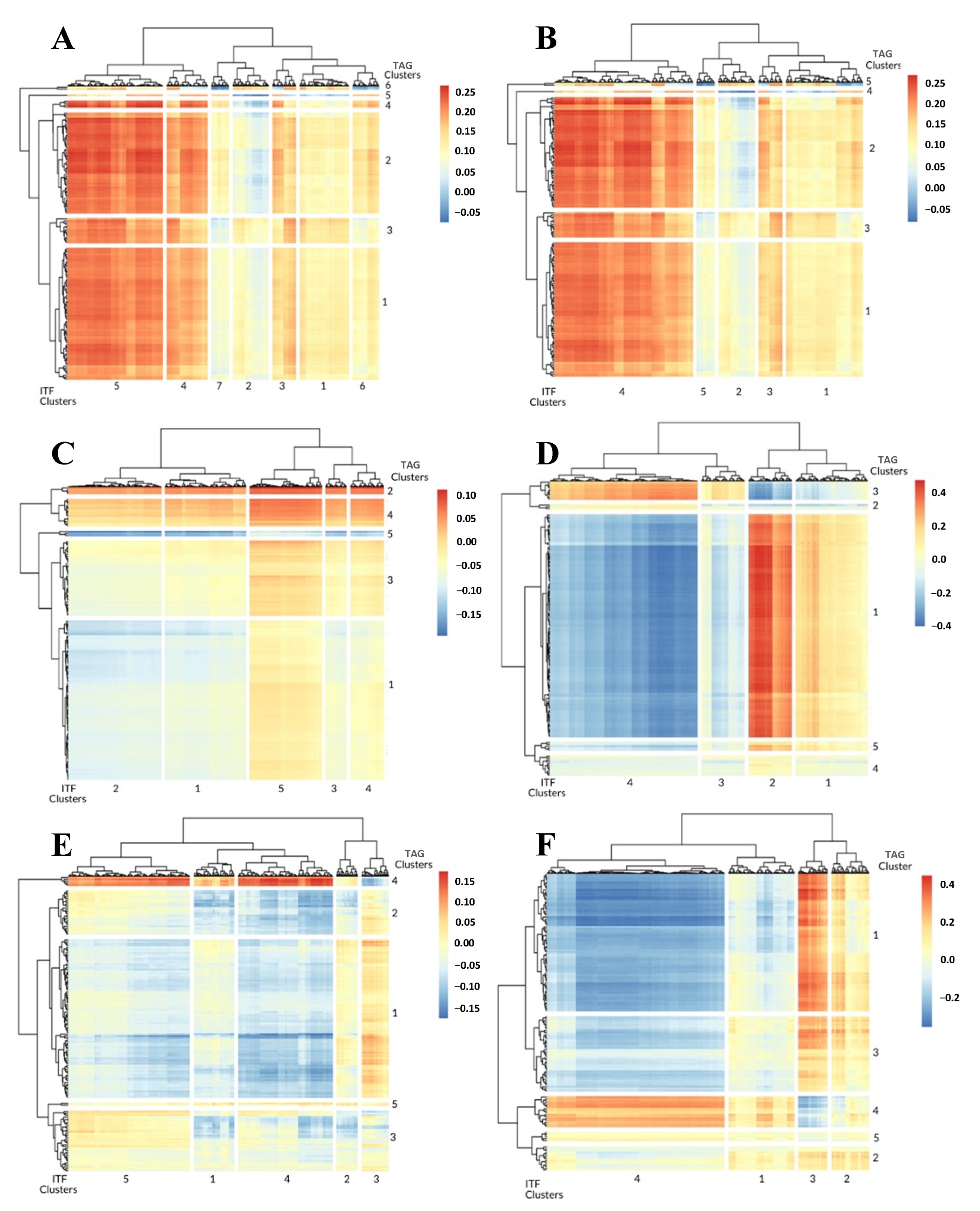
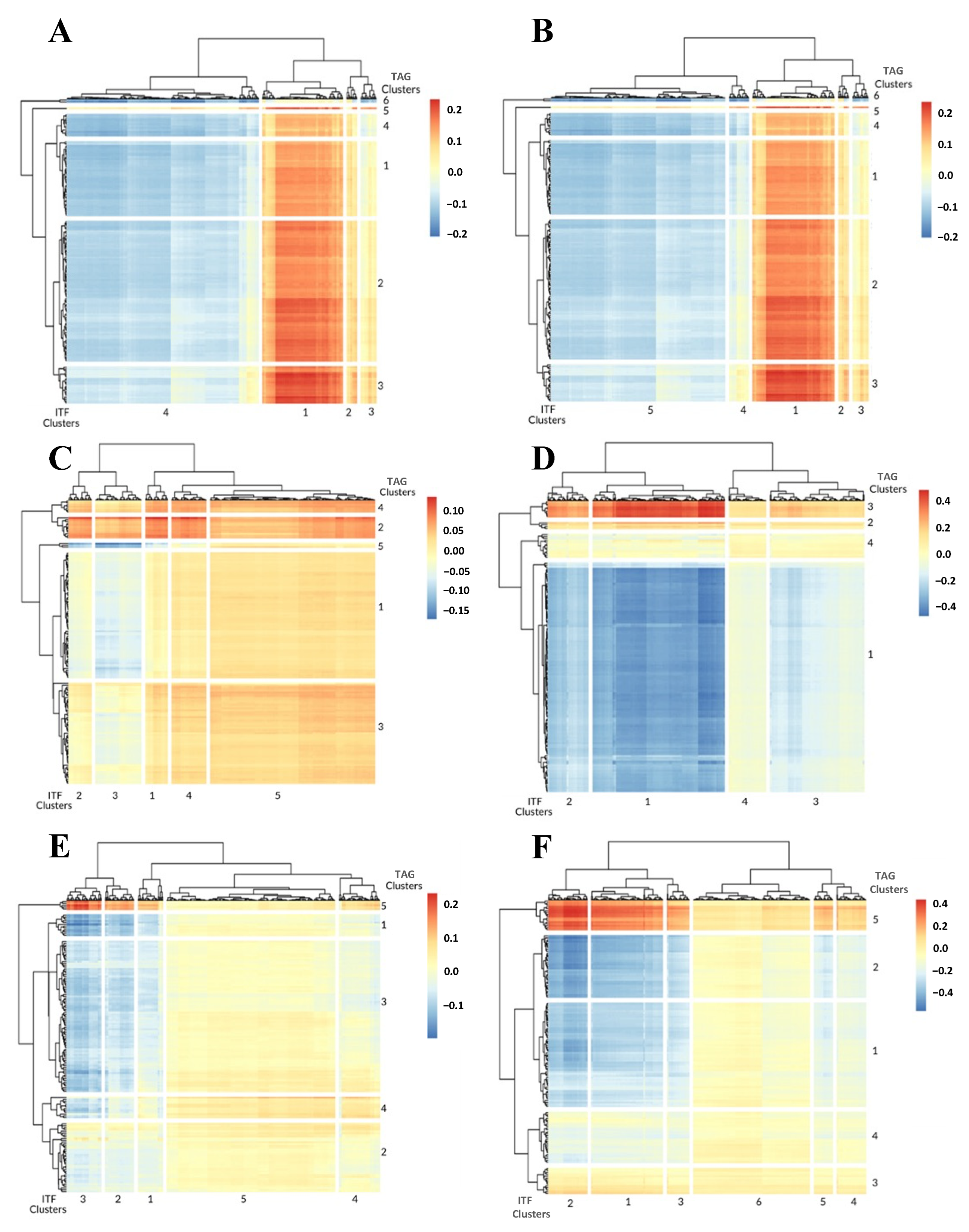
3.1. 1-Dimensional ITFs Correlate with Gene Sets and Functional Enrichment
3.2. 0-Dimensional ITFs Correlate with Gene Sets and Functional Enrichment
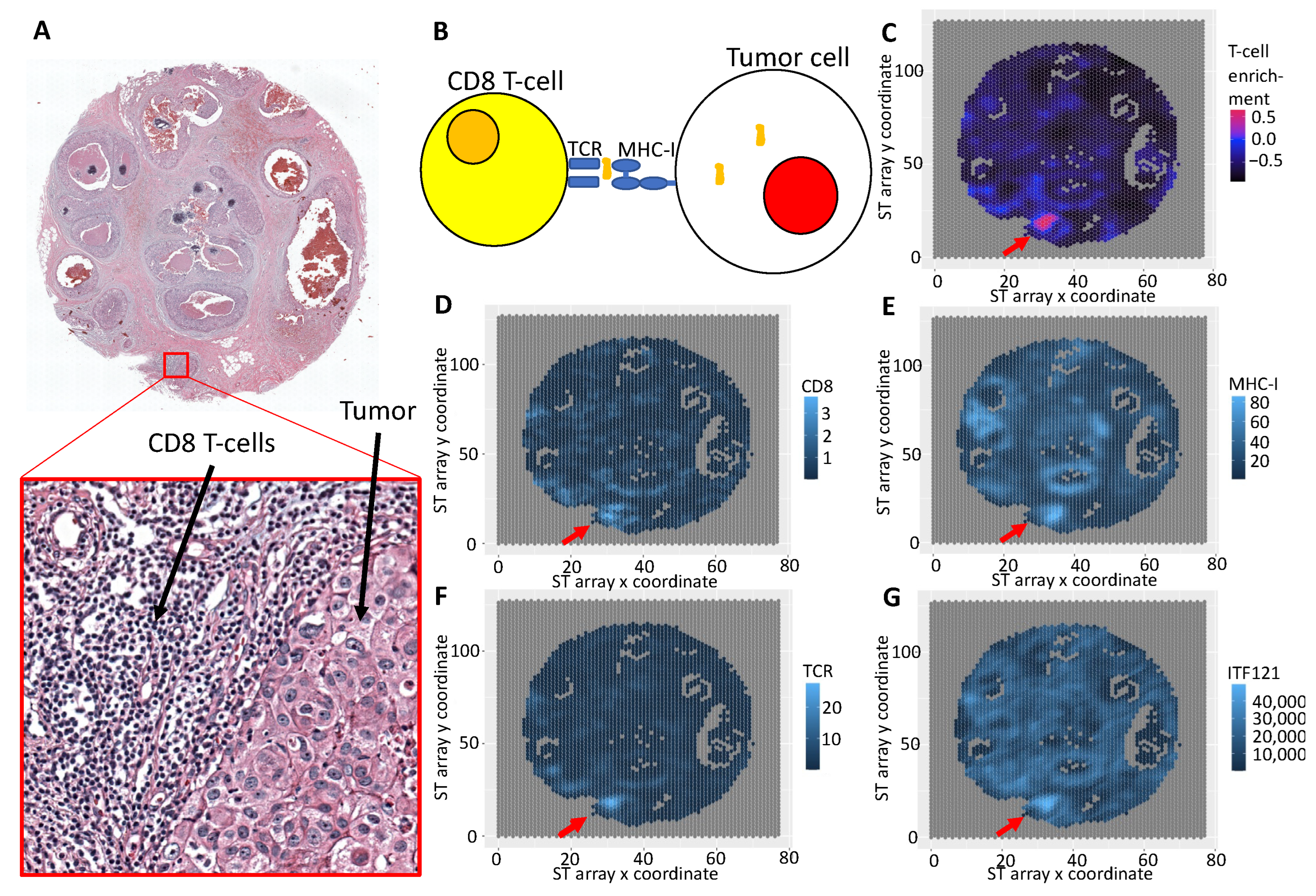
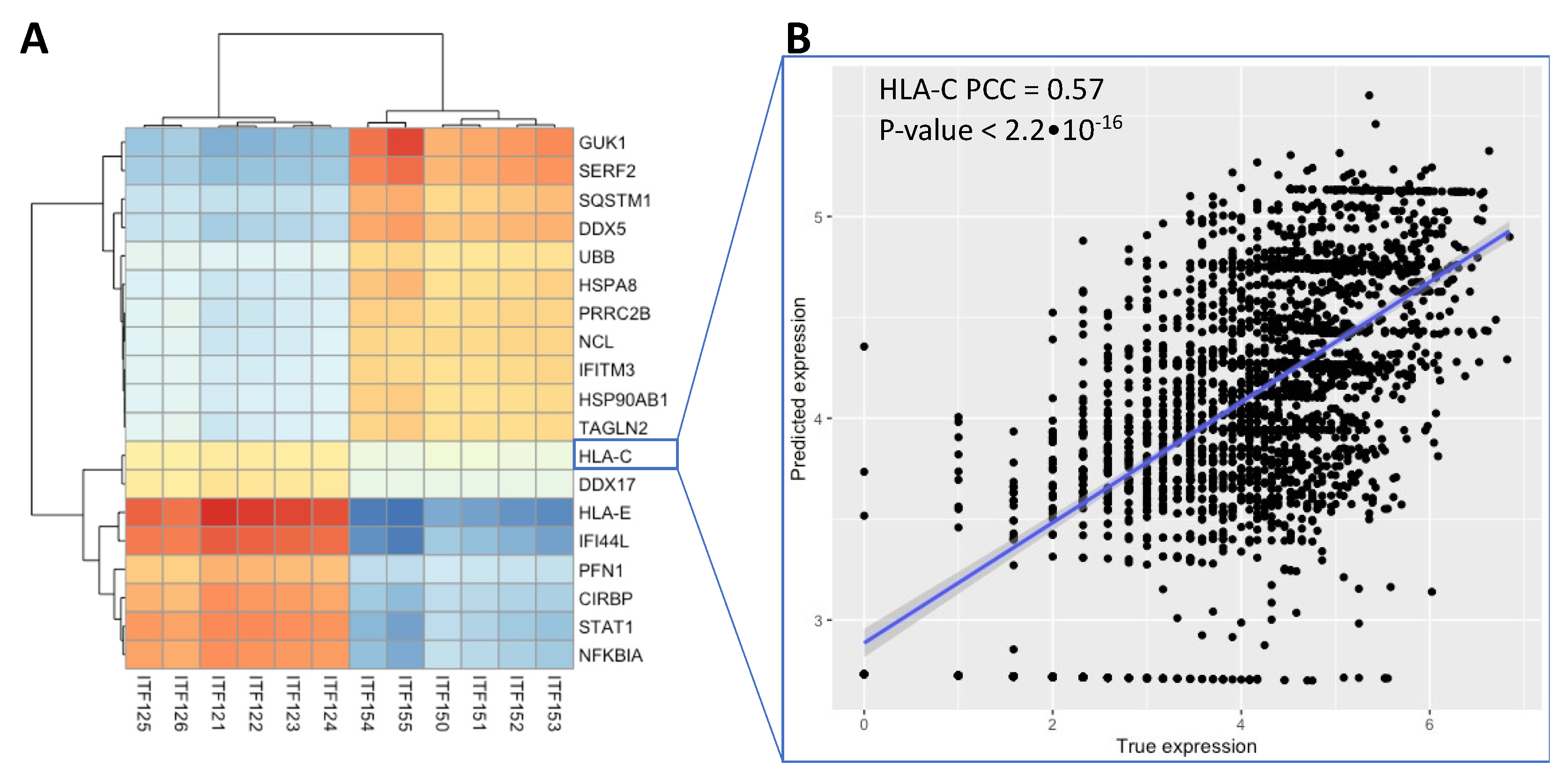
3.3. Integrative Analysis Reveals Immune Signaling in the FFPE Human Breast Cancer Slide
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- NCI. Public Health Research and Cancer; National Cancer Institute: Bethesda, MD, USA, 2020.
- WHO. Cancer; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- Henke, E.; Nandigama, R.; Ergün, S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front. Mol. Biosci. 2020, 6, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brassart-Pasco, S.; Brézillon, S.; Brassart, B.; Ramont, L.; Oudart, J.-B.; Monboisse, J.C. Tumor Microenvironment: Extracellular Matrix Alterations Influence Tumor Progression. Front. Oncol. 2020, 10, 397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Z.; Xu, S.; Shao, W.; Wu, Y.; Zhang, J.; Han, Z.; Feng, Q.; Huang, K. Deep-Learning–Based Characterization of Tumor-Infiltrating Lymphocytes in Breast Cancers From Histopathology Images and Multiomics Data. JCO Clin. Cancer Inform. 2020, 4, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, G. Topology and data. Bull. Am. Math. Soc. 2009, 46, 255–308. [Google Scholar] [CrossRef] [Green Version]
- Edelsbrunner, H.; Harer, J.L. Computational Topology: An Introduction; American Mathematical Society: Providence, RI, USA, 2022. [Google Scholar]
- Loughrey, C.F.; Fitzpatrick, P.; Orr, N.; Jurek-Loughrey, A. The topology of data: Opportunities for cancer research. Bioinformatics 2021, 37, 3091–3098. [Google Scholar] [CrossRef]
- Singer, J.; Irmisch, A.; Ruscheweyh, H.-J.; Singer, F.; Toussaint, N.C.; Levesque, M.P.; Stekhoven, D.J.; Beerenwinkel, N. Bioinformatics for precision oncology. Brief. Bioinform. 2019, 20, 778–788. [Google Scholar] [CrossRef] [Green Version]
- Aukerman, A.; Carrière, M.; Chen, C.; Gardner, K.; Rabadán, R.; Vanguri, R. Persistent homology based characterization of the breast cancer immune microenvironment: A feasibility study. In Proceedings of the 36th International Symposium on Computational Geometry (SoCG), Zürich, Switzerland, 23–26 June 2020. [Google Scholar]
- Lawson, P.; Sholl, A.B.; Brown, J.Q.; Fasy, B.T.; Wenk, C. Persistent Homology for the Quantitative Evaluation of Architectural Features in Prostate Cancer Histology. Sci. Rep. 2019, 9, 1139. [Google Scholar] [CrossRef] [Green Version]
- Bergenstråhle, L.; He, B.; Bergenstråhle, J.; Abalo, X.; Mirzazadeh, R.; Thrane, K.; Ji, A.L.; Andersson, A.; Larsson, L.; Stakenborg, N.; et al. Super-resolved spatial transcriptomics by deep data fusion. Nat. Biotechnol. 2022, 40, 476–479. [Google Scholar] [CrossRef]
- Chelebian, E.; Avenel, C.; Kartasalo, K.; Marklund, M.; Tanoglidi, A.; Mirtti, T.; Colling, R.; Erickson, A.; Lamb, A.D.; Lundeberg, J.; et al. Morphological Features Extracted by AI Associated with Spatial Transcriptomics in Prostate Cancer. Cancers 2021, 13, 4837. [Google Scholar] [CrossRef]
- Ash, J.T.; Darnell, G.; Munro, D.; Engelhardt, B.E. Joint analysis of expression levels and histological images identifies genes associated with tissue morphology. Nat. Commun. 2021, 12, 1609. [Google Scholar] [CrossRef]
- He, B.; Bergenstråhle, L.; Stenbeck, L.; Abid, A.; Andersson, A.; Borg, Å.; Maaskola, J.; Lundeberg, J.; Zou, J. Integrating spatial gene expression and breast tumour morphology via deep learning. Nat. Biomed. Eng. 2020, 4, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ye, X.; Yu, C.Y.; Shao, W.; Hou, J.; Feng, W.; Zhang, J.; Huang, K. TPSC: A module detection method based on topology potential and spectral clustering in weighted networks and its application in gene co-expression module discovery. BMC Bioinform. 2021, 22, 111. [Google Scholar] [CrossRef] [PubMed]
- Thanati, F.; Karatzas, E.; Baltoumas, F.; Stravopodis, D.; Eliopoulos, A.; Pavlopoulos, G. FLAME: A Web Tool for Functional and Literature Enrichment Analysis of Multiple Gene Lists. Biology 2021, 10, 665. [Google Scholar] [CrossRef] [PubMed]
- Kolde, R. Pheatmap: Pretty Heatmaps; CRAN: Vienna, Austria, 2019. [Google Scholar]
- GUDHI. GUDHI Library for TDA. 2021. Available online: https://gudhi.inria.fr/ (accessed on 1 September 2022).
- Levy, J.; Haudenschild, C.; Barwick, C.; Christensen, B.; Vaickus, L. Topological Feature Extraction and Visualization of Whole Slide Images using Graph Neural Networks. In Biocomputing 2021, Proceedings of the Pacific Symposium, Kohala Coast, HI, USA, 3–7 January 2021; World Scientific: Singapore, 2021. [Google Scholar]
- Clustering Metrics Better than the Elbow-Method. Available online: https://towardsdatascience.com/clustering-metrics-better-than-the-elbow-method-6926e1f723a6 (accessed on 1 September 2022).
- Kolberg, L.; Raudvere, U.; Kuzmin, I.; Vilo, J.; Peterson, H. Gprofiler2—An R package for gene list functional enrichment analysis and namespace conversion toolset g: Profiler. F1000Research 2020, 9, ELIXIR-709. [Google Scholar] [CrossRef]
- Johnson, T.S.; Yu, C.Y.; Huang, Z.; Xu, S.; Wang, T.; Dong, C.; Shao, W.; Zaid, M.A.; Huang, X.; Wang, Y.; et al. Diagnostic Evidence GAuge of Single cells (DEGAS): A flexible deep transfer learning framework for prioritizing cells in relation to disease. Genome Med. 2022, 14, 11. [Google Scholar] [CrossRef]
- Karaayvaz, M.; Cristea, S.; Gillespie, S.M.; Patel, A.P.; Mylvaganam, R.; Luo, C.C.; Specht, M.C.; Bernstein, B.E.; Michor, F.; Ellisen, L.W. Unravelling subclonal heterogeneity and aggressive disease states in TNBC through single-cell RNA-seq. Nat. Commun. 2018, 9, 3588. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Guestrin, C. Xgboost: A scalable tree boosting system. In Proceedings of the 22nd Acm Sigkdd International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016. [Google Scholar]
- Ke, G.; Meng, Q.; Finley, T.; Wang, T.; Chen, W.; Ma, W.; Ye, Q.; Liu, T.-Y. Lightgbm: A highly efficient gradient boosting decision tree. In Proceedings of the Advances in Neural Information Processing Systems 30, Long Beach, CA, USA, 4–9 December 2017. [Google Scholar]
- Darvishian, F.; Ozerdem, U.; Adams, S.; Chun, J.; Pirraglia, E.; Kaplowitz, E.; Guth, A.; Axelrod, D.; Shapiro, R.; Price, A.; et al. Tumor-Infiltrating Lymphocytes in a Contemporary Cohort of Women with Ductal Carcinoma In Situ (DCIS). Ann. Surg. Oncol. 2019, 26, 3337–3343. [Google Scholar] [CrossRef]
- Acs, B.; Ahmed, F.S.; Gupta, S.; Wong, P.F.; Gartrell, R.D.; Pradhan, J.S.; Rizk, E.M.; Rothberg, B.G.; Saenger, Y.; Rimm, D.L. An open source automated tumor infiltrating lymphocyte algorithm for prognosis in melanoma. Nat. Commun. 2019, 10, 5440. [Google Scholar] [CrossRef] [Green Version]
- Failmezger, H.; Muralidhar, S.; Rullan, A.; de Andrea, C.E.; Sahai, E.; Yuan, Y. Topological Tumor Graphs: A Graph-Based Spatial Model to Infer Stromal Recruitment for Immuno-suppression in Melanoma Histology. Cancer Res 2020, 80, 1199–1209. [Google Scholar] [CrossRef] [Green Version]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef]
- National Cancer Institute. What Is Cancer? National Cancer Institute: Bethesda, MD, USA, 2021.
- Nallanthighal, S.; Heiserman, J.P.; Cheon, D.-J. The Role of the Extracellular Matrix in Cancer Stemness. Front. Cell Dev. Biol. 2019, 7, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Zheng, X.; Zheng, Y.; Chen, Y.; Fei, W.; Wang, F.; Zheng, C. Extracellular Matrix: Emerging Roles and Potential Therapeutic Targets for Breast Cancer. Front. Oncol. 2021, 11, 650453. [Google Scholar] [CrossRef] [PubMed]
- Vlaeminck-Guillem, V. Extracellular Vesicles in Prostate Cancer Carcinogenesis, Diagnosis, and Management. Front. Oncol. 2018, 8, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Xu, H.; Wang, W.; Li, S.; Li, H.; Li, T.; Zhang, W.; Yu, X.; Liu, L. The role of collagen in cancer: From bench to bedside. J. Transl. Med. 2019, 17, 309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, J.A.F.; Biancardi, M.F.; Stach-Machado, D.R.; Reis, L.O.; Sant’Anna, O.A.; Carvalho, H.F. The origin of prostate gland-secreted IgA and IgG. Sci. Rep. 2017, 7, 16488. [Google Scholar] [CrossRef] [Green Version]
- Cui, M.; Huang, J.; Zhang, S.; Liu, Q.; Liao, Q.; Qiu, X. Immunoglobulin Expression in Cancer Cells and Its Critical Roles in Tumorigenesis. Front. Immunol. 2021, 12, 613530. [Google Scholar] [CrossRef]
- Zhong, Z.; Nan, K.; Weng, M.; Yue, Y.; Zhou, W.; Wang, Z.; Chu, Y.; Liu, R.; Miao, C. Pro- and Anti- Effects of Immunoglobulin A- Producing B Cell in Tumors and Its Triggers. Front. Immunol. 2021, 12, 765044. [Google Scholar] [CrossRef]
- Cao, R.; Ke, M.; Wu, Q.; Tian, Q.; Liu, L.; Dai, Z.; Lu, S.; Liu, P. AZGP1 is androgen responsive and involved in AR-induced prostate cancer cell proliferation and metastasis. J. Cell Physiol. 2019, 234, 17444–17458. [Google Scholar] [CrossRef]
- Edechi, C.A.; Ikeogu, N.M.; Akaluka, G.N.; Terceiro, L.E.L.; Machado, M.; Salako, E.S.; Barazandeh, A.F.; Kung, S.K.P.; Uzonna, J.E.; Myal, Y. The Prolactin Inducible Protein Modulates Antitumor Immune Responses and Metastasis in a Mouse Model of Triple Negative Breast Cancer. Front. Oncol. 2021, 11, 639859. [Google Scholar] [CrossRef]
- Blanchard, A.; Nistor, A.; Castaneda, F.; Martin, D.; Hicks, G.; Amara, F.; Shiu, R.; Myal, Y. Generation and initial characterization of the prolactin-inducible protein (PIP) null mouse: Accompanying global changes in gene expression in the submandibular gland. Can. J. Physiol. Pharmacol. 2009, 87, 859–872. [Google Scholar] [CrossRef]
- Cheng, J.; Han, Z.; Mehra, R.; Shao, W.; Cheng, M.; Feng, Q.; Ni, D.; Huang, K.; Cheng, L.; Zhang, J. Computational analysis of pathological images enables a better diagnosis of TFE3 Xp11.2 translocation renal cell carcinoma. Nat. Commun. 2020, 11, 1778. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Zhang, J.; Han, Y.; Wang, X.; Ye, X.; Meng, Y.; Parwani, A.; Han, Z.; Feng, Q.; Huang, K. Integrative Analysis of Histopathological Images and Genomic Data Predicts Clear Cell Renal Cell Carcinoma Prognosis. Cancer Res. 2017, 77, e91–e100. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alsaleh, L.; Li, C.; Couetil, J.L.; Ye, Z.; Huang, K.; Zhang, J.; Chen, C.; Johnson, T.S. Spatial Transcriptomic Analysis Reveals Associations between Genes and Cellular Topology in Breast and Prostate Cancers. Cancers 2022, 14, 4856. https://doi.org/10.3390/cancers14194856
Alsaleh L, Li C, Couetil JL, Ye Z, Huang K, Zhang J, Chen C, Johnson TS. Spatial Transcriptomic Analysis Reveals Associations between Genes and Cellular Topology in Breast and Prostate Cancers. Cancers. 2022; 14(19):4856. https://doi.org/10.3390/cancers14194856
Chicago/Turabian StyleAlsaleh, Lujain, Chen Li, Justin L. Couetil, Ze Ye, Kun Huang, Jie Zhang, Chao Chen, and Travis S. Johnson. 2022. "Spatial Transcriptomic Analysis Reveals Associations between Genes and Cellular Topology in Breast and Prostate Cancers" Cancers 14, no. 19: 4856. https://doi.org/10.3390/cancers14194856