
Novel Therapies for Unmet Clinical Needs in Myelodysplastic Syndromes


Abstract
:Simple Summary
Abstract
1. Introduction
2. Diagnosis and Classification and Their Drawbacks
3. Unmet Clinical Needs of Current Therapies in Lower-Risk Patients
4. Unmet Clinical Needs and Current Therapies in Higher-Risk Patients
5. New Approaches for Low-Risk MDS Patients
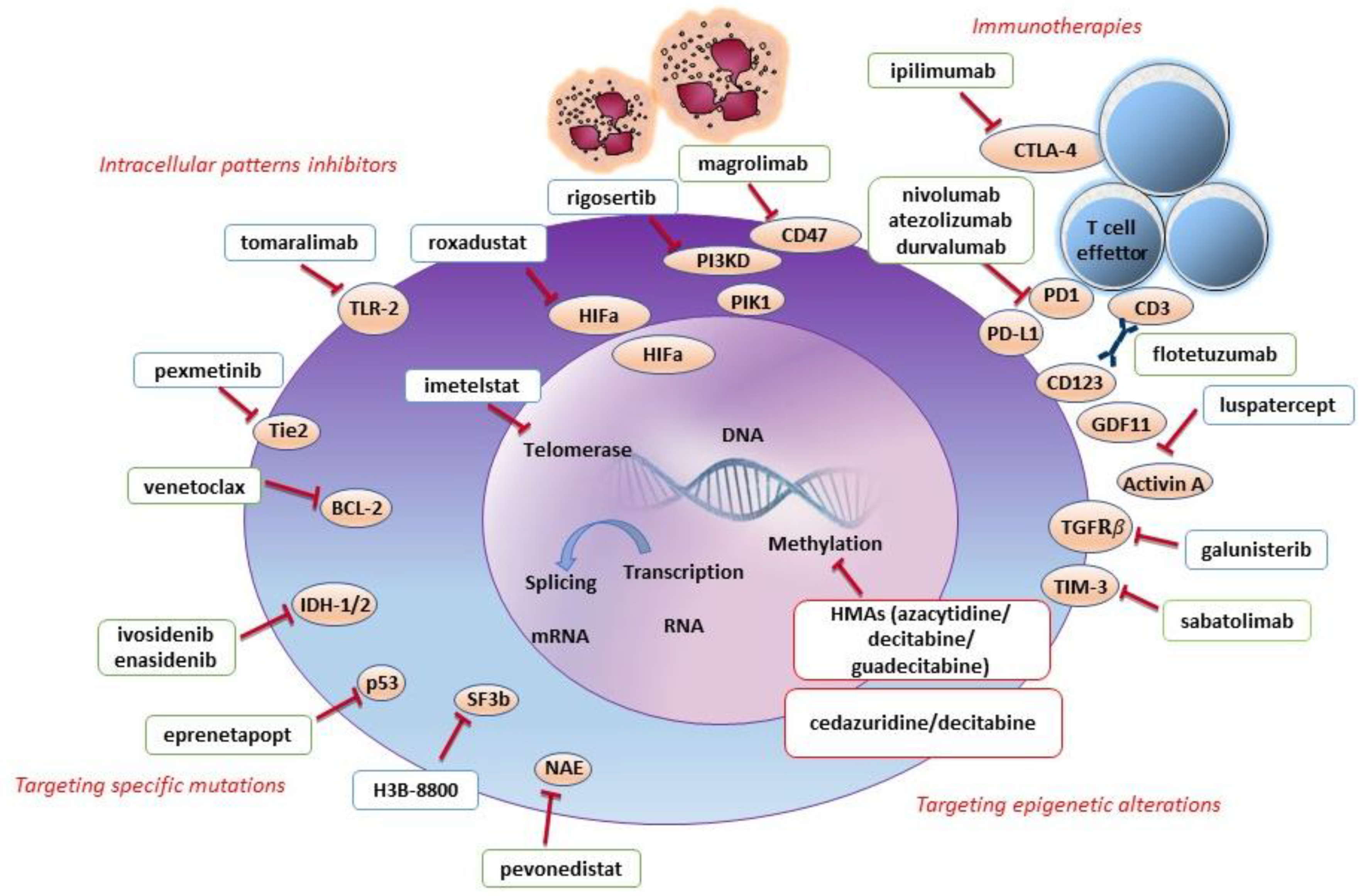
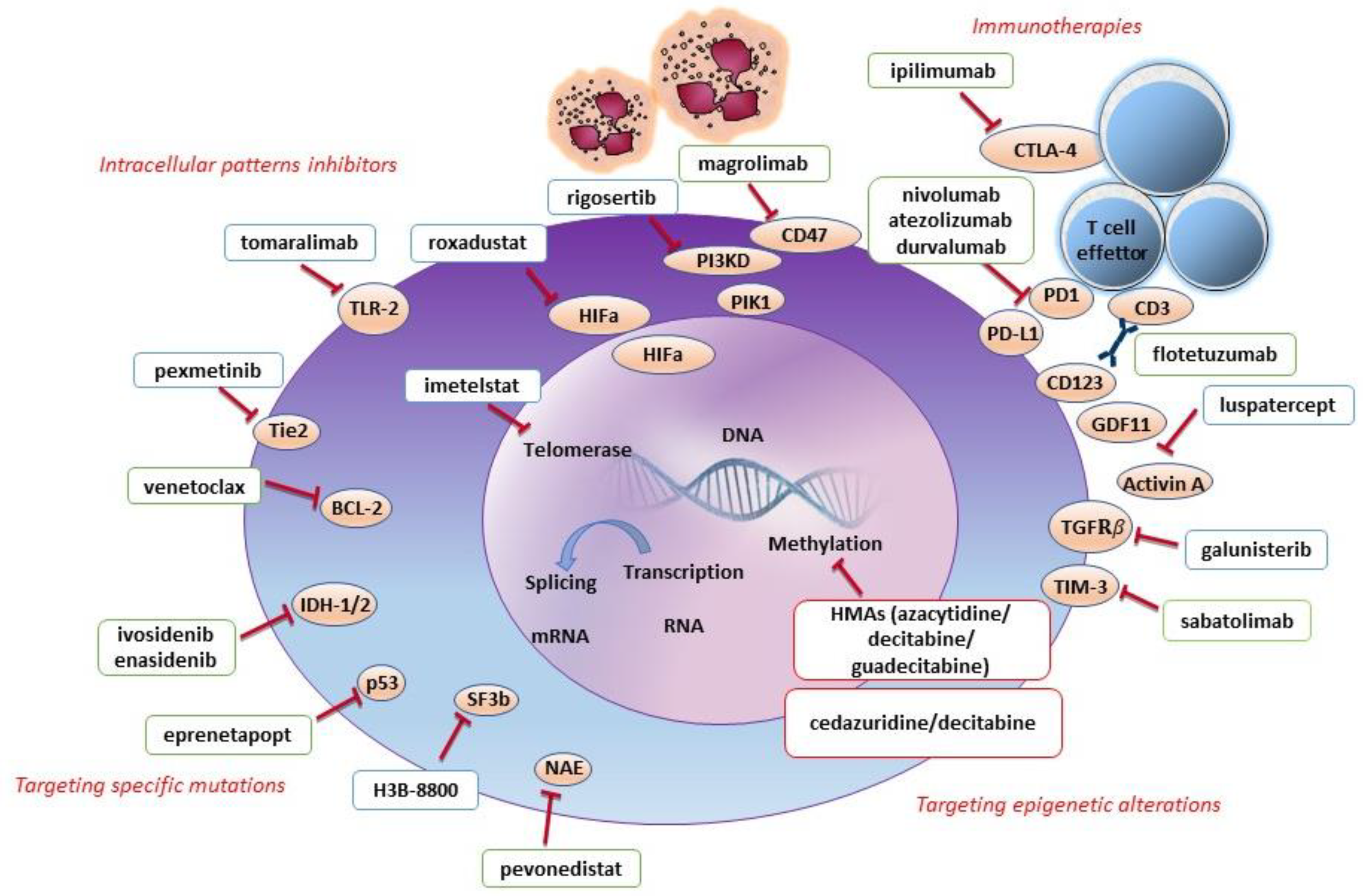
5.1. Targeting Epigenetic Alterations
5.2. Targeting Intracellular Pathways
5.3. Targeting Immunity
5.4. Targeting Specific Mutations
6. New Approaches for High-Risk MDS Patients
6.1. Targeting Epigenetic Alterations
6.2. Targeting Intracellular Pathways
6.3. Targeting Immunity
6.4. Targeting Specific Mutations
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Will, B.; Zhou, L.; Vogler, T.O.; Ben-Neriah, S.; Schinke, C.; Tamari, R.; Yu, Y.; Bhagat, T.D.; Bhattacharyya, S.; Barreyro, L.; et al. Stem and Progenitor Cells in Myelodysplastic Syndromes Show Aberrant Stage-Specific Expansion and Harbor Genetic and Epigenetic Alterations. Blood 2012, 120, 2076–2086. [Google Scholar] [CrossRef]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical Effect of Point Mutations in Myelodysplastic Syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Qian, Y.; Eksioglu, E.; Epling-Burnette, P.K.; Wei, S. The Inflammatory Microenvironment in MDS. Cell. Mol. Life Sci. 2015, 72, 1959–1966. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Dunbar, A.; Gondek, L.P.; Mohan, S.; Rataul, M.; O’Keefe, C.; Sekeres, M.; Saunthararajah, Y.; Maciejewski, J.P. Aberrant DNA Methylation Is a Dominant Mechanism in MDS Progression to AML. Blood 2009, 113, 1315–1325. [Google Scholar] [CrossRef] [PubMed]
- Neukirchen, J.; Schoonen, W.M.; Strupp, C.; Gattermann, N.; Aul, C.; Haas, R.; Germing, U. Incidence and Prevalence of Myelodysplastic Syndromes: Data from the Düsseldorf MDS-Registry. Leuk. Res. 2011, 35, 1591–1596. [Google Scholar] [CrossRef] [PubMed]
- Adès, L.; Itzykson, R.; Fenaux, P. Myelodysplastic Syndromes. Lancet 2014, 383, 2239–2252. [Google Scholar] [CrossRef]
- Greenberg, P.; Cox, C.; LeBeau, M.M.; Fenaux, P.; Morel, P.; Sanz, G.; Sanz, M.; Vallespi, T.; Hamblin, T.; Oscier, D.; et al. International Scoring System for Evaluating Prognosis in Myelodysplastic Syndromes. Blood 1997, 89, 2079–2088. [Google Scholar] [CrossRef]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Solé, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised International Prognostic Scoring System for Myelodysplastic Syndromes. Blood 2012, 120, 2454–2465. [Google Scholar] [CrossRef] [Green Version]
- Fattizzo, B.; Levati, G.V.; Giannotta, J.A.; Cassanello, G.; Cro, L.M.; Zaninoni, A.; Barbieri, M.; Croci, G.A.; Revelli, N.; Barcellini, W. Low-Risk Myelodysplastic Syndrome Revisited: Morphological, Autoimmune, and Molecular Features as Predictors of Outcome in a Single Center Experience. Front. Oncol. 2022, 12, 891. [Google Scholar] [CrossRef]
- Bersanelli, M.; Travaglino, E.; Meggendorfer, M.; Matteuzzi, T.; Sala, C.; Mosca, E.; Chiereghin, C.; Di Nanni, N.; Gnocchi, M.; Zampini, M.; et al. Classification and Personalized Prognostic Assessment on the Basis of Clinical and Genomic Features in Myelodysplastic Syndromes. J. Clin. Oncol. 2021, 39, 1223–1233. [Google Scholar] [CrossRef]
- Bernard, E.; Tuechler, H.; Greenberg, P.L.; Hasserjian, R.P.; Ossa, J.E.A.; Nannya, Y.; Devlin, S.M.; Creignou, M.; Pinel, P.; Monnier, L.; et al. Molecular International Prognostic Scoring System for Myelodysplastic Syndromes. NEJM Evid. 2022, 1, EVIDoa2200008. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Malcovati, L.; Karimi, M.; Papaemmanuil, E.; Ambaglio, I.; Jädersten, M.; Jansson, M.; Elena, C.; Gallì, A.; Walldin, G.; Porta, M.G.D.; et al. SF3B1 Mutation Identifies a Distinct Subset of Myelodysplastic Syndrome with Ring Sideroblasts. Blood 2015, 126, 233–241. [Google Scholar] [CrossRef] [Green Version]
- Durrani, J.; Maciejewski, J.P. Idiopathic Aplastic Anemia vs. Hypocellular Myelodysplastic Syndrome. Hematology 2019, 2019, 97–104. [Google Scholar] [CrossRef]
- Buesche, G.; Teoman, H.; Wilczak, W.; Ganser, A.; Hecker, H.; Wilkens, L.; Göhring, G.; Schlegelberger, B.; Bock, O.; Georgii, A.; et al. Marrow Fibrosis Predicts Early Fatal Marrow Failure in Patients with Myelodysplastic Syndromes. Leukemia 2008, 22, 313–322. [Google Scholar] [CrossRef] [Green Version]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th Edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal Hematopoiesis of Indeterminate Potential and Its Distinction from Myelodysplastic Syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Shlush, L.I. Age-Related Clonal Hematopoiesis. Blood 2018, 131, 496–504. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, J.A.; Ebert, B.L. Clinical Implications of Genetic Mutations in Myelodysplastic Syndrome. J. Clin. Oncol. 2017, 35, 968–974. [Google Scholar] [CrossRef] [Green Version]
- Hellström-Lindberg, E.; Gulbrandsen, N.; Lindberg, G.; Ahlgren, T.; Dahl, I.M.S.; Dybedal, I.; Grimfors, G.; Hesse-Sundin, E.; Hjorth, M.; Kanter-Lewensohn, L.; et al. A Validated Decision Model for Treating the Anaemia of Myelodysplastic Syndromes with Erythropoietin + Granulocyte Colony-Stimulating Factor: Significant Effects on Quality of Life. Br. J. Haematol. 2003, 120, 1037–1046. [Google Scholar] [CrossRef]
- Fenaux, P.; Santini, V.; Spiriti, M.A.A.; Giagounidis, A.; Schlag, R.; Radinoff, A.; Gercheva-Kyuchukova, L.; Anagnostopoulos, A.; Oliva, E.N.; Symeonidis, A.; et al. A Phase 3 Randomized, Placebo-Controlled Study Assessing the Efficacy and Safety of Epoetin-α in Anemic Patients with Low-Risk MDS. Leukemia 2018, 32, 2648. [Google Scholar] [CrossRef] [Green Version]
- Cermak, J.; Kacirkova, P.; Mikulenkova, D.; Michalova, K. Impact of Transfusion Dependency on Survival in Patients with Early Myelodysplastic Syndrome without Excess of Blasts. Leuk. Res. 2009, 33, 1469–1474. [Google Scholar] [CrossRef]
- Fattizzo, B.; Rizzo, L.; Giannotta, J.A.; Mazzon, F.; Cecchi, N.; Frangi, C.; Barcellini, W.; Riva, M. Switching to an Alternative Recombinant Erythropoietin Agent in Patients with Myelodysplastic Syndromes: A Second Honeymoon? Br. J. Haematol. 2021, 195, e147–e150. [Google Scholar] [CrossRef]
- Kelaidi, C.; Park, S.; Brechignac, S.; Mannone, L.; Vey, N.; Dombret, H.; Aljassem, L.; Stamatoullas, A.; Adès, L.; Giraudier, S.; et al. Treatment of Myelodysplastic Syndromes with 5q Deletion before the Lenalidomide Era; the GFM Experience with EPO and Thalidomide. Leuk. Res. 2008, 32, 1049–1053. [Google Scholar] [CrossRef]
- Santini, V.; Almeida, A.; Giagounidis, A.; Gröpper, S.; Jonasova, A.; Vey, N.; Mufti, G.J.; Buckstein, R.; Mittelman, M.; Platzbecker, U.; et al. Randomized Phase III Study of Lenalidomide Versus Placebo in RBC Transfusion-Dependent Patients with Lower-Risk Non-Del(5q) Myelodysplastic Syndromes and Ineligible for or Refractory to Erythropoiesis-Stimulating Agents. J. Clin. Oncol. 2016, 34, 2988–2996. [Google Scholar] [CrossRef]
- Jädersten, M.; Saft, L.; Smith, A.; Kulasekararaj, A.; Pomplun, S.; Göhring, G.; Hedlund, A.; Hast, R.; Schlegelberger, B.; Porwit, A.; et al. TP53 Mutations in Low-Risk Myelodysplastic Syndromes with Del(5q) Predict Disease Progression. J. Clin. Oncol. 2011, 29, 1971–1979. [Google Scholar] [CrossRef]
- Bewersdorf, J.P.; Zeidan, A.M. Transforming Growth Factor (TGF)-β Pathway as a Therapeutic Target in Lower Risk Myelodysplastic Syndromes. Leukemia 2019, 33, 1303–1312. [Google Scholar] [CrossRef]
- Fenaux, P.; Platzbecker, U.; Mufti, G.J.; Garcia-Manero, G.; Buckstein, R.; Santini, V.; Díez-Campelo, M.; Finelli, C.; Cazzola, M.; Ilhan, O.; et al. Luspatercept in Patients with Lower-Risk Myelodysplastic Syndromes. N. Engl. J. Med. 2020, 382, 140–151. [Google Scholar] [CrossRef]
- Stahl, M.; DeVeaux, M.; De Witte, T.; Neukirchen, J.; Sekeres, M.A.; Brunner, A.M.; Roboz, G.J.; Steensma, D.P.; Bhatt, V.R.; Platzbecker, U.; et al. The Use of Immunosuppressive Therapy in MDS: Clinical Outcomes and Their Predictors in a Large International Patient Cohort. Blood Adv. 2018, 2, 1765–1772. [Google Scholar] [CrossRef] [Green Version]
- Parikh, A.R.; Olnes, M.J.; Barrett, A.J. Immunomodulatory Treatment of Myelodysplastic Syndromes: Antithymocyte Globulin, Cyclosporine, and Alemtuzumab. Semin. Hematol. 2012, 49, 304–311. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Giles, F.J.; Greenberg, P.L.; Paquette, R.L.; Wang, E.S.; Gabrilove, J.L.; Garcia-Manero, G.; Hu, K.; Franklin, J.L.; Berger, D.P. Phase 2 Study of Romiplostim in Patients with Low- or Intermediate-Risk Myelodysplastic Syndrome Receiving Azacitidine Therapy. Blood 2010, 116, 3163–3170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantarjian, H.M.; Fenaux, P.; Sekeres, M.A.; Szer, J.; Platzbecker, U.; Kuendgen, A.; Gaidano, G.; Wiktor-Jedrzejczak, W.; Carpenter, N.; Mehta, B.; et al. Long-Term Follow-up for up to 5 Years on the Risk of Leukaemic Progression in Thrombocytopenic Patients with Lower-Risk Myelodysplastic Syndromes Treated with Romiplostim or Placebo in a Randomised Double-Blind Trial. Lancet Haematol. 2018, 5, e117–e126. [Google Scholar] [CrossRef]
- Oliva, E.N.; Alati, C.; Santini, V.; Poloni, A.; Molteni, A.; Niscola, P.; Salvi, F.; Sanpaolo, G.; Balleari, E.; Germing, U.; et al. Eltrombopag versus Placebo for Low-Risk Myelodysplastic Syndromes with Thrombocytopenia (EQoL-MDS): Phase 1 Results of a Single-Blind, Randomised, Controlled, Phase 2 Superiority Trial. Lancet. Haematol. 2017, 4, e127–e136. [Google Scholar] [CrossRef]
- Colunga-Pedraza, P.R.; Colunga-Pedraza, J.E.; Garza-Ledezma, M.A.; Jaime-Pérez, J.C.; Cantú-Rodríguez, O.G.; Gutiérrez-Aguirre, C.H.; Rendón-Ramírez, E.J.; López-García, Y.K.; Lozano-Morales, R.E.; Gómez-De León, A.; et al. Danazol as First-Line Therapy for Myelodysplastic Syndrome. Clin. Lymphoma. Myeloma Leuk. 2018, 18, e109–e113. [Google Scholar] [CrossRef]
- Qu, W.Y.; Zhao, L.; Tan, X.C.; Zhao, Y.H. Stanozolol for the Treatment of Anemic Lower-Risk Myelodysplastic Syndromes without Del(5q) after Failure of Epoetin Alfa: Findings from a Retrospective Study. Ann. Hematol. 2021, 100, 1451–1457. [Google Scholar] [CrossRef]
- Angelucci, E.; Li, J.; Greenberg, P.; Wu, D.; Hou, M.; Figueroa, E.H.M.; Rodriguez, M.G.; Dong, X.; Ghosh, J.; Izquierdo, M.; et al. Iron Chelation in Transfusion-Dependent Patients with Low- To Intermediate-1-Risk Myelodysplastic Syndromes: A Randomized Trial. Ann. Intern. Med. 2020, 172, 513–522. [Google Scholar] [CrossRef]
- Temraz, S.; Santini, V.; Musallam, K.; Taher, A. Iron Overload and Chelation Therapy in Myelodysplastic Syndromes. Crit. Rev. Oncol. Hematol. 2014, 91, 64–73. [Google Scholar] [CrossRef]
- Janusz, K.; Izquierdo, M.M.; Cadenas, F.L.; Ramos, F.; Sánchez, J.M.H.; Lumbreras, E.; Robledo, C.; del Real, J.S.; Caballero, J.C.; Collado, R.; et al. Clinical, Biological, and Prognostic Implications of SF3B1 Co-Occurrence Mutations in Very Low/Low- and Intermediate-Risk MDS Patients. Ann. Hematol. 2021, 100, 1995–2004. [Google Scholar] [CrossRef]
- Fenaux, P.; Mufti, G.J.; Hellstrom-Lindberg, E.; Santini, V.; Finelli, C.; Giagounidis, A.; Schoch, R.; Gattermann, N.; Sanz, G.; List, A.; et al. Efficacy of Azacitidine Compared with That of Conventional Care Regimens in the Treatment of Higher-Risk Myelodysplastic Syndromes: A Randomised, Open-Label, Phase III Study. Lancet. Oncol. 2009, 10, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Lübbert, M.; Suciu, S.; Baila, L.; Rüter, B.H.; Platzbecker, U.; Giagounidis, A.; Selleslag, D.; Labar, B.; Germing, U.; Salih, H.R.; et al. Low-Dose Decitabine versus Best Supportive Care in Elderly Patients with Intermediate- or High-Risk Myelodysplastic Syndrome (MDS) Ineligible for Intensive Chemotherapy: Final Results of the Randomized Phase III Study of the European Organisation for Research and Treatment of Cancer Leukemia Group and the German MDS Study Group. J. Clin. Oncol. 2011, 29, 1987–1996. [Google Scholar] [CrossRef]
- De Witte, T.; Bowen, D.; Robin, M.; Malcovati, L.; Niederwieser, D.; Yakoub-Agha, I.; Mufti, G.J.; Fenaux, P.; Sanz, G.; Martino, R.; et al. Allogeneic Hematopoietic Stem Cell Transplantation for MDS and CMML: Recommendations from an International Expert Panel. Blood 2017, 129, 1753–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santini, V. How I Treat MDS after Hypomethylating Agent Failure. Blood 2019, 133, 521–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apuri, S.; Al Ali, N.; Padron, E.; Lancet, J.E.; List, A.F.; Komrokji, R.S. Evidence for Selective Benefit of Sequential Treatment with Hypomethylating Agents in Patients with Myelodysplastic Syndrome. Clin. Lymphoma. Myeloma Leuk. 2017, 17, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Saber, W.; Cutler, C.S.; Nakamura, R.; Zhang, M.J.; Atallah, E.; Rizzo, J.D.; Maziarz, R.T.; Cortes, J.; Kalaycio, M.E.; Horowitz, M.M. Impact of Donor Source on Hematopoietic Cell Transplantation Outcomes for Patients with Myelodysplastic Syndromes (MDS). Blood 2013, 122, 1974. [Google Scholar] [CrossRef] [Green Version]
- Kindwall-Keller, T.; Isola, L.M. The Evolution of Hematopoietic SCT in Myelodysplastic Syndrome. Bone Marrow Transplant. 2009, 43, 597–609. [Google Scholar] [CrossRef]
- Mendez, L.M.; Posey, R.R.; Pandolfi, P.P. The Interplay between the Genetic and Immune Landscapes of AML: Mechanisms and Implications for Risk Stratification and Therapy. Front. Oncol. 2019, 9, 1162. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Yang, Y.; Gao, S.; Chen, J.; Yu, J.; Zhang, H.; Li, M.; Zhan, X.; Li, W. Immune Dysregulation in Myelodysplastic Syndrome: Clinical Features, Pathogenesis and Therapeutic Strategies. Crit. Rev. Oncol. Hematol. 2018, 122, 123–132. [Google Scholar] [CrossRef]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of Genetic Lesions in 944 Patients with Myelodysplastic Syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and Biological Implications of Driver Mutations in Myelodysplastic Syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef] [Green Version]
- Della Porta, M.G.; Gallì, A.; Bacigalupo, A.; Zibellini, S.; Bernardi, M.; Rizzo, E.; Allione, B.; Van Lint, M.T.; Pioltelli, P.; Marenco, P.; et al. Clinical Effects of Driver Somatic Mutations on the Outcomes of Patients with Myelodysplastic Syndromes Treated With Allogeneic Hematopoietic Stem-Cell Transplantation. J. Clin. Oncol. 2016, 34, 3627–3637. [Google Scholar] [CrossRef]
- Lindsley, R.C.; Saber, W.; Mar, B.G.; Redd, R.; Wang, T.; Haagenson, M.D.; Grauman, P.V.; Hu, Z.-H.; Spellman, S.R.; Lee, S.J.; et al. Prognostic Mutations in Myelodysplastic Syndrome after Stem-Cell Transplantation. N. Engl. J. Med. 2017, 376, 536–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tobiasson, M.; Dybedahl, I.; Holm, M.S.; Karimi, M.; Brandefors, L.; Garelius, H.; Grövdal, M.; Högh-Dufva, I.; Grønbæk, K.; Jansson, M.; et al. Limited Clinical Efficacy of Azacitidine in Transfusion-Dependent, Growth Factor-Resistant, Low- and Int-1-Risk MDS: Results from the Nordic NMDSG08A Phase II Trial. Blood Cancer J. 2014, 4, e189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thépot, S.; Abdelali, R.B.; Chevret, S.; Renneville, A.; Beyne-Rauzy, O.; Prébet, T.; Park, S.; Stamatoullas, A.; Guerci-Bresler, A.; Cheze, S.; et al. A Randomized Phase II Trial of Azacitidine +/- Epoetin-β in Lower-Risk Myelodysplastic Syndromes Resistant to Erythropoietic Stimulating Agents. Haematologica 2016, 101, 918–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabbour, E.; Short, N.J.; Montalban-Bravo, G.; Huang, X.; Bueso-Ramos, C.; Qiao, W.; Yang, H.; Zhao, C.; Kadia, T.; Borthakur, G.; et al. Randomized Phase 2 Study of Low-Dose Decitabine vs. Low-Dose Azacitidine in Lower-Risk MDS and MDS/MPN. Blood 2017, 130, 1514–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Manero, G.; Santini, V.; Almeida, A.; Platzbecker, U.; Jonasova, A.; Silverman, L.R.; Falantes, J.; Reda, G.; Buccisano, F.; Fenaux, P.; et al. Phase III, Randomized, Placebo-Controlled Trial of CC-486 (Oral Azacitidine) in Patients with Lower-Risk Myelodysplastic Syndromes. J. Clin. Oncol. 2021, 39, 1426–1436. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; McCloskey, J.K.; Griffiths, E.A.; Yee, K.; Zeidan, A.M.; Al-Kali, A.; Deeg, H.J.; Patel, P.; Sabloff, M.; Keating, M.-M.; et al. Oral Decitabine/Cedazuridine in Patients with Lower Risk Myelodysplastic Syndrome: A Longer-Term Follow-up of from the Ascertain Study. Blood 2021, 138, 66. [Google Scholar] [CrossRef]
- Komrokji, R.; Garcia-Manero, G.; Ades, L.; Prebet, T.; Steensma, D.P.; Jurcic, J.G.; Sekeres, M.A.; Berdeja, J.; Savona, M.R.; Beyne-Rauzy, O.; et al. Sotatercept with Long-Term Extension for the Treatment of Anaemia in Patients with Lower-Risk Myelodysplastic Syndromes: A Phase 2, Dose-Ranging Trial. Lancet. Haematol. 2018, 5, e63–e72. [Google Scholar] [CrossRef]
- Santini, V.; Valcarcel, D.; Platzbecker, U.; Komrokji, R.S.; Cleverly, A.L.; Lahn, M.M.; Janssen, J.; Zhao, Y.; Chiang, A.; Giagounidis, A.; et al. Phase II Study of the ALK5 Inhibitor Galunisertib in Very Low-, Low-, and Intermediate-Risk Myelodysplastic Syndromes. Clin. Cancer Res. 2019, 25, 6976–6985. [Google Scholar] [CrossRef] [Green Version]
- Henry, D.H.; Glaspy, J.; Harrup, R.; Mittelman, M.; Zhou, A.; Carraway, H.E.; Bradley, C.; Saha, G.; Modelska, K.; Bartels, P.; et al. Roxadustat for the Treatment of Anemia in Patients with Lower-Risk Myelodysplastic Syndrome: Open-Label, Dose-Selection, Lead-in Stage of a Phase 3 Study. Am. J. Hematol. 2022, 97, 174–184. [Google Scholar] [CrossRef]
- Dong, W.; Wu, L.; Sun, H.; Ren, X.; Epling-Burnette, P.K.; Yang, L. MDS Shows a Higher Expression of HTERT and Alternative Splice Variants in Unactivated T-Cells. Oncotarget 2016, 7, 71904. [Google Scholar] [CrossRef]
- Steensma, D.P.; Fenaux, P.; van Eygen, K.; Raza, A.; Santini, V.; Germing, U.; Font, P.; Diez-Campelo, M.; Thepot, S.; Vellenga, E.; et al. Imetelstat Achieves Meaningful and Durable Transfusion Independence in High Transfusion-Burden Patients with Lower-Risk Myelodysplastic Syndromes in a Phase II Study. J. Clin. Oncol. 2021, 39, 48–56. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Khoury, H.J.; Jabbour, E.; Lancet, J.; Winski, S.L.; Cable, L.A.; Rush, S.; Maloney, L.; Hogeland, G.; Ptaszynski, M.; et al. A Phase I Study of Oral ARRY-614, a P38 MAPK/Tie2 Dual Inhibitor, in Patients with Low or Intermediate-1 Risk Myelodysplastic Syndromes. Clin. Cancer Res. 2015, 21, 985–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Mooij, C.E.M.; Netea, M.G.; Van Der Velden, W.J.F.M.; Blijlevens, N.M.A. Targeting the Interleukin-1 Pathway in Patients with Hematological Disorders. Blood 2017, 129, 3155–3164. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Manero, G.; Jabbour, E.J.; Konopleva, M.Y.; Daver, N.G.; Borthakur, G.; DiNardo, C.D.; Bose, P.; Patel, P.; Komrokji, R.S.; Shastri, A.; et al. A Clinical Study of Tomaralimab (OPN-305), a Toll-like Receptor 2 (TLR-2) Antibody, in Heavily Pre-Treated Transfusion Dependent Patients with Lower Risk Myelodysplastic Syndromes (MDS) That Have Received and Failed on Prior Hypomethylating Agent (HMA) Therapy. Blood 2018, 132, 798. [Google Scholar] [CrossRef]
- Steensma, D.P.; Wermke, M.; Klimek, V.M.; Greenberg, P.L.; Font, P.; Komrokji, R.S.; Yang, J.; Brunner, A.M.; Carraway, H.E.; Ades, L.; et al. Phase I First-in-Human Dose Escalation Study of the Oral SF3B1 Modulator H3B-8800 in Myeloid Neoplasms. Leukemia 2021, 35, 3542–3550. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G.; Roboz, G.; Walsh, K.; Kantarjian, H.; Ritchie, E.; Kropf, P.; O’Connell, C.; Tibes, R.; Lunin, S.; Rosenblat, T.; et al. Guadecitabine (SGI-110) in Patients with Intermediate or High-Risk Myelodysplastic Syndromes: Phase 2 Results from a Multicentre, Open-Label, Randomised, Phase 1/2 Trial. Lancet Haematol. 2019, 6, e317–e327. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; McCloskey, J.; Griffiths, E.A.; Yee, K.W.L.; Zeidan, A.M.; Al-Kali, A.; Dao, K.-H.; Deeg, H.J.; Patel, P.A.; Sabloff, M.; et al. Pharmacokinetic Exposure Equivalence and Preliminary Efficacy and Safety from a Randomized Cross over Phase 3 Study (ASCERTAIN Study) of an Oral Hypomethylating Agent ASTX727 (Cedazuridine/Decitabine) Compared to IV Decitabine. Blood 2019, 134, 846. [Google Scholar] [CrossRef]
- Sekeres, M.A.; Watts, J.; Radinoff, A.; Sangerman, M.A.; Cerrano, M.; Lopez, P.F.; Zeidner, J.F.; Campelo, M.D.; Graux, C.; Liesveld, J.; et al. Randomized Phase 2 Trial of Pevonedistat plus Azacitidine versus Azacitidine for Higher-Risk MDS/CMML or Low-Blast AML. Leukemia 2021, 35, 2119–2124. [Google Scholar] [CrossRef] [PubMed]
- Adès, L.; Girshova, L.; Doronin, V.A.; Díez-Campelo, M.; Valcárcel, D.; Kambhampati, S.; Viniou, N.-A.; Woszczyk, D.; De Paz Arias, R.; Symeonidis, A.; et al. Pevonedistat plus Azacitidine vs. Azacitidine Alone in Higher-Risk MDS/Chronic Myelomonocytic Leukemia or Low-Blast-Percentage AML. Blood Adv. 2022, 6, 5132–5145. [Google Scholar] [CrossRef]
- Platzbecker, U. Treatment of MDS. Blood 2019, 133, 1096–1107. [Google Scholar] [CrossRef]
- Aldoss, I.; Yang, D.; Aribi, A.; Ali, H.; Sandhu, K.; Al Malki, M.M.; Mei, M.; Salhotra, A.; Khaled, S.; Nakamura, R.; et al. Efficacy of the Combination of Venetoclax and Hypomethylating Agents in Relapsed/Refractory Acute Myeloid Leukemia. Haematologica 2018, 103, e404–e407. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Garcia, J.S.; Borate, U.; Fong, C.Y.; Baer, M.R.; Nowak, D.; Peterlin, P.; Jurcic, J.; Jacoby, M.A.; Platzbecker, U.; et al. MDS-158: Updated Safety and Efficacy of Venetoclax in Combination with Azacitidine for the Treatment of Patients with Treatment-Naïve, Higher-Risk Myelodysplastic Syndromes: Phase 1b Results. Clin. Lymphoma Myeloma Leuk. 2021, 21, S343. [Google Scholar] [CrossRef]
- Bazinet, A.; Darbaniyan, F.; Jabbour, E.; Montalban-Bravo, G.; Ohanian, M.; Chien, K.; Kadia, T.; Takahashi, K.; Masarova, L.; Short, N.; et al. Azacitidine plus Venetoclax in Patients with High-Risk Myelodysplastic Syndromes or Chronic Myelomonocytic Leukaemia: Phase 1 Results of a Single-Centre, Dose-Escalation, Dose-Expansion, Phase 1-2 Study. Lancet Haematol. 2022, 9, e756–e765. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Fenaux, P.; Al-Kali, A.; Baer, M.R.; Sekeres, M.A.; Roboz, G.J.; Gaidano, G.; Scott, B.L.; Greenberg, P.; Platzbecker, U.; et al. Rigosertib versus Best Supportive Care for Patients with High-Risk Myelodysplastic Syndromes after Failure of Hypomethylating Drugs (ONTIME): A Randomised, Controlled, Phase 3 Trial. Lancet Oncol. 2016, 17, 496–508. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Fenaux, P.; Al-Kali, A.; Baer, M.R.; Sekeres, M.A.; Roboz, G.J.; Gaidano, G.; Scott, B.L.; Greenberg, P.L.; Platzbecker, U.; et al. Overall Survival and Subgroup Analysis from a Randomized Phase III Study of Intravenous Rigosertib Versus Best Supportive Care (BSC) in Patients (Pts) with Higher-Risk Myelodysplastic Syndrome (HR-MDS) after Failure of Hypomethylating Agents (HMAs). Blood 2014, 124, 163. [Google Scholar] [CrossRef]
- Daver, N.; Kantarjian, H.; Ravandi, F.; Estey, E.; Wang, X.; Garcia-Manero, G.; Jabbour, E.; Konopleva, M.; O’Brien, S.; Verstovsek, S.; et al. A Phase II Study of Decitabine and Gemtuzumab Ozogamicin in Newly Diagnosed and Relapsed Acute Myeloid Leukemia and High-Risk Myelodysplastic Syndrome. Leukemia 2016, 30, 268–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubasch, A.S.; Schulze, F.; Giagounidis, A.; Götze, K.S.; Krönke, J.; Sockel, K.; Middeke, J.M.; Chermat, F.; Gloaguen, S.; Puttrich, M.; et al. Single Agent Talacotuzumab Demonstrates Limited Efficacy but Considerable Toxicity in Elderly High-Risk MDS or AML Patients Failing Hypomethylating Agents. Leukemia 2019, 34, 1182–1186. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Bueso-Ramos, C.; Dinardo, C.; Estecio, M.R.; Davanlou, M.; Geng, Q.R.; Fang, Z.; Nguyen, M.; Pierce, S.; Wei, Y.; et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in Myelodysplastic Syndromes Is Enhanced by Treatment with Hypomethylating Agents. Leukemia 2014, 28, 1280–1288. [Google Scholar] [CrossRef] [Green Version]
- Morita, K.; Kantarjian, H.M.; Montalban Bravo, G.; Sasaki, K.; Daver, N.; Jabbour, E.; Alvarado, Y.; Chien, K.S.; DiNardo, C.D.; Ravandi, F.; et al. A Phase II Study of Double Immune Checkpoint Inhibitor Blockade with Nivolumab and Ipilimumab with or without Azacitidine in Patients with Myelodysplastic Syndrome (MDS). Blood 2020, 136, 7–9. [Google Scholar] [CrossRef]
- Zeidan, A.M.; Cavenagh, J.; Voso, M.T.; Taussig, D.; Tormo, M.; Boss, I.; Copeland, W.B.; Gray, V.E.; Previtali, A.; O’Connor, T.; et al. Efficacy and Safety of Azacitidine (AZA) in Combination with the Anti-PD-L1 Durvalumab (Durva) for the Front-Line Treatment of Older Patients (Pts) with Acute Myeloid Leukemia (AML) Who Are Unfit for Intensive Chemotherapy (IC) and Pts with Higher-Risk Myelodysplastic Syndromes (HR-MDS): Results from a Large, International, Randomized Phase 2 Study. Blood 2019, 134, 829. [Google Scholar] [CrossRef]
- Gerds, A.T.; Scott, B.L.; Greenberg, P.L.; Khaled, S.K.; Lin, T.L.; Pollyea, D.A.; Verma, A.; Dail, M.; Green, C.; Ma, C.; et al. PD-L1 Blockade with Atezolizumab in Higher-Risk Myelodysplastic Syndrome: An Initial Safety and Efficacy Analysis. Blood 2018, 132, 466. [Google Scholar] [CrossRef]
- Borate, U.; Esteve, J.; Porkka, K.; Knapper, S.; Vey, N.; Scholl, S.; Garcia-Manero, G.; Wermke, M.; Janssen, J.; Traer, E.; et al. Phase Ib Study of the Anti-TIM-3 Antibody MBG453 in Combination with Decitabine in Patients with High-Risk Myelodysplastic Syndrome (MDS) and Acute Myeloid Leukemia (AML). Blood 2019, 134, 570. [Google Scholar] [CrossRef]
- Sallman, D.; Malki, M.A.; Asch, A.; Wang, E.; Jurcic, J.; Bradley, T.; Flinn, I.; Pollyea, D.; Kambhampati, S.; Tanaka, T.; et al. MDS-445 Magrolimab in Combination With Azacitidine for Patients With Untreated Higher-Risk Myelodysplastic Syndromes (HR MDS): 5F9005 Phase 1b Study Results. Clin. Lymphoma. Myeloma Leuk. 2022, 22 (Suppl. 2), S314–S315. [Google Scholar] [CrossRef]
- Uy, G.L.; Godwin, J.; Rettig, M.P.; Vey, N.; Foster, M.; Arellano, M.L.; Rizzieri, D.A.; Topp, M.S.; Huls, G.; Lowenberg, B.; et al. Preliminary Results of a Phase 1 Study of Flotetuzumab, a CD123 x CD3 Bispecific Dart® Protein, in Patients with Relapsed/Refractory Acute Myeloid Leukemia and Myelodysplastic Syndrome. Blood 2017, 130, 637. [Google Scholar] [CrossRef]
- Watts, J.M.; Lin, T.; Wang, E.S.; Mims, A.S.; Cull, E.H.; Patel, P.A.; Shami, P.J.; Walter, R.B.; Cogle, C.R.; Chenault, R.A.; et al. Preliminary Results from a Phase 1 Study of APVO436, a Novel Anti-CD123 x Anti-CD3 Bispecific Molecule, in Relapsed/Refractory Acute Myeloid Leukemia and Myelodysplastic Syndrome. Blood 2020, 136, 11–12. [Google Scholar] [CrossRef]
- Mardiana, S.; Gill, S. CAR T Cells for Acute Myeloid Leukemia: State of the Art and Future Directions. Front. Oncol. 2020, 10, 697. [Google Scholar] [CrossRef] [PubMed]
- Marvin-Peek, J.; Savani, B.N.; Olalekan, O.O.; Dholaria, B. Challenges and Advances in Chimeric Antigen Receptor Therapy for Acute Myeloid Leukemia. Cancers 2022, 14, 497. [Google Scholar] [CrossRef]
- Al-Homsi, A.S.; Purev, E.; Lewalle, P.; Abdul-Hay, M.; Pollyea, D.A.; Salaroli, A.; Lequertier, T.; Alcantar-Orozco, E.; Borghese, F.; Lonez, C.; et al. Interim Results from the Phase I Deplethink Trial Evaluating the Infusion of a NKG2D CAR T-Cell Therapy Post a Non-Myeloablative Conditioning in Relapse or Refractory Acute Myeloid Leukemia and Myelodysplastic Syndrome Patients. Blood 2019, 134, 3844. [Google Scholar] [CrossRef]
- Sebert, M.; Cluzeau, T.; Beyne Rauzy, O.; Stamatoulas Bastard, A.; Dimicoli-Salazar, S.; Thepot, S.; Peterlin, P.; Park, S.; Gourin, M.-P.; Brehar, O.; et al. Ivosidenib Monotherapy Is Effective in Patients with IDH1 Mutated Myelodysplastic Syndrome (MDS): The Idiome Phase 2 Study by the GFM Group. Blood 2021, 138, 62. [Google Scholar] [CrossRef]
- Stein, E.M.; Fathi, A.T.; DiNardo, C.D.; Pollyea, D.A.; Roboz, G.J.; Collins, R.; Sekeres, M.A.; Stone, R.M.; Attar, E.C.; Frattini, M.G.; et al. Enasidenib in Patients with Mutant IDH2 Myelodysplastic Syndromes: A Phase 1 Subgroup Analysis of the Multicentre, AG221-C-001 Trial. Lancet. Haematol. 2020, 7, e309–e319. [Google Scholar] [CrossRef]
- Cluzeau, T.; Sebert, M.; Rahmé, R.; Cuzzubbo, S.; Lehmann-Che, J.; Madelaine, I.; Peterlin, P.; Bève, B.; Attalah, H.; Chermat, F.; et al. Eprenetapopt Plus Azacitidine in TP53-Mutated Myelodysplastic Syndromes and Acute Myeloid Leukemia: A Phase II Study by the Groupe Francophone des Myélodysplasies (GFM). J. Clin. Oncol. 2021, 39, 1575–1583. [Google Scholar] [CrossRef] [PubMed]
- Suragani, R.N.V.S.; Cadena, S.M.; Cawley, S.M.; Sako, D.; Mitchell, D.; Li, R.; Davies, M.V.; Alexander, M.J.; Devine, M.; Loveday, K.S.; et al. Transforming Growth Factor-β Superfamily Ligand Trap ACE-536 Corrects Anemia by Promoting Late-Stage Erythropoiesis. Nat. Med. 2014, 20, 408–414. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| New Active Principle | Mechanism of Action | Efficacy in Evaluable Patients | Clinical Trial |
|---|---|---|---|
| Imetelstat | Telomerase inhibitor | N = 38 R/R to ESAs 8-week TI rate of 37% for a median duration of 20 months | NCT02598661 phase II/III |
| Pexmetinib | p38/Tie2 inhibitor | N = 44 HI rate of 32% | NCT00916227 phase I |
| Galunisertib | TGF-β receptor type 1 kinase (ALK5) oral inhibitor |
N = 41 HI rate of 24.4% | NCT02008318 phase II |
| Oral azacytidine (CC-486) | HMA DNA/RNA methyltransferases inhibitor | N = 216 RBC-TI rate of 31% with a median duration of 11.1 months | NCT01566695 phase III |
| Tomaralimab (OPN-305) | TLR-2 inhibitor | N = 22 TI for at least 2 cycles in 27% (major responders) | NCT02363491 phase I/II |
| Roxadustat | HIF-PH inhibitor | N = 24 TI rate of 37.5% for ≥56 consecutive days within the first 28 weeks | NCT03263091 phase III |
| Cedazuridine/decitabine (ASTX727) | Fixed-dose combination of the HMA decitabine and the novel cytidine deaminase inhibitor cedazuridine | N = 27 RBC-TI rate of 48% | NCT03502668 phase I/II |
| New Active Principle | Mechanism of Action | Efficacy in Evaluable Patients | Clinical Trial |
|---|---|---|---|
| Guadecitabine | HMA, inhibits DNA/RNA methyltransferases | N = 105 ORR 51% treatment naïve patients ORR 43% R/R patients | NCT01261312 phase I/II |
| Pevonedistat + azacytidine | NAE first inhibitor | N = 58 CR rate of 51% with a median duration of response of 34 months | NCT02610777 phase II |
| Venetoclax + azacytidine | BCL-2 inhibitor | N = 78 CR rate of 40% | NCT02942290 phase Ib |
| Nivolumab + ipilimumab +/− azacytidine | Anti PD1 and anti CTLA4 immune checkpoint inhibitors | N = 26 CR rate of 18% in HMA failure cohort (N = 11) CR rate of 33% in HMA naïve cohort (N = 15) | NCT02530463 phase II |
| Sabatolimab + decitabine | Humanized anti-TIM-3 antibody | N = 16 CR rate of 50% | NCT03066648 phase I |
| Magrolimab + azacytidine | Anti CD47 immune checkpoint inhibitor | N = 33 ORR 91%, CR rate of 42% | NCT03248479 phase I |
| Flotetuzumab | CD123 X CD3 bispecific antibody | N = 14 Patients with either R/R AML or MDS ORR 43% | NCT02152956 phase I |
| Ivosidenib | mutant IDH1 inhibitor | N = 26 ORR 69%, CR rate of 46% | NCT03503409 phase II |
| Enasidenib | Mutant IDH2 inhibitor | N = 17 ORR 53% | NCT01915498 phase I |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cassanello, G.; Pasquale, R.; Barcellini, W.; Fattizzo, B. Novel Therapies for Unmet Clinical Needs in Myelodysplastic Syndromes. Cancers 2022, 14, 4941. https://doi.org/10.3390/cancers14194941
Cassanello G, Pasquale R, Barcellini W, Fattizzo B. Novel Therapies for Unmet Clinical Needs in Myelodysplastic Syndromes. Cancers. 2022; 14(19):4941. https://doi.org/10.3390/cancers14194941
Chicago/Turabian StyleCassanello, Giulio, Raffaella Pasquale, Wilma Barcellini, and Bruno Fattizzo. 2022. "Novel Therapies for Unmet Clinical Needs in Myelodysplastic Syndromes" Cancers 14, no. 19: 4941. https://doi.org/10.3390/cancers14194941
APA StyleCassanello, G., Pasquale, R., Barcellini, W., & Fattizzo, B. (2022). Novel Therapies for Unmet Clinical Needs in Myelodysplastic Syndromes. Cancers, 14(19), 4941. https://doi.org/10.3390/cancers14194941