
Early Mechanisms of Chemoresistance in Retinoblastoma

 , , ,
, , ,  , , and
, , and 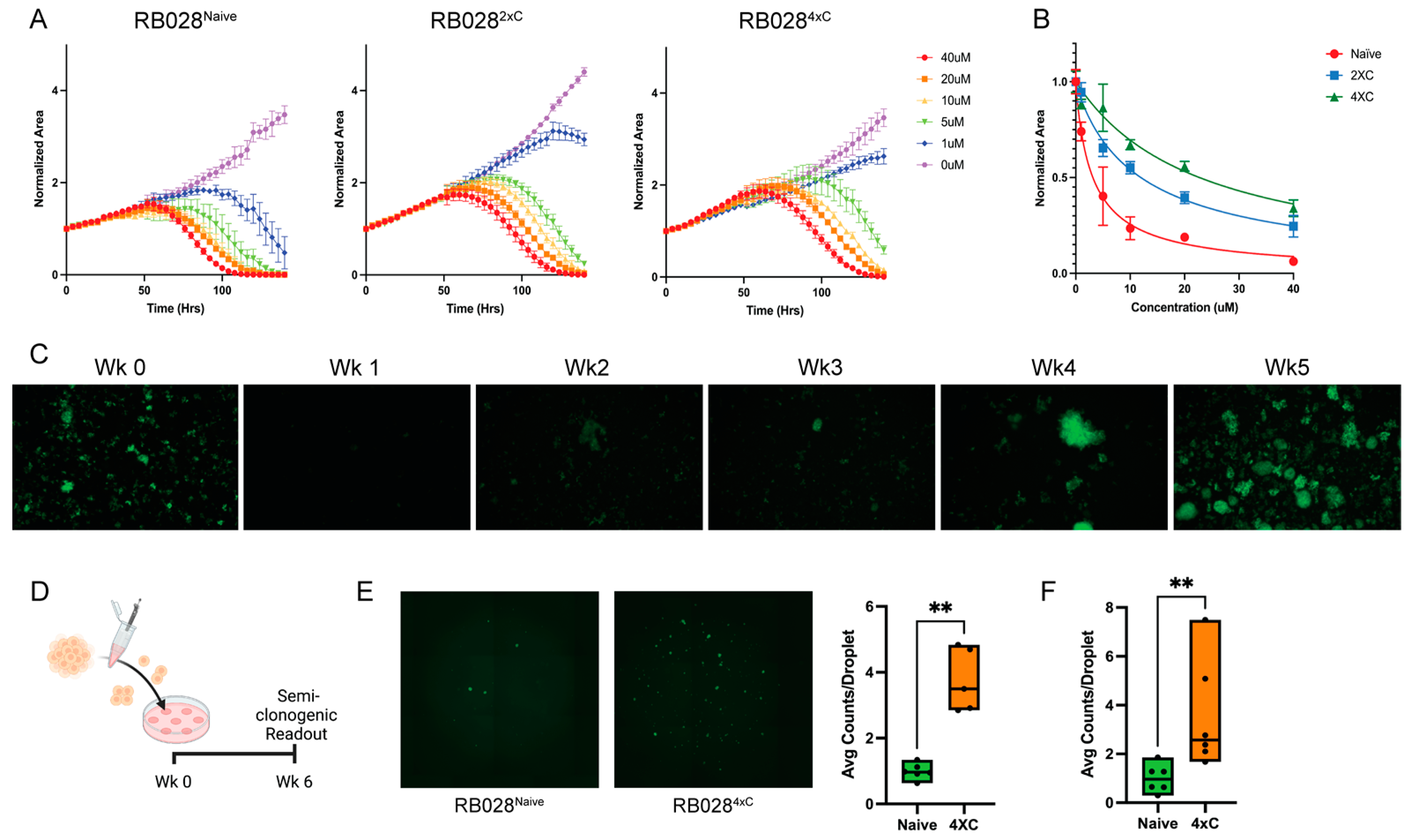{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Preparation of Cell Lines
2.2. Cell Viability Analysis
2.3. RNA Sequencing
2.4. Seeding Assay
2.5. Barcoding and Single-Cell RNA Sequencing (scRNA-Seq)
2.6. scRNA-Seq Analysis
2.7. Quantitative PCR (qPCR)
3. Results
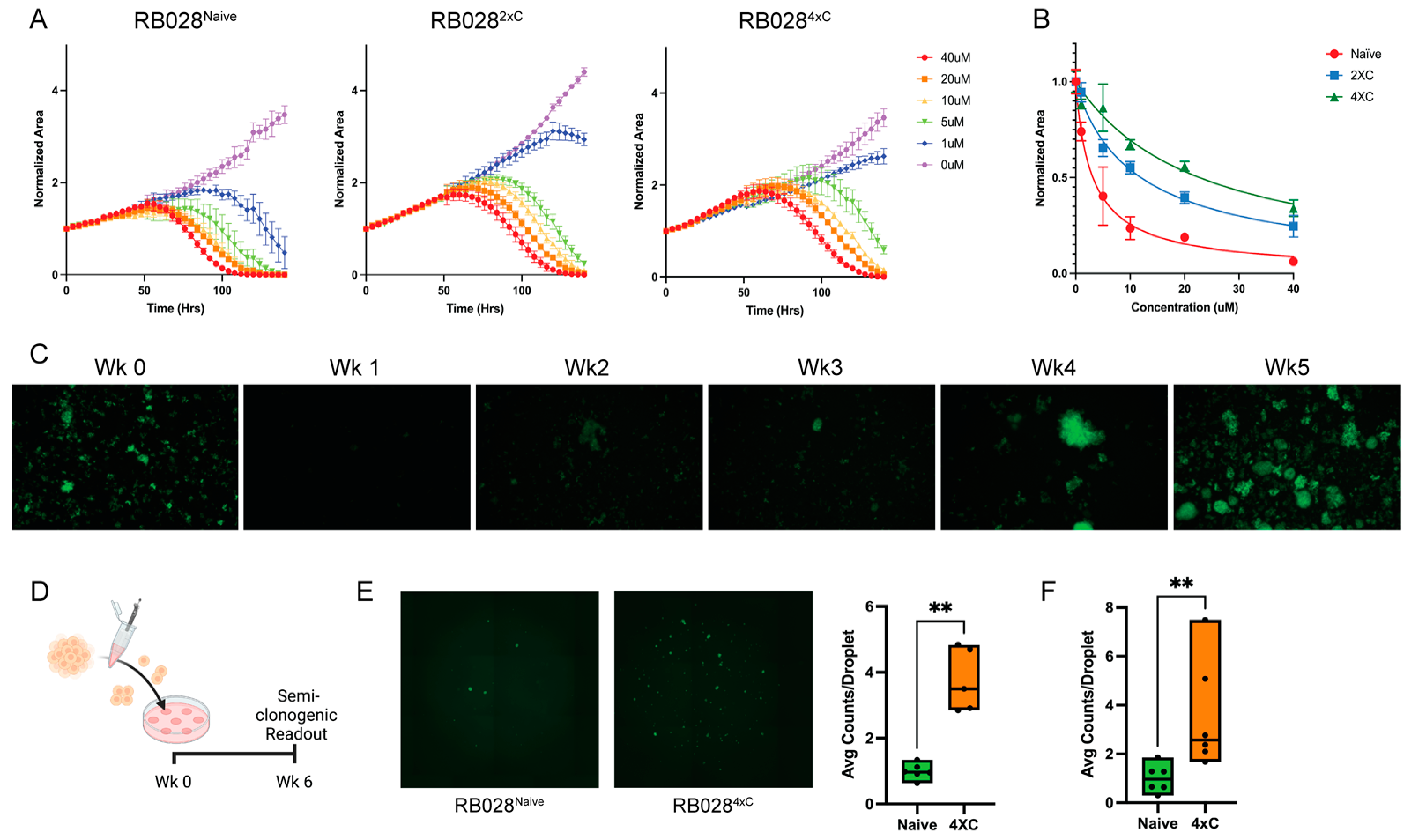
3.1. Characterization of Carboplatin-Resistant Retinoblastoma Cells
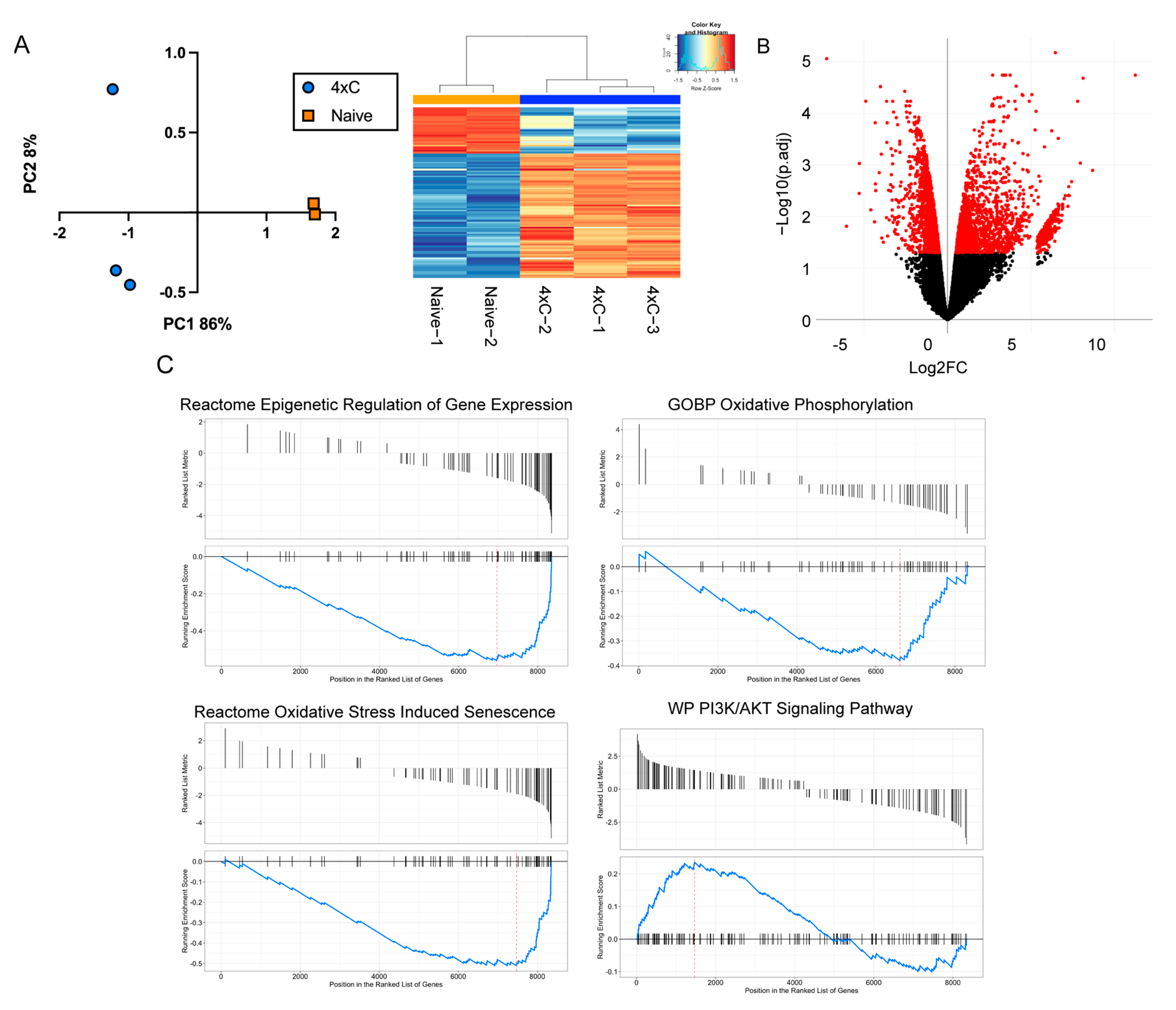
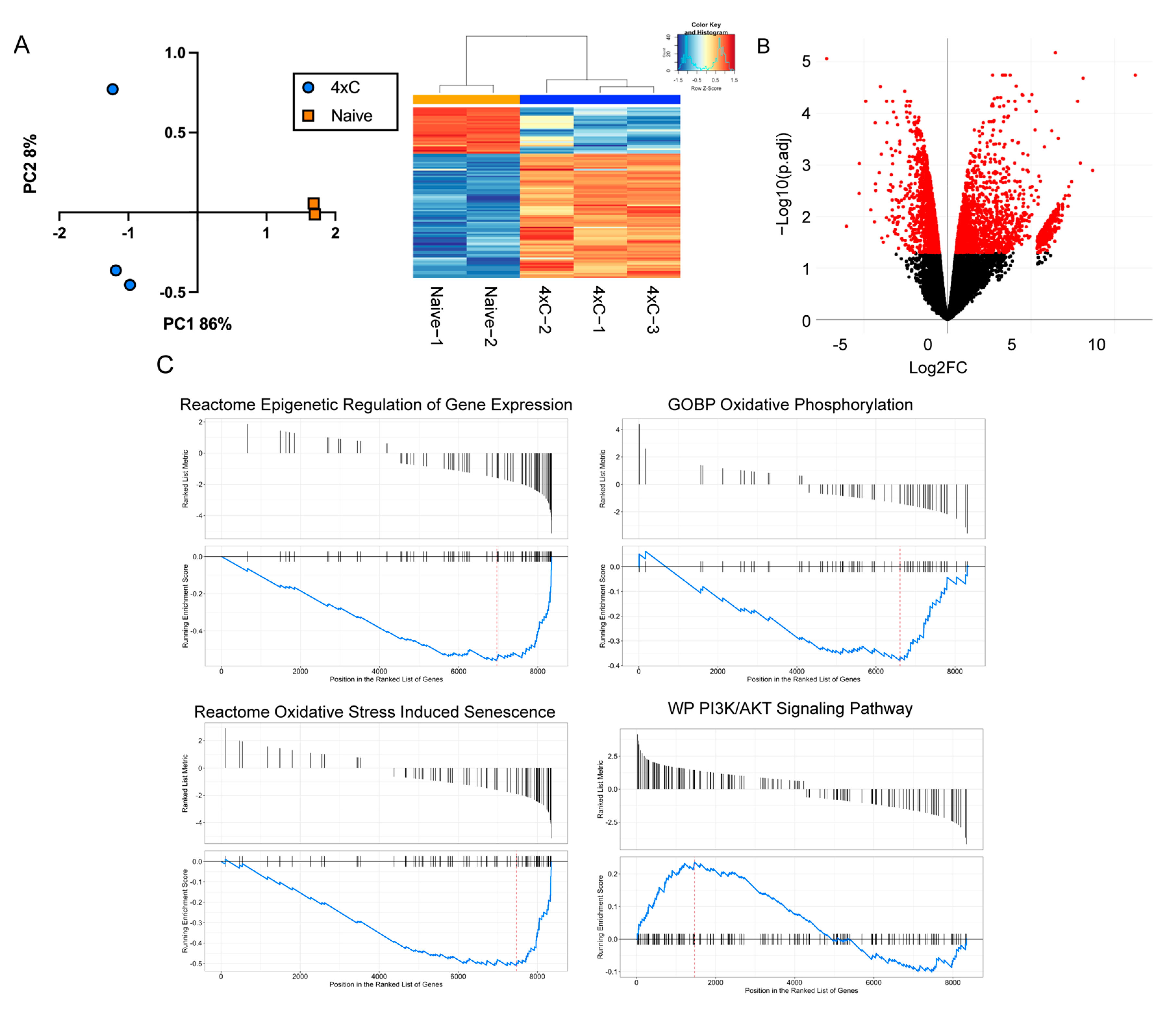
3.2. Transcriptomic Reprogramming of Carboplatin-Resistant Retinoblastoma Cells
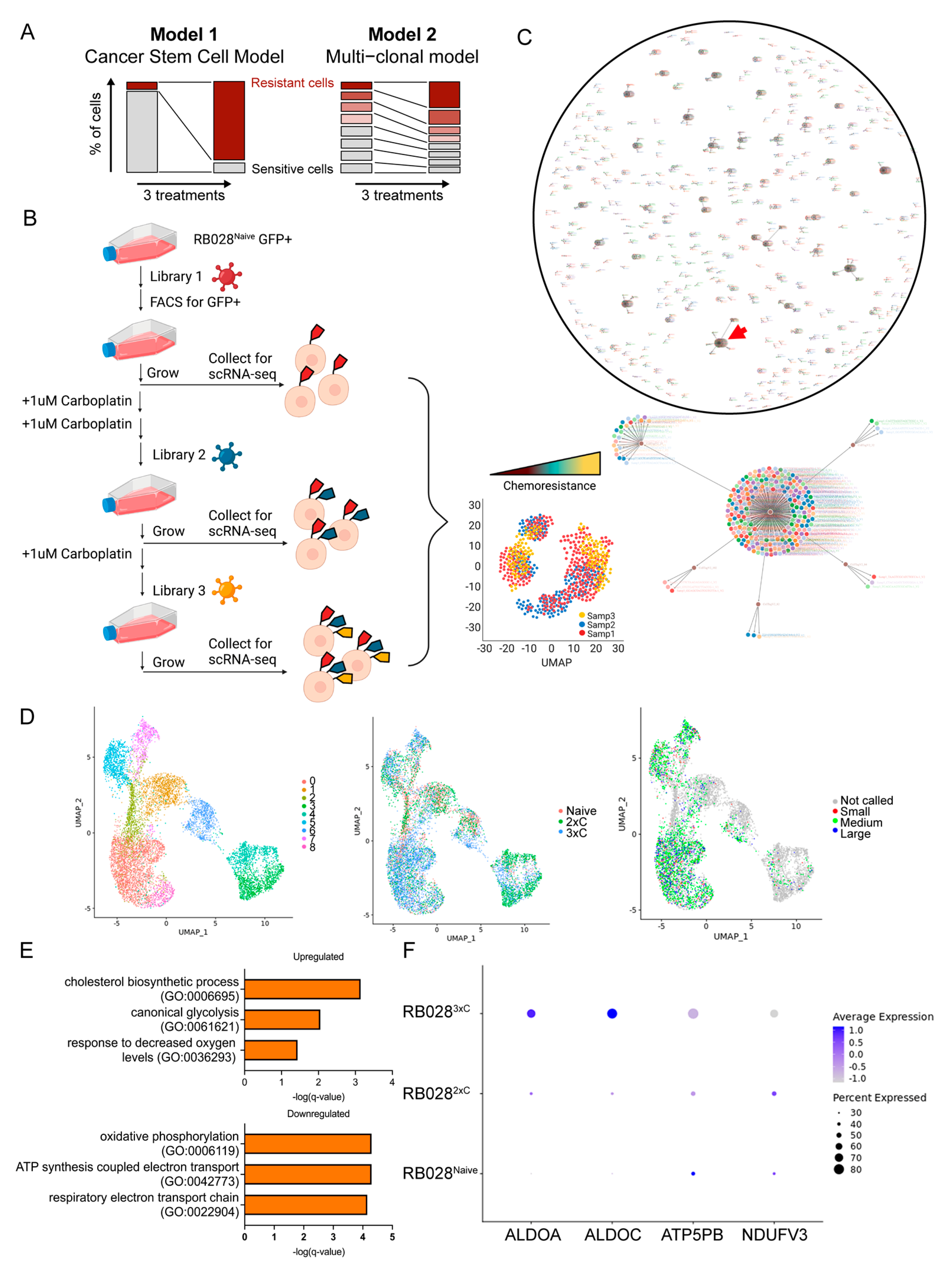
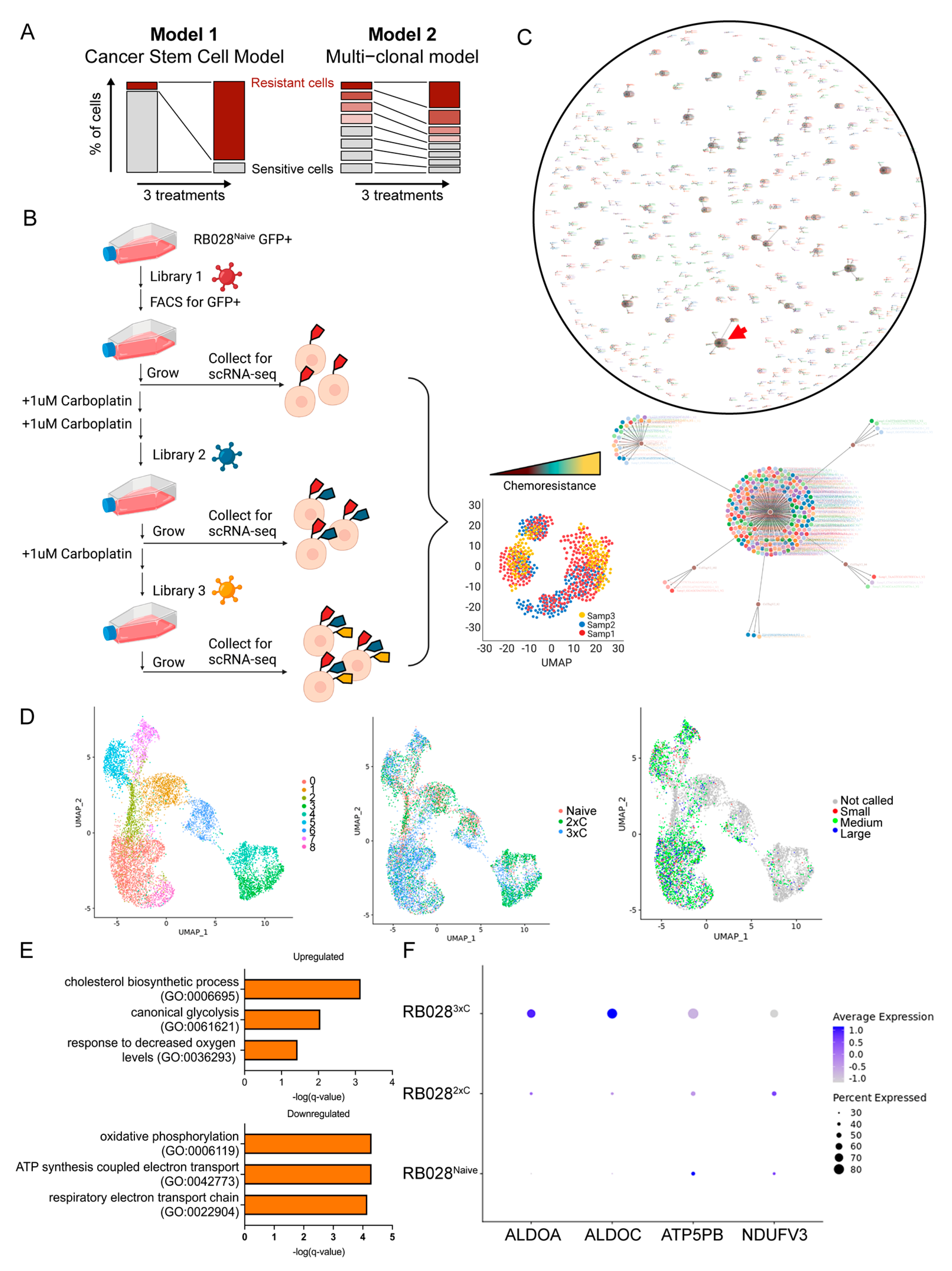
3.3. Single-Cell Analysis Reveals Early Mechanisms of Carboplatin Resistance
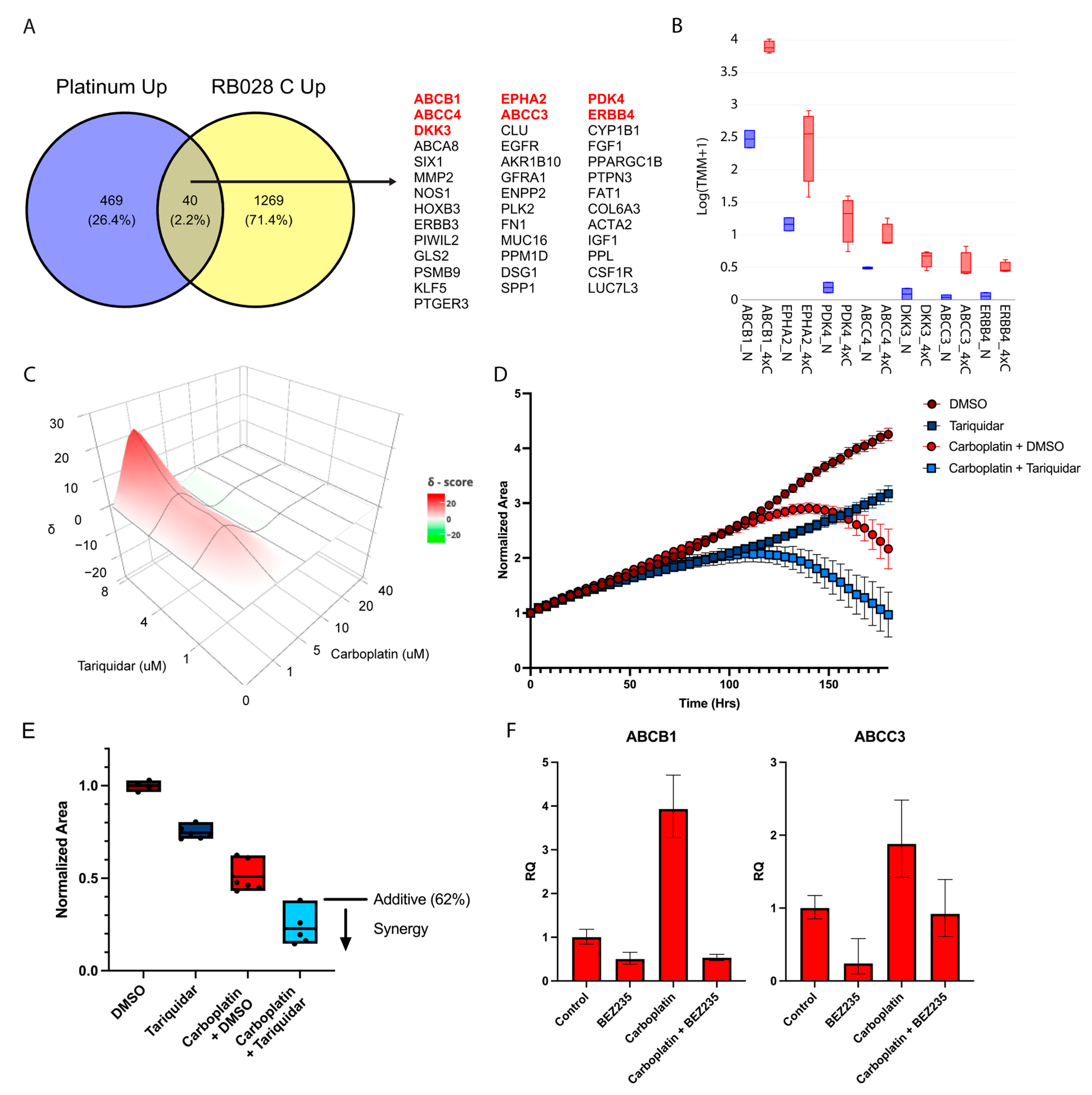
3.4. Pharmacologic Inhibition of ABCB1 Reverses Resistance to Carboplatin
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fabian, I.D.; Abdallah, E.; Abdullahi, S.U.; Abdulqader, R.A.; Adamou Boubacar, S.; Ademola-Popoola, D.S.; Adio, A.; Afshar, A.R.; Aggarwal, P.; Aghaji, A.E.; et al. Global Retinoblastoma Presentation and Analysis by National Income Level. JAMA Oncol. 2020, 6, 685–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, J.R.; Tucker, M.A.; Kleinerman, R.A.; Devesa, S.S. Retinoblastoma incidence patterns in the US Surveillance, Epidemiology, and End Results program. JAMA Ophthalmol. 2014, 132, 478–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabian, I.D.; Onadim, Z.; Karaa, E.; Duncan, C.; Chowdhury, T.; Scheimberg, I.; Ohnuma, S.I.; Reddy, M.A.; Sagoo, M.S. The management of retinoblastoma. Oncogene 2018, 37, 1551–1560. [Google Scholar] [CrossRef]
- Shields, C.L.; Honavar, S.G.; Meadows, A.T.; Shields, J.A.; Demirci, H.; Singh, A.; Friedman, D.L.; Naduvilath, T.J. Chemoreduction plus focal therapy for retinoblastoma: Factors predictive of need for treatment with external beam radiotherapy or enucleation. Am. J. Ophthalmol. 2002, 133, 657–664. [Google Scholar] [CrossRef]
- Sanati-Mehrizy, A.; Ghosh, T.; Peterson, E.; Starke, R.M.; Harbour, J.W.; Corrales-Medina, F.F. Hematologic Complications Associated With Intra-arterial Chemotherapy for Retinoblastoma Treatment: A Single Institution Experience. J. Pediatr. Hematol./Oncol. 2022, 44, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Murry, D.J. Comparative clinical pharmacology of cisplatin and carboplatin. Pharmacotherapy 1997, 17, 140s–145s. [Google Scholar] [PubMed]
- Wilson, M.W.; Fraga, C.H.; Rodriguez-Galindo, C.; Hagedorn, N.; Leggas, M.L.; Stewart, C. Expression of the multi-drug resistance proteins and the pregnane X receptor in treated and untreated retinoblastoma. Curr. Eye Res. 2009, 34, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, B.; Bai, S.W.; Wang, H.J. Constitutively active Akt contributes to vincristine resistance in human retinoblastoma cells. Cancer Investig. 2010, 28, 156–165. [Google Scholar] [CrossRef]
- Field, M.G.; Kuznetsoff, J.N.; Zhang, M.G.; Dollar, J.J.; Durante, M.A.; Sayegh, Y.; Decatur, C.L.; Kurtenbach, S.; Pelaez, D.; Harbour, J.W. RB1 loss triggers dependence on ESRRG in retinoblastoma. Sci. Adv. 2022, 8, eabm8466. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Savage, S.R.; Calinawan, A.P.; Lin, C.; Zhang, B.; Wang, P.; Starr, T.K.; Birrer, M.J.; Paulovich, A.G. A highly annotated database of genes associated with platinum resistance in cancer. Oncogene 2021, 40, 6395–6405. [Google Scholar] [CrossRef] [PubMed]
- Saengwimol, D.; Rojanaporn, D.; Chaitankar, V.; Chittavanich, P.; Aroonroch, R.; Boontawon, T.; Thammachote, W.; Jinawath, N.; Hongeng, S.; Kaewkhaw, R. A three-dimensional organoid model recapitulates tumorigenic aspects and drug responses of advanced human retinoblastoma. Sci. Rep. 2018, 8, 15664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biddy, B.A.; Kong, W.; Kamimoto, K.; Guo, C.; Waye, S.E.; Sun, T.; Morris, S.A. Single-cell mapping of lineage and identity in direct reprogramming. Nature 2018, 564, 219–224. [Google Scholar] [CrossRef]
- Kong, W.; Biddy, B.A.; Kamimoto, K.; Amrute, J.M.; Butka, E.G.; Morris, S.A. CellTagging: Combinatorial indexing to simultaneously map lineage and identity at single-cell resolution. Nat. Protoc. 2020, 15, 750–772. [Google Scholar] [CrossRef]
- Zheng, G.X.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef] [Green Version]
- McGinnis, C.S.; Murrow, L.M.; Gartner, Z.J. DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst. 2019, 8, 329–337.e4. [Google Scholar] [CrossRef]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M., 3rd; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Amram, A.L.; Rico, G.; Kim, J.W.; Chintagumpala, M.; Herzog, C.E.; Gombos, D.S.; Chevez-Barrios, P. Vitreous Seeds in Retinoblastoma: Clinicopathologic Classification and Correlation. Ophthalmology 2017, 124, 1540–1547. [Google Scholar] [CrossRef] [PubMed]
- Francis, J.H.; Marr, B.P.; Abramson, D.H. Classification of Vitreous Seeds in Retinoblastoma: Correlations with Patient, Tumor, and Treatment Characteristics. Ophthalmology 2016, 123, 1601–1605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, C.; Freeman, M.J.; Lu, H.; Wang, X.; Forster, C.L.; Sarver, A.L.; Hallstrom, T.C. Retinoblastoma cells activate the AKT pathway and are vulnerable to the PI3K/mTOR inhibitor NVP-BEZ235. Oncotarget 2017, 8, 38084–38098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphree, A.L.; Villablanca, J.G.; Deegan, W.F., 3rd; Sato, J.K.; Malogolowkin, M.; Fisher, A.; Parker, R.; Reed, E.; Gomer, C.J. Chemotherapy plus local treatment in the management of intraocular retinoblastoma. Arch. Ophthalmol. 1996, 114, 1348–1356. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, A.; Suzuki, S. Eye-preservation treatment of retinoblastoma with vitreous seeding. Jpn. J. Clin. Oncol. 2003, 33, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Abramson, D.H.; Dunkel, I.J.; Brodie, S.E.; Kim, J.W.; Gobin, Y.P. A phase I/II study of direct intraarterial (ophthalmic artery) chemotherapy with melphalan for intraocular retinoblastoma initial results. Ophthalmology 2008, 115, 1398–1404.e1. [Google Scholar] [CrossRef]
- Harbour, J.W.; Murray, T.G.; Hamasaki, D.; Cicciarelli, N.; Hernandez, E.; Smith, B.; Windle, J.; O’Brien, J.M. Local carboplatin therapy in transgenic murine retinoblastoma. Investig. Ophthalmol. Vis. Sci. 1996, 37, 1892–1898. [Google Scholar] [CrossRef]
- Munier, F.L.; Gaillard, M.C.; Balmer, A.; Soliman, S.; Podilsky, G.; Moulin, A.P.; Beck-Popovic, M. Intravitreal chemotherapy for vitreous disease in retinoblastoma revisited: From prohibition to conditional indications. Br. J. Ophthalmol. 2012, 96, 1078–1083. [Google Scholar] [CrossRef]
- Munier, F.L.; Mosimann, P.; Puccinelli, F.; Gaillard, M.C.; Stathopoulos, C.; Houghton, S.; Bergin, C.; Beck-Popovic, M. First-line intra-arterial versus intravenous chemotherapy in unilateral sporadic group D retinoblastoma: Evidence of better visual outcomes, ocular survival and shorter time to success with intra-arterial delivery from retrospective review of 20 years of treatment. Br. J. Ophthalmol. 2016, 101, 1086–1093. [Google Scholar] [CrossRef]
- Leng, T.; Cebulla, C.M.; Schefler, A.C.; Murray, T.G. Focal periocular carboplatin chemotherapy avoids systemic chemotherapy for unilateral, progressive retinoblastoma. Retina 2010, 30, S66–S68. [Google Scholar] [CrossRef]
- Wang, N.; Fan, H.; Fu, S.; Li, S.; Zhou, B.; Jin, Q.; You, Z. Long noncoding RNA UCA1 promotes carboplatin resistance in retinoblastoma cells by acting as a ceRNA of miR-206. Am. J. Cancer Res. 2022, 12, 2160–2172. [Google Scholar] [PubMed]
- Narayana, R.V.L.; Jana, P.; Tomar, N.; Prabhu, V.; Nair, R.M.; Manukonda, R.; Kaliki, S.; Coupland, S.E.; Alexander, J.; Kalirai, H.; et al. Carboplatin- and Etoposide-Loaded Lactoferrin Protein Nanoparticles for Targeting Cancer Stem Cells in Retinoblastoma In Vitro. Investig. Ophthalmol. Vis. Sci. 2021, 62, 13. [Google Scholar] [CrossRef] [PubMed]
- Sazonova, E.V.; Kopeina, G.S.; Imyanitov, E.N.; Zhivotovsky, B. Platinum drugs and taxanes: Can we overcome resistance? Cell Death Discov. 2021, 7, 155. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Jin, Y.; Fan, Z. The Mechanism of Warburg Effect-Induced Chemoresistance in Cancer. Front. Oncol. 2021, 11, 698023. [Google Scholar] [CrossRef]
- Jiang, T.; Zhou, M.L.; Fan, J. Inhibition of GLUT-1 expression and the PI3K/Akt pathway to enhance the chemosensitivity of laryngeal carcinoma cells in vitro. OncoTargets Ther. 2018, 11, 7865–7872. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Xia, L.; Oyang, L.; Liang, J.; Tan, S.; Wu, N.; Yi, P.; Pan, Q.; Rao, S.; Han, Y.; et al. The POU2F1-ALDOA axis promotes the proliferation and chemoresistance of colon cancer cells by enhancing glycolysis and the pentose phosphate pathway activity. Oncogene 2022, 41, 1024–1039. [Google Scholar] [CrossRef]
- Wen, J.F.; Jiang, Y.Q.; Li, C.; Dai, X.K.; Wu, T.; Yin, W.Z. LncRNA-SARCC sensitizes osteosarcoma to cisplatin through the miR-143-mediated glycolysis inhibition by targeting Hexokinase 2. Cancer Biomark. 2020, 28, 231–246. [Google Scholar] [CrossRef]
- Sradhanjali, S.; Tripathy, D.; Rath, S.; Mittal, R.; Reddy, M.M. Overexpression of pyruvate dehydrogenase kinase 1 in retinoblastoma: A potential therapeutic opportunity for targeting vitreous seeds and hypoxic regions. PLoS ONE 2017, 12, e0177744. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020, 11, 797. [Google Scholar] [CrossRef]
- Spangle, J.M.; Roberts, T.M.; Zhao, J.J. The emerging role of PI3K/AKT-mediated epigenetic regulation in cancer. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Hamon, Y.; Chimini, G. The human ATP-binding cassette (ABC) transporter superfamily. J. Lipid Res. 2001, 42, 1007–1017. [Google Scholar] [CrossRef]
- Chan, H.S.; Thorner, P.S.; Haddad, G.; Gallie, B.L. Multidrug-resistant phenotype in retinoblastoma correlates with P-glycoprotein expression. Ophthalmology 1991, 98, 1425–1431. [Google Scholar] [CrossRef]
- Sethi, S.; Malik, M.A.; Goswami, S.; Saxena, P.; Srivastava, A.; Kashyap, S.; Pushker, N.; Bajaj, M.S.; Bakhshi, S.; Kaur, J. Expression of P-glycoprotein in human retinoblastoma and its clinical significance. Tumour Biol. 2014, 35, 11735–11740. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, L.; Lu, L.; Wang, Y. miR-214-3p Regulates Multi-Drug Resistance and Apoptosis in Retinoblastoma Cells by Targeting ABCB1 and XIAP. OncoTargets Ther. 2020, 13, 803–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souza Filho, J.P.; Martins, M.C.; Caissie, A.L.; Torres, V.L.; Fernandes, L.H.; Erwenne, C.M.; Burnier, M.N., Jr. Relationship between histopathological features of chemotherapy treated retinoblastoma and P-glycoprotein expression. Clin. Exp. Ophthalmol. 2005, 33, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.S.; DeBoer, G.; Thiessen, J.J.; Budning, A.; Kingston, J.E.; O’Brien, J.M.; Koren, G.; Giesbrecht, E.; Haddad, G.; Verjee, Z.; et al. Combining cyclosporin with chemotherapy controls intraocular retinoblastoma without requiring radiation. Clin. Cancer Res. 1996, 2, 1499–1508. [Google Scholar]
- Munoz, M.; Rosso, M.; Gonzalez, A.; Saenz, J.; Covenas, R. The broad-spectrum antitumor action of cyclosporin A is due to its tachykinin receptor antagonist pharmacological profile. Peptides 2010, 31, 1643–1648. [Google Scholar] [CrossRef]
- Fox, E.; Widemann, B.C.; Pastakia, D.; Chen, C.C.; Yang, S.X.; Cole, D.; Balis, F.M. Pharmacokinetic and pharmacodynamic study of tariquidar (XR9576), a P-glycoprotein inhibitor, in combination with doxorubicin, vinorelbine, or docetaxel in children and adolescents with refractory solid tumors. Cancer Chemother. Pharmacol. 2015, 76, 1273–1283. [Google Scholar] [CrossRef] [PubMed]
- Mohan, A.; Kandalam, M.; Ramkumar, H.L.; Gopal, L.; Krishnakumar, S. Stem cell markers: ABCG2 and MCM2 expression in retinoblastoma. Br. J. Ophthalmol. 2006, 90, 889–893. [Google Scholar] [CrossRef] [Green Version]
- Nair, R.M.; Balla, M.M.; Khan, I.; Kalathur, R.K.R.; Kondaiah, P.; Vemuganti, G.K. In vitro characterization of CD133(lo) cancer stem cells in Retinoblastoma Y79 cell line. BMC Cancer 2017, 17, 779. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.G.; Kuznetsoff, J.N.; Owens, D.A.; Gallo, R.A.; Kalahasty, K.; Cruz, A.M.; Kurtenbach, S.; Correa, Z.M.; Pelaez, D.; Harbour, J.W. Early Mechanisms of Chemoresistance in Retinoblastoma. Cancers 2022, 14, 4966. https://doi.org/10.3390/cancers14194966
Zhang MG, Kuznetsoff JN, Owens DA, Gallo RA, Kalahasty K, Cruz AM, Kurtenbach S, Correa ZM, Pelaez D, Harbour JW. Early Mechanisms of Chemoresistance in Retinoblastoma. Cancers. 2022; 14(19):4966. https://doi.org/10.3390/cancers14194966
Chicago/Turabian StyleZhang, Michelle G., Jeffim N. Kuznetsoff, Dawn A. Owens, Ryan A. Gallo, Karthik Kalahasty, Anthony M. Cruz, Stefan Kurtenbach, Zelia M. Correa, Daniel Pelaez, and J. William Harbour. 2022. "Early Mechanisms of Chemoresistance in Retinoblastoma" Cancers 14, no. 19: 4966. https://doi.org/10.3390/cancers14194966
APA StyleZhang, M. G., Kuznetsoff, J. N., Owens, D. A., Gallo, R. A., Kalahasty, K., Cruz, A. M., Kurtenbach, S., Correa, Z. M., Pelaez, D., & Harbour, J. W. (2022). Early Mechanisms of Chemoresistance in Retinoblastoma. Cancers, 14(19), 4966. https://doi.org/10.3390/cancers14194966