
Regulation of Bcl-2 Family Proteins in Estrogen Receptor-Positive Breast Cancer and Their Implications in Endocrine Therapy



Simple Summary
Abstract
1. Introduction
2. Estrogen Receptor Signaling
2.1. The Estrogen Receptor
2.2. Classical and Non-Classical Genomic ER Signaling Pathways
2.3. Alternative ER Signaling Pathways
3. Mechanism of Anti-Estrogen Therapy
4. Bcl-2 Proteins in the Regulation of the Intrinsic Cell Death Pathway
5. Receptor Tyrosine Kinase (RTK)-Regulated Signaling in Anti-Estrogen-Induced Apoptosis
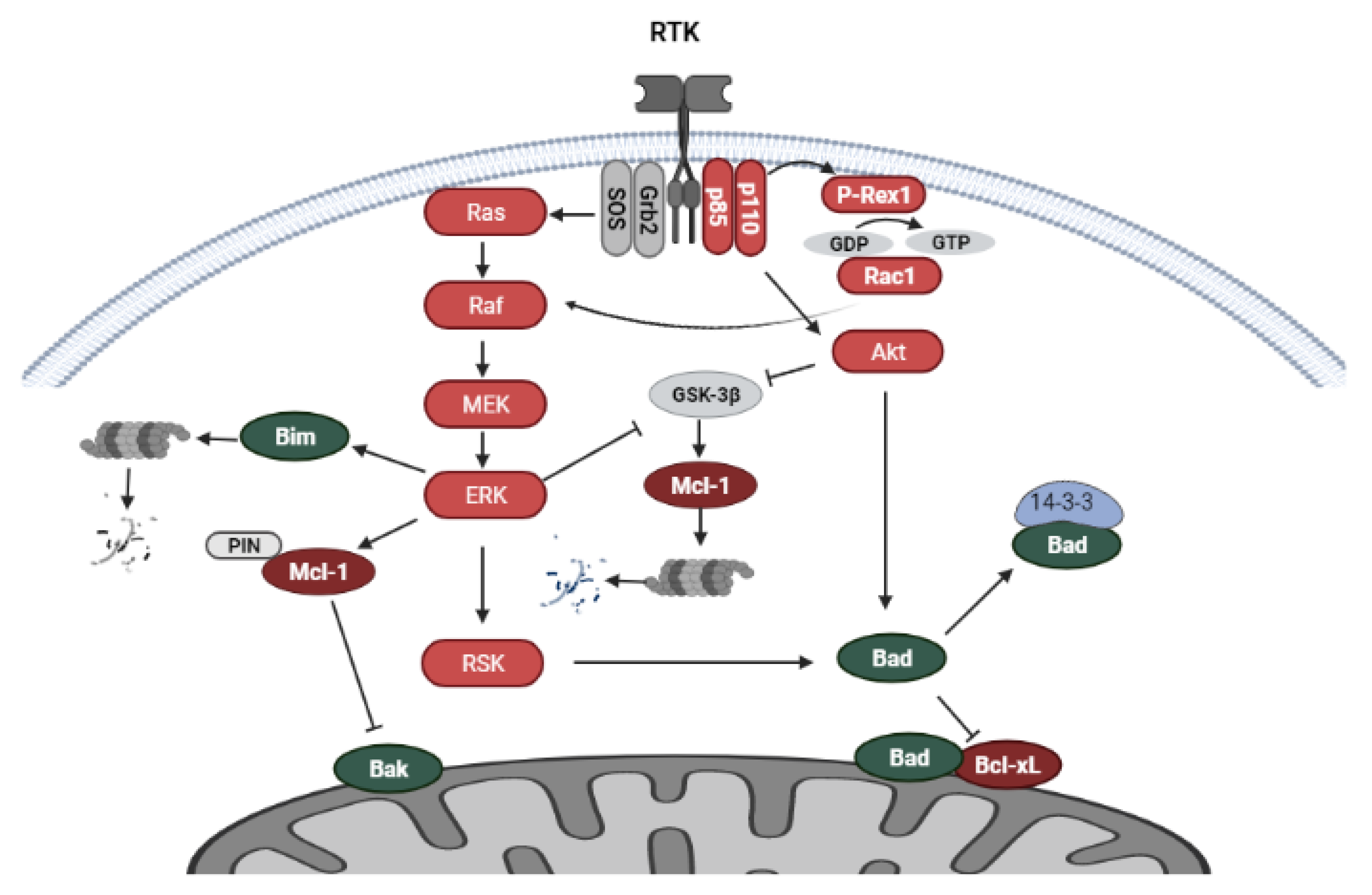
5.1. PI3K/Akt and MAPK Signaling
5.2. Regulation of Bcl-2 Family Proteins by RTK-Mediated Signaling
5.3. Endocrine Therapy and RTK-Mediated Signaling
6. Endoplasmic Reticulum in the Regulation of Anti-Estrogen Induced Apoptosis
6.1. Regulation of Bcl-2 Family Proteins at the Endoplasmic Reticulum
6.2. GRP78-Mediated Sensitivity of ER+ Breast Cancer to Endocrine Therapy
7. Bcl-2 Family Proteins in Breast Cancer
7.1. Bcl-2
7.2. Bcl-xL
7.3. Mcl-1
8. Targeting Bcl-2 in Breast Cancer with BH3 Mimetics
8.1. BH3 Mimetics
8.2. Mitochondrial Priming for BH3 Mimetic Activity
8.3. Venetoclax and ER+ Breast Cancer
9. Targeting Pro-Survival Bcl-2 Family Proteins in Breast Cancer
9.1. Targeting Mcl-1 in ER+ Breast Cancer
9.2. Compensatory Role Mcl-1 and Bcl-xL in ER+ Breast Cancer
9.3. Potential Drawbacks of BH3 Mimetics
10. Role of Pro-Apoptotic Proteins in Breast Cancer-Mediated Cell Death
10.1. The Pro-Apoptotic Proteins Bax and Bak
10.2. The Pro-Apoptotic Protein Bik
10.3. Tumor Suppressor p53
11. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Heer, E.; Harper, A.; Escandor, N.; Sung, H.; McCormack, V.; Fidler-Benaoudia, M.M. Global burden and trends in premenopausal and postmenopausal breast cancer: A population-based study. Lancet Glob. Health 2020, 8, e1027–e1037. [Google Scholar] [CrossRef]
- Pereira, B.; Chin, S.F.; Rueda, O.M.; Vollan, H.K.M.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.J.; et al. The somatic mutation profiles of 2433 breast cancers refine their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef] [PubMed]
- Waks, A.G.; Winer, E.P. Breast cancer treatment: A review. JAMA-J. Am. Med. Assoc. 2019, 321, 288–300. [Google Scholar] [CrossRef]
- Gao, J.J.; Swain, S.M. Luminal a breast cancer and molecular assays: A review. Oncologist 2018, 23, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Fragomeni, S.M.; Sciallis, A.; Jeruss, J.S. Molecular subtypes and local-regional control of breast cancer. Surg. Oncol. Clin. N. Am. 2018, 27, 95–120. [Google Scholar] [CrossRef]
- Feng, Y.; Spezia, M.; Huang, S.; Yuan, C.; Zeng, Z.; Zhang, L.; Ji, X.; Liu, W.; Huang, B.; Luo, W.; et al. Breast cancer development and progression: Risk factors, cancer stem cells, signaling pathways, genomics, and molecular pathogenesis. Genes Dis. 2018, 5, 77–106. [Google Scholar] [CrossRef]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Weigelt, B.; Geyer, F.C.; Reis-Filho, J.S. Histological types of breast cancer: How special are they? Mol. Oncol. 2010, 4, 192–208. [Google Scholar] [CrossRef]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed]
- Cheang, M.C.U.; Chia, S.K.; Voduc, D.; Gao, D.; Leung, S.; Snider, J.; Watson, M.; Davies, S.; Bernard, P.S.; Parker, J.S.; et al. Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J. Natl. Cancer Inst. 2009, 101, 736–750. [Google Scholar] [CrossRef]
- Tran, B.; Bedard, P.L. Luminal-B breast cancer and novel therapeutic targets. Breast Cancer Res. 2011, 13, 221. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.H.; Hu, P.H.; Tu, J.H.; Yu, N.S. Luminal B breast cancer: Patterns of recurrence and clinical outcome. Oncotarget 2016, 7, 65024–65033. [Google Scholar] [CrossRef]
- Ignatiadis, M.; Sotiriou, C. Luminal breast cancer: From biology to treatment. Nat. Rev. Clin. Oncol. 2013, 10, 494–506. [Google Scholar] [CrossRef]
- Patel, H.K.; Bihani, T. Selective estrogen receptor modulators (SERMs) and selective estrogen receptor degraders (SERDs) in cancer treatment. Pharmacol. Ther. 2018, 186, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Le Romancer, M.; Poulard, C.; Cohen, P.; Sentis, S.P.; Renoir, J.M.; Corbo, L. Cracking the estrogen receptor’s posttranslational code in breast tumors. Endocr. Rev. 2011, 32, 597–622. [Google Scholar] [CrossRef] [PubMed]
- Dawson, S.J.; Makretsov, N.; Blows, F.M.; Driver, K.E.; Provenzano, E.; Le Quesne, J.; Baglietto, L.; Severi, G.; Giles, G.G.; McLean, C.A.; et al. BCL2 in breast cancer: A favourable prognostic marker across molecular subtypes and independent of adjuvant therapy received. Br. J. Cancer 2010, 103, 668–675. [Google Scholar] [CrossRef]
- Merino, D.; Lok, S.W.; Visvader, J.E.; Lindeman, G.J. Targeting BCL-2 to enhance vulnerability to therapy in estrogen receptor-positive breast cancer. Oncogene 2016, 35, 1877–1887. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Finger, L.R.; Yunis, J.; Nowell, P.C.; Croce, C.M. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science 1984, 226, 1097–1099. [Google Scholar] [CrossRef]
- Lipponen, P.; Pietiläinen, T.; Kosma, V.-M.; Aaltomaa, S.; Eskelinen, M.; Syrjäunen, K. Apoptosis suppressing protein bcl-2 is expressed in well-differentiated breast carcinomas with favourable prognosis. J. Pathol. 1995, 177, 49–55. [Google Scholar] [CrossRef]
- Leung, L.K.; Wang, T.T.Y. Paradoxical regulation of Bcl-2 family proteins by 17β-oestradiol in human breast cancer cells MCF-7. Br. J. Cancer 1999, 81, 387–392. [Google Scholar] [CrossRef]
- Fuentes, N.; Silveyra, P. Estrogen receptor signaling mechanisms. Adv. Protein Chem. Struct. Biol. 2019, 116, 135–170. [Google Scholar]
- Weikum, E.R.; Liu, X.; Ortlund, E.A. The nuclear receptor superfamily: A structural perspective. Protein Sci. 2018, 27, 1876–1892. [Google Scholar] [CrossRef] [PubMed]
- Dey, P.; Barros, R.P.A.; Warner, M.; Strom, A.; Gustafsson, J.-Å. Insight into the mechanisms of action of estrogen receptor beta. J. Mol. Endocrinol. 2013, 51, T61–T74. [Google Scholar] [CrossRef] [PubMed]
- Heldring, N.; Pike, A.; Andersson, S.; Matthews, J.; Cheng, G.; Hartman, J.; Tujague, M.; Ström, A.; Treuter, E.; Warner, M.; et al. Estrogen receptors: How do they signal and what are their targets. Physiol. Rev. 2007, 87, 905–931. [Google Scholar] [CrossRef] [PubMed]
- Tee, M.K.; Rogatsky, I.; Tzagarakis-Foster, C.; Cvoro, A.; An, J.; Christy, R.J.; Yamamoto, K.R.; Leitman, D.C. Estradiol and selective estrogen receptor modulators differentially regulate target genes with estrogen receptors α and β. Mol. Biol. Cell 2004, 15, 1262–1272. [Google Scholar] [PubMed]
- Paruthiyil, S.; Parmar, H.; Kerekatte, V.; Cunha, G.R.; Firestone, G.L.; Leitmant, D.C. Estrogen receptor β inhibits human breast cancer cell proliferation and tumor formation by causing a G2 cell cycle arrest. Cancer Res. 2004, 64, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Gougelet, A.; Mueller, S.O.; Korach, K.S.; Renoir, J.M. Oestrogen receptors pathways to oestrogen responsive elements: The transactivation function-1 acts as the keystone of oestrogen receptor (ER)β-mediated transcriptional repression of ERα. J. Steroid Biochem. Mol. Biol. 2007, 104, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.M.; Albanese, C.; Anderson, C.M.; Hilty, K.; Webb, P.; Uht, R.M.; Price, R.H.; Pestell, R.G.; Kushn, P.J. Opposing action of estrogen receptors α and β on cyclin D1 gene expression. J. Biol. Chem. 2002, 277, 24353–24360. [Google Scholar] [CrossRef]
- Papoutsi, Z.; Zhao, C.; Putnik, M.; Gustafsson, J.Å.; Dahlman-Wright, K. Binding of estrogen receptor α/β heterodimers to chromatin in MCF-7 cells. J. Mol. Endocrinol. 2009, 43, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Gougelet, A.; Bouclier, C.; Marsaud, V.; Maillard, S.; Mueller, S.O.; Korach, K.S.; Renoir, J.M. Estrogen receptor α and β subtype expression and transactivation capacity are differentially affected by receptor-, hsp90- and immunophilin-ligands in human breast cancer cells. J. Steroid Biochem. Mol. Biol. 2005, 94, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Karmakar, S.; Foster, E.A.; Smith, C.L. Unique roles of p160 coactivators for regulation of breast cancer cell proliferation and estrogen receptor-α transcriptional activity. Endocrinology 2009, 150, 1588–1596. [Google Scholar] [CrossRef]
- Dobrzycka, K.M.; Townson, S.M.; Jiang, S.; Oesterreich, S. Estrogen receptor corepressors—A role in human breast cancer? Endocr. Relat. Cancer 2003, 10, 517–536. [Google Scholar] [CrossRef]
- O’Lone, R.; Frith, M.C.; Karlsson, E.K.; Hansen, U. Genomic targets of nuclear estrogen receptors. Mol. Endocrinol. 2004, 18, 1859–1875. [Google Scholar] [CrossRef] [PubMed]
- Kalaitzidis, D.; Gilmore, T.D. Transcription factor cross-talk: The estrogen receptor and NF-κB. Trends Endocrinol. Metab. 2005, 16, 46–52. [Google Scholar] [CrossRef]
- Umayahara, Y.; Kawamori, R.; Watada, H.; Imano, E.; Iwama, N.; Morishima, T.; Yamasaki, Y.; Kajimoto, Y.; Kamada, T. Estrogen regulation of the insulin-like growth factor I gene transcription involves an AP-1 enhancer. J. Biol. Chem. 1994, 269, 16433–16442. [Google Scholar] [CrossRef]
- Webb, P.; Nguyen, P.; Valentine, C.; Lopez, G.N.; Kwok, G.R.; McInerney, E.; Katzenellenbogen, B.S.; Enmark, E.; Gustafsson, J.Å.; Nilsson, S.; et al. The estrogen receptor enhances AP-1 activity by two distinct mechanisms with different requirements for receptor transactivation functions. Mol. Endocrinol. 1999, 13, 1672–1685. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yau, C.; Gray, J.W.; Chew, K.; Dairkee, S.H.; Moore, D.H.; Eppenberger, U.; Eppenberger-Castori, S.; Benz, C.C. Enhanced NFκB and AP-1 transcriptional activity associated with antiestrogen resistant breast cancer. BMC Cancer 2007, 7, 59. [Google Scholar] [CrossRef]
- Sas, L.; Lardon, F.; Vermeulen, P.B.; Hauspy, J.; Van Dam, P.; Pauwels, P.; Dirix, L.Y.; Van Laere, S.J. The interaction between ER and NFκB in resistance to endocrine therapy. Breast Cancer Res. 2012, 14, 212. [Google Scholar] [CrossRef]
- Schiff, R.; Massarweh, S.A.; Shou, J.; Bharwani, L.; Mohsin, S.K.; Osborne, C.K.; Nicholson, R.; Ellis, M.; Santen, R.; Brown, M. Cross-talk between estrogen receptor and growth factor pathways as a molecular target for overcoming endocrine resistance. Clin. Cancer Res. 2004, 10, 331–336. [Google Scholar] [CrossRef]
- Martin, M.B.; Franke, T.F.; Stoica, G.E.; Chambon, P.; Katzenellenbogen, B.S.; Stoica, B.A.; McLemore, M.S.; Olivo, S.E.; Stoica, A. A role for Akt in mediating the estrogenic functions of epidermal growth factor and insulin-like growth factor I. Endocrinology 2000, 141, 4503–4511. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Endoh, H.; Masuhiro, Y.; Kitamoto, T.; Uchiyama, S.; Sasaki, H.; Masushige, S.; Gotoh, Y.; Nishida, E.; Kawashima, H.; et al. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science 1995, 270, 1491–1494. [Google Scholar] [CrossRef]
- Osborne, C.K.; Schiff, R. Estrogen-receptor biology: Continuing progress and therapeutic implications. J. Clin. Oncol. 2005, 23, 1616–1622. [Google Scholar] [CrossRef] [PubMed]
- Björnström, L.; Sjöberg, M. Mechanisms of estrogen receptor signaling: Convergence of genomic and nongenomic actions on target genes. Mol. Endocrinol. 2005, 19, 833–842. [Google Scholar] [CrossRef]
- Poulard, C.; Jacquemetton, J.; Trédan, O.; Cohen, P.A.; Vendrell, J.; Ghayad, S.E.; Treilleux, I.; Marangoni, E.; Le Romancer, M. Oestrogen non-genomic signalling is activated in tamoxifen-resistant breast cancer. Int. J. Mol. Sci. 2019, 20, 2773. [Google Scholar] [CrossRef] [PubMed]
- Song, R.X.D.; Chen, Y.; Zhang, Z.; Bao, Y.; Yue, W.; Wang, J.P.; Fan, P.; Santen, R.J. Estrogen utilization of IGF-1-R and EGF-R to signal in breast cancer cells. J. Steroid Biochem. Mol. Biol. 2010, 118, 219–230. [Google Scholar] [CrossRef]
- Moriarty, K.; Kim, K.H.; Bender, J.R. Minireview: Estrogen receptor-mediated rapid signaling. Endocrinology 2006, 147, 5557–5563. [Google Scholar] [CrossRef]
- Chung, Y.L.; Sheu, M.L.; Yang, S.C.; Lin, C.H.; Yen, S.H. Resistance to tamoxifen-induced apoptosis is associated with direct interaction between Her2/neu and cell membrane estrogen receptor in breast cancer. Int. J. Cancer 2002, 97, 306–312. [Google Scholar] [CrossRef]
- Arpino, G.; Wiechmann, L.; Osborne, C.K.; Schiff, R. Crosstalk between the estrogen receptor and the HER tyrosine kinase receptor family: Molecular mechanism and clinical implications for endocrine therapy resistance. Endocr. Rev. 2008, 29, 217–233. [Google Scholar] [CrossRef]
- Flouriot, G.; Brand, H.; Denger, S.; Metivier, È.; Kos, M.; Reid, G.; Sonntag-buck, V.; Gannon, F. Identification of a new isoform of the human estrogen receptor-alpha (hER-alpha) that is encoded by distinct transcripts and that is able to repress hER- a activation function 1. EMBO J. 2000, 19, 4688–4700. [Google Scholar] [CrossRef]
- Thiebaut, C.; Konan, H.P.; Guerquin, M.J.; Chesnel, A.; Livera, G.; Le Romancer, M.; Dumond, H. The role of erα36 in development and tumor malignancy. Int. J. Mol. Sci. 2020, 21, 4116. [Google Scholar] [CrossRef]
- Lazennec, G.; Bresson, D.; Lucas, A.; Chauveau, C.; Vignon, F. ERβ inhibits proliferation and invasion of breast cancer cells. Endocrinology 2001, 142, 4120–4130. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, X. The role of estrogen receptor beta in breast cancer. Biomark. Res. 2020, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Dahlman-Wright, K.; Cavailles, V.; Fuqua, S.A.; Jordan, V.C.; Katzenellenbogen, J.A.; Korach, K.S.; Maggi, A.; Muramatsu, M.; Parker, M.G.; Gustafsson, J.Å. International union of pharmacology. LXIV. Estrogen receptors. Pharmacol. Rev. 2006, 58, 773–781. [Google Scholar] [CrossRef]
- Wu, V.S.; Kanaya, N.; Lo, C.; Mortimer, J.; Chen, S. From bench to bedside: What do we know about hormone receptor-positive and human epidermal growth factor receptor 2-positive breast cancer? J. Steroid Biochem. Mol. Biol. 2015, 153, 45–53. [Google Scholar] [CrossRef]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Prim. 2019, 5, 66. [Google Scholar] [CrossRef]
- Frasor, J.; Stossi, F.; Danes, J.M.; Komm, B.; Lyttle, C.R.; Katzenellenbogen, B.S. Selective estrogen receptor modulators: Discrimination of agonistic versus antagonistic activities by gene expression profiling in breast cancer cells. Cancer Res. 2004, 64, 1522–1533. [Google Scholar] [CrossRef] [PubMed]
- Shao, W.; Brown, M. Advances in estrogen receptor biology: Prospects for improvements in targeted breast cancer therapy. Breast Cancer Res. 2004, 6, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.L.; O’Malley, B.W. Coregulator Function: A Key to Understanding Tissue Specificity of Selective Receptor Modulators. Endocr. Rev. 2004, 25, 45–71. [Google Scholar] [CrossRef]
- Rondón-Lagos, M.; Villegas, V.E.; Rangel, N.; Sánchez, M.C.; Zaphiropoulos, P.G. Tamoxifen resistance: Emerging molecular targets. Int. J. Mol. Sci. 2016, 17, 1357. [Google Scholar] [CrossRef] [PubMed]
- Dowsett, M.; Nicholson, R.I.; Pietras, R.J. Biological characteristics of the pure antiestrogen fulvestrant: Overcoming endocrine resistance. Breast Cancer Res. Treat. 2005, 93, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Howell, A. Pure oestrogen antagonists for the treatment of advanced breast cancer. Endocr. Relat. Cancer 2006, 13, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.R.D.; Dowsett, M. Aromatase inhibitors for breast cancer: Lessons from the laboratory. Nat. Rev. Cancer 2003, 3, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Shamas-Din, A.; Brahmbhatt, H.; Leber, B.; Andrews, D.W. BH3-only proteins: Orchestrators of apoptosis. Biochim. Biophys. Acta-Mol. Cell Res. 2011, 1813, 508–520. [Google Scholar] [CrossRef] [PubMed]
- Lomonosova, E.; Chinnadurai, G. BH3-only proteins in apoptosis and beyond: An overview. Oncogene 2008, 27, 2–19. [Google Scholar] [CrossRef] [PubMed]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef]
- Kutuk, O.; Letai, A. Regulation of Bcl-2 family proteins by posttranslational modifications. Curr. Mol. Med. 2008, 8, 102–118. [Google Scholar]
- Kalkavan, H.; Green, D.R. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018, 25, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.E.; Du, F.; Fang, M.; Wang, X. Formation of apoptosome is initiated by cytochrome c-induced dATP hydrolysis and subsequent nucleotide exchange on Apaf-1. Proc. Natl. Acad. Sci. USA 2005, 102, 17545–17550. [Google Scholar] [CrossRef]
- Pihán, P.; Carreras-Sureda, A.; Hetz, C. BCL-2 family: Integrating stress responses at the ER to control cell demise. Cell Death Differ. 2017, 24, 1478–1487. [Google Scholar] [CrossRef]
- Butti, R.; Das, S.; Gunasekaran, V.P.; Yadav, A.S.; Kumar, D.; Kundu, G.C. Receptor tyrosine kinases (RTKs) in breast cancer: Signaling, therapeutic implications and challenges. Mol. Cancer 2018, 17, 34. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar]
- Wu, P.K.; Becker, A.; Park, J.I. Growth inhibitory signaling of the raf/mek/erk pathway. Int. J. Mol. Sci. 2020, 21, 5436. [Google Scholar] [CrossRef]
- Pérez-Tenorio, G.; Alkhori, L.; Olsson, B.; Waltersson, M.A.; Nordenskjöld, B.; Rutqvist, L.E.; Skoog, L.; Stål, O. PIK3CA mutations and PTEN loss correlate with similar prognostic factors and are not mutually exclusive in breast cancer. Clin. Cancer Res. 2007, 13, 3577–3584. [Google Scholar] [CrossRef]
- Zardavas, D.; Phillips, W.A.; Loi, S. PIK3CA mutations in breast cancer: Reconciling findings from preclinical and clinical data. Breast Cancer Res. 2014, 16, 201. [Google Scholar] [CrossRef]
- Luciano, F.; Jacquel, A.; Colosetti, P.; Herrant, M.; Cagnol, S.; Pages, G.; Auberger, P. Phosphorylation of Bim-EL by Erk1/2 on serine 69 promotes its degradation via the proteasome pathway and regulates its proapoptotic function. Oncogene 2003, 22, 6785–6793. [Google Scholar] [CrossRef]
- Ding, Q.; Huo, L.; Yang, J.Y.; Xia, W.; Wei, Y.; Liao, Y.; Chang, C.J.; Yang, Y.; Lai, C.C.; Lee, D.F.; et al. Down-regulation of myeloid cell leukemia-1 through inhibiting Erk/Pin 1 pathway by sorafenib facilitates chemosensitization in breast cancer. Cancer Res. 2008, 68, 6109–6117. [Google Scholar] [CrossRef]
- Kawiak, A.; Domachowska, A.; Krolicka, A.; Smolarska, M.; Lojkowska, E. 3-Chloroplumbagin induces cell death in breast cancer cells through MAPK-mediated MCL-1 inhibition. Front. Pharmacol. 2019, 10, 784. [Google Scholar] [CrossRef]
- Maurer, U.; Charvet, C.; Wagman, A.S.; Dejardin, E.; Green, D.R. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol. Cell 2006, 21, 749–760. [Google Scholar] [CrossRef]
- Ding, Q.; He, X.; Xia, W.; Hsu, J.-M.; Chen, C.-T.; Li, L.-Y.; Lee, D.-F.; Yang, J.-Y.; Xie, X.; Liu, J.-C.; et al. Myeloid cell leukemia-1 inversely correlates with glycogen synthase kinase-3β activity and associates with poor prognosis in human breast cancer. Cancer Res. 2007, 67, 4564–4571. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Hsu, J.L.; Hung, M.C. Regulation of ubiquitination-mediated protein degradation by survival kinases in cancer. Front. Oncol. 2012, 2, 15. [Google Scholar] [CrossRef] [PubMed]
- Hirai, I.; Wang, H.G. Survival-factor-induced phosphorylation of bad results in its dissociation from Bcl-xL but not Bcl-2. Biochem. J. 2001, 359, 345–352. [Google Scholar] [CrossRef]
- Fernando, R.I.; Wimalasena, J. Estradiol abrogates apoptosis in breast cancer cells through inactivation of BAD: Ras-dependent nongenomic pathways requiring signaling through ERK and Akt. Mol. Biol. Cell 2004, 15, 3266–3284. [Google Scholar] [CrossRef]
- Ebi, H.; Costa, C.; Faber, A.C.; Nishtala, M.; Kotani, H.; Juric, D.; Della Pelle, P.; Song, Y.; Yano, S.; Mino-Kenudson, M.; et al. PI3K regulates MEK/ERK signaling in breast cancer via the Rac-GEF, P-Rex1. Proc. Natl. Acad. Sci. USA 2013, 110, 21124–21129. [Google Scholar] [CrossRef] [PubMed]
- Saal, L.H.; Holm, K.; Maurer, M.; Memeo, L.; Su, T.; Wang, X.; Yu, J.S.; Malmström, P.O.; Mansukhani, M.; Enoksson, J.; et al. PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Res. 2005, 65, 2554–2559. [Google Scholar] [CrossRef] [PubMed]
- Sabnis, G.J.; Jelovac, D.; Long, B.; Brodie, A. The role of growth factor receptor pathways in human breast cancer cells adapted to long-term estrogen deprivation. Cancer Res. 2005, 65, 3903–3910. [Google Scholar] [CrossRef]
- Sabnis, G.; Goloubeva, O.; Jelovac, D.; Schayowitz, A.; Brodie, A. Inhibition of the phosphatidylinositol 3-kinase/Akt pathway improves response of long-term estrogen-deprived breast cancer xenografts to antiestrogens. Clin. Cancer Res. 2007, 13, 2751–2757. [Google Scholar] [CrossRef]
- Miller, T.W.; Hennessy, B.T.; González-Angulo, A.M.; Fox, E.M.; Mills, G.B.; Chen, H.; Higham, C.; García-Echeverría, C.; Shyr, Y.; Arteaga, C.L. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor-positive human breast cancer. J. Clin. Investig. 2010, 120, 2406–2413. [Google Scholar] [CrossRef]
- Yue, W.; Wang, J.P.; Conaway, M.R.; Li, Y.; Santen, R.J. Adaptive hypersensitivity following long-term estrogen deprivation: Involvement of multiple signal pathways. J. Steroid Biochem. Mol. Biol. 2003, 86, 265–274. [Google Scholar] [CrossRef]
- Jeng, M.H.; Yue, W.; Eischeid, A.; Wang, J.P.; Santen, R.J. Role of MAP kinase in the enhanced cell proliferation of long term estrogen deprived human breast cancer cells. Breast Cancer Res. Treat. 2000, 62, 167–175. [Google Scholar] [CrossRef]
- Fan, P.; Wang, J.; Santen, R.J.; Yue, W. Long-term treatment with tamoxifen facilitates translocation of estrogen receptor α out of the nucleus and enhances its interaction with EGFR in MCF-7 breast cancer cells. Cancer Res. 2007, 67, 1352–1360. [Google Scholar] [CrossRef]
- Jelovac, D.; Sabnis, G.; Long, B.J.; Macedo, L.; Goloubeva, O.G.; Brodie, A.M.H. Activation of mitogen-activated protein kinase in xenografts and cells during prolonged treatment with aromatase inhibitor letrozole. Cancer Res. 2005, 65, 5380–5389. [Google Scholar] [CrossRef]
- Brachmann, S.M.; Hofmann, I.; Schnell, C.; Fritsch, C.; Wee, S.; Lane, H.; Wang, S.; Garcia-Echeverria, C.; Maira, S.M. Specific apoptosis induction by the dual PI3K/mTor inhibitor NVP-BEZ235 in HER2 amplified and PIK3CA mutant breast cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 22299–22304. [Google Scholar] [CrossRef]
- Stratikopoulos, E.E.; Kiess, N.; Szabolcs, M.; Pegno, S.; Kakit, C.; Wu, X.; Poulikakos, P.I.; Cheung, P.; Schmidt, H.; Parsons, R. Mouse ER+/PIK3CA H1047R breast cancers caused by exogenous estrogen are heterogeneously dependent on estrogen and undergo BIM-dependent apoptosis with BH3 and PI3K agents. Oncogene 2019, 38, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Hur, J.; Chesnes, J.; Coser, K.R.; Lee, R.S.; Geck, P.; Isselbacher, K.J.; Shioda, T. The Bik BH3-only protein is induced in estrogen-starved and antiestrogen-exposed breast cancer cells and provokes apoptosis. Proc. Natl. Acad. Sci. USA 2004, 101, 2351–2356. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Li, J.; Lee, A.S. GRP78/BiP inhibits endoplasmic reticulum BIK and protects human breast cancer cells against estrogen starvation-induced apoptosis. Cancer Res. 2007, 67, 3734–3740. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. Glucose-regulated proteins in cancer: Molecular mechanisms and therapeutic potential. Nat. Rev. Cancer 2014, 14, 263–276. [Google Scholar] [CrossRef]
- Fernandez, P.M.; Tabbara, S.O.; Jacobs, L.K.; Manning, F.C.; Tsangaris, T.N.; Schwartz, A.M.; Kennedy, K.A.; Patierno, S.R. Overexpression of the glucose-regulated stress gene GRP78 in malignant but not benign human breast lesions. Breast Cancer Res. Treat. 2000, 59, 15–26. [Google Scholar] [CrossRef]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid. Redox Signal. 2009, 11, 2307–2316. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhang, Y.; Fu, Y.; Chan, L.; Lee, A.S. Novel mechanism of anti-apoptotic function of 78-kDa glucose-regulated protein (GRP78). J. Biol. Chem. 2011, 286, 25687–25696. [Google Scholar] [CrossRef]
- Kawiak, A.; Domachowska, A.; Jaworska, A.; Lojkowska, E. Plumbagin sensitizes breast cancer cells to tamoxifen-induced cell death through GRP78 inhibition and Bik upregulation. Sci. Rep. 2017, 7, 43781. [Google Scholar] [CrossRef]
- Cook, K.L.; Shajahan, A.N.; Wärri, A.; Jin, L.; Hilakivi-Clarke, L.A.; Clarke, R. Glucose-regulated protein 78 controls cross-talk between apoptosis and autophagy to determine antiestrogen responsiveness. Cancer Res. 2012, 72, 3337–3339. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.L.; Clarke, R. Role of GRP78 in promoting therapeutic-resistant breast cancer. Future Med. Chem. 2015, 7, 1529–1534. [Google Scholar] [CrossRef] [PubMed]
- Hosford, S.R.; Shee, K.; Wells, J.D.; Traphagen, N.A.; Fields, J.L.; Hampsch, R.A.; Kettenbach, A.N.; Demidenko, E.; Miller, T.W. Estrogen therapy induces an unfolded protein response to drive cell death in ER+ breast cancer. Mol. Oncol. 2019, 13, 1778–1794. [Google Scholar] [CrossRef] [PubMed]
- Urano, F.; Wang, X.Z.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef]
- Logue, S.E.; Cleary, P.; Saveljeva, S.; Samali, A. New directions in ER stress-induced cell death. Apoptosis 2013, 18, 537–546. [Google Scholar] [CrossRef]
- Dhanasekaran, D.N.; Reddy, E.P. JNK signaling in apoptosis. Oncogene 2008, 2, 6245–6251. [Google Scholar] [CrossRef] [PubMed]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER stress triggers apoptosis by activating BH3-only protein bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef] [PubMed]
- Pincus, D.; Chevalier, M.W.; Aragón, T.; van Anken, E.; Vidal, S.E.; El-Samad, H.; Walter, P. BiP binding to the ER-stress sensor Ire1 tunes the homeostatic behavior of the unfolded protein response. PLoS Biol. 2010, 8, 1000415. [Google Scholar] [CrossRef]
- Kota, K.; Brufsky, A.; Oesterreich, S.; Lee, A. Estradiol as a targeted, late-line therapy in metastatic breast cancer with estrogen receptor amplification. Cureus 2017, 9, 1434. [Google Scholar] [CrossRef]
- Li, S.; Shen, D.; Shao, J.; Crowder, R.; Liu, W.; Prat, A.; He, X.; Liu, S.; Hoog, J.; Lu, C.; et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 2013, 4, 1116–1130. [Google Scholar] [CrossRef]
- Holst, F. Estrogen receptor alpha gene amplification in breast cancer: 25 years of debate. World J. Clin. Oncol. 2016, 7, 160–173. [Google Scholar] [CrossRef]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Vaillant, F.; Merino, D.; Lee, L.; Breslin, K.; Pal, B.; Ritchie, M.E.; Smyth, G.K.; Christie, M.; Phillipson, L.J.; Burns, C.J.; et al. Targeting BCL-2 with the BH3 mimetic ABT-199 in estrogen receptor-positive breast cancer. Cancer Cell 2013, 24, 120–129. [Google Scholar] [CrossRef]
- Joensuu, H.; Pylkkänen, L.; Toikkanen, S. Bcl-2 protein expression and long-term survival in breast cancer. Am. J. Pathol. 1994, 145, 1191–1198. [Google Scholar]
- Callagy, G.M.; Webber, M.J.; Pharoah, P.D.P.; Caldas, C. Meta-analysis confirms BCL2 is an independent prognostic marker in breast cancer. BMC Cancer 2008, 8, 153. [Google Scholar] [CrossRef]
- Perillo, B.; Sasso, A.; Abbondanza, C.; Palumbo, G. 17β-estradiol inhibits apoptosis in MCF-7 cells, inducing bcl-2 expression via two estrogen-responsive elements present in the coding sequence. Mol. Cell. Biol. 2000, 20, 2890–2901. [Google Scholar] [CrossRef] [PubMed]
- Olopade, O.I.; Adeyanju, M.O.; Safa, A.R.; Hagos, F.; Mick, R.; Thompson, C.B.; Recant, W.M. Overexpression of BCL-x protein in primary breast cancer is associated with high tumor grade and nodal metastases. Cancer J. Sci. Am. 1997, 3, 230–237. [Google Scholar] [PubMed]
- Martin, S.S.; Ridgeway, A.G.; Pinkas, J.; Lu, Y.; Reginato, M.J.; Koh, E.Y.; Michelman, M.; Daley, G.Q.; Brugge, J.S.; Leder, P. A cytoskeleton-based functional genetic screen identifies Bcl-xL as an enhancer of metastasis, but not primary tumor growth. Oncogene 2004, 23, 4641–4645. [Google Scholar] [CrossRef][Green Version]
- Fernández, Y.; España, L.; Mañas, S.; Fabra, A.; Sierra, A. Bcl-x(L) promotes metastasis of breast cancer cells by induction of cytokines resistance. Cell Death Differ. 2000, 7, 350–359. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Choi, S.; Chen, Z.; Tang, L.H.; Fang, Y.; Shin, S.J.; Panarelli, N.C.; Chen, Y.T.; Li, Y.; Jiang, X.; Du, Y.C.N. Bcl-xL promotes metastasis independent of its anti-apoptotic activity. Nat. Commun. 2016, 7, 10384. [Google Scholar] [CrossRef]
- Bessou, M.; Lopez, J.; Gadet, R.; Deygas, M.; Popgeorgiev, N.; Poncet, D.; Nougarède, A.; Billard, P.; Mikaelian, I.; Gonzalo, P.; et al. The apoptosis inhibitor Bcl-xL controls breast cancer cell migration through mitochondria-dependent reactive oxygen species production. Oncogene 2020, 39, 3056–3074. [Google Scholar] [CrossRef]
- Williams, M.M.; Lee, L.; Hicks, D.J.; Joly, M.M.; Elion, D.; Rahman, B.; McKernan, C.; Sanchez, V.; Balko, J.M.; Stricker, T.; et al. Key survival factor, Mcl-1, correlates with sensitivity to combined Bcl-2/Bcl-xL blockade. Mol. Cancer Res. 2017, 15, 259–268. [Google Scholar] [CrossRef]
- Campbell, K.J.; Dhayade, S.; Ferrari, N.; Sims, A.H.; Johnson, E.; Mason, S.M.; Dickson, A.; Ryan, K.M.; Kalna, G.; Edwards, J.; et al. MCL-1 is a prognostic indicator and drug target in breast cancer article. Cell Death Dis. 2018, 9, 19. [Google Scholar] [CrossRef]
- Xiao, Y.; Nimmer, P.; Sheppard, G.S.; Bruncko, M.; Hessler, P.; Lu, X.; Roberts-Rapp, L.; Pappano, W.N.; Elmore, S.W.; Souers, A.J.; et al. MCL-1 is a key determinant of breast cancer cell survival: Validation of MCL-1 dependency utilizing a highly selective small molecule inhibitor. Mol. Cancer Ther. 2015, 14, 1837–1847. [Google Scholar] [CrossRef]
- Schacter, J.L.; Henson, E.S.; Gibson, S.B. Estrogen regulation of anti-apoptotic Bcl-2 family member Mcl-1 expression in breast cancer cells. PLoS ONE 2014, 9, e100364. [Google Scholar] [CrossRef] [PubMed]
- Davids, M.S.; Letai, A. Targeting the B-cell lymphoma/leukemia 2 family in cancer. J. Clin. Oncol. 2012, 30, 3127–3135. [Google Scholar] [CrossRef]
- Billard, C. BH3 mimetics: Status of the field and new developments. Mol. Cancer Ther. 2013, 12, 1691–1700. [Google Scholar] [CrossRef] [PubMed]
- Azmi, A.S.; Wang, Z.; Philip, P.A.; Mohammad, R.M.; Sarkar, F.H. Emerging Bcl-2 inhibitors for the treatment of cancer. Expert Opin. Emerg. Drugs 2011, 16, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.R.; Vaillant, F.; Lim, E.; Lee, L.; Breslin, K.; Feleppa, F.; Deb, S.; Ritchie, M.E.; Takano, E.; Ward, T.; et al. Sensitization of BCL-2-expressing breast tumors to chemotherapy by the BH3 mimetic ABT-737. Proc. Natl. Acad. Sci. USA 2012, 109, 2766–2771. [Google Scholar] [CrossRef]
- Chen, J.; Jin, S.; Abraham, V.; Huang, X.; Liu, B.; Mitten, M.J.; Nimmer, P.; Lin, X.; Smith, M.; Shen, Y.; et al. The Bcl-2/Bcl-X L/Bcl-w inhibitor, navitoclax, enhances the activity of chemotherapeutic agents in vitro and in vivo. Mol. Cancer Ther. 2011, 10, 2340–2349. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Xia, J.; Zou, J.; Wang, Q.; Ma, Q.; Sun, R.; Liao, H.; Xu, L.; Wang, D.; Guo, X. The PI3K/mTOR dual inhibitor NVP-BEZ235 stimulates mutant p53 degradation to exert anti-tumor effects on triple-negative breast cancer cells. FEBS Open Bio 2020, 10, 535–545. [Google Scholar] [CrossRef]
- Muranen, T.; Selfors, L.M.; Worster, D.T.; Iwanicki, M.P.; Song, L.; Morales, F.C.; Gao, S.; Mills, G.B.; Brugge, J.S. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell 2012, 21, 227–239. [Google Scholar] [CrossRef]
- Kutuk, O.; Letai, A. Alteration of the mitochondrial apoptotic pathway is key to acquired paclitaxel resistance and can be reversed by ABT-737. Cancer Res. 2008, 68, 7985–7994. [Google Scholar] [CrossRef]
- Séveno, C.; Loussouarn, D.; Bréchet, S.; Campone, M.; Juin, P.; Barillé-Nion, S. γ-Secretase inhibition promotes cell death, Noxa upregulation, and sensitization to BH3 mimetic ABT-737 in human breast cancer cells. Breast Cancer Res. 2012, 14, R96. [Google Scholar] [CrossRef] [PubMed]
- Lok, S.W.; Whittle, J.R.; Vaillant, F.; Teh, C.E.; Lo, L.L.; Policheni, A.N.; Bergin, A.R.T.; Desai, J.; Ftouni, S.; Gandolfo, L.C.; et al. A phase Ib dose-escalation and expansion study of the BCL2 inhibitor venetoclax combined with tamoxifen in ER and BCL2– positive metastatic breast cancer. Cancer Discov. 2019, 9, 354–369. [Google Scholar] [CrossRef] [PubMed]
- Lindeman, G.J.; Bowen, R.; Jerzak, K.J.; Song, X.; Decker, T.; Boyle, F.M.; McCune, S.L.; Armstrong, A.; Shannon, C.M.; Bertelli, G.; et al. Results from VERONICA: A randomized, phase II study of second-/third-line venetoclax (VEN) + fulvestrant (F) versus F alone in estrogen receptor (ER)-positive, HER2-negative, locally advanced, or metastatic breast cancer (LA/MBC). J. Clin. Oncol. 2021, 39 (Suppl. 15), 1004. [Google Scholar] [CrossRef]
- Williams, M.M.; Elion, D.L.; Rahman, B.; Hicks, D.J.; Sanchez, V.; Cook, R.S. Therapeutic inhibition of Mcl-1 blocks cell survival in estrogen receptor-positive breast cancers. Oncotarget 2019, 10, 5389–5402. [Google Scholar] [CrossRef]
- Kotschy, A.; Szlavik, Z.; Murray, J.; Davidson, J.; Maragno, A.L.; Le Toumelin-Braizat, G.; Chanrion, M.; Kelly, G.L.; Gong, J.N.; Moujalled, D.M.; et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 2016, 538, 477–482. [Google Scholar] [CrossRef]
- Campbell, K.J.; Mason, S.M.; Winder, M.L.; Willemsen, R.B.E.; Cloix, C.; Lawson, H.; Rooney, N.; Dhayade, S.; Sims, A.H.; Blyth, K.; et al. Breast cancer dependence on MCL-1 is due to its canonical anti-apoptotic function. Cell Death Differ. 2021, 28, 2589–2600. [Google Scholar] [CrossRef]
- Alcon, C.; Zañudo, J.G.T.; Albert, R.; Wagle, N.; Scaltriti, M.; Letai, A.; Samitier, J.; Montero, J. Er+ breast cancer strongly depends on mcl-1 and bcl-xl anti-apoptotic proteins. Cells 2021, 10, 1659. [Google Scholar] [CrossRef] [PubMed]
- Wuillème-Toumi, S.; Trichet, V.; Gomez-Bougie, P.; Gratas, C.; Bataille, R.; Amiot, M. Reciprocal protection of Mcl-1 and Bim from ubiquitin-proteasome degradation. Biochem. Biophys. Res. Commun. 2007, 361, 865–869. [Google Scholar] [CrossRef]
- Oudenaarden, C.R.L.; van de Ven, R.A.H.; Derksen, P.W.B. Re-inforcing the cell death army in the fight against breast cancer. J. Cell Sci. 2018, 131, jcs212563. [Google Scholar] [CrossRef]
- Lam, L.; Hu, X.; Aktary, Z.; Andrews, D.W.; Pasdar, M. Tamoxifen and ICI 182,780 increase Bcl-2 levels and inhibit growth of breast carcinoma cells by modulating PI3K/AKT, ERK and IGF-1R pathways independent of ERα. Breast Cancer Res. Treat. 2009, 118, 605–621. [Google Scholar] [CrossRef]
- Moore, V.D.G.; Brown, J.R.; Certo, M.; Love, T.M.; Novina, C.D.; Letai, A. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J. Clin. Investig. 2007, 117, 112–121. [Google Scholar] [CrossRef]
- Tanos, R.; Karmali, D.; Nalluri, S.; Goldsmith, K.C. Select Bcl-2 antagonism restores chemotherapy sensitivity in high-risk neuroblastoma. BMC Cancer 2016, 16, 97. [Google Scholar] [CrossRef]
- de Vos, S.; Leonard, J.P.; Friedberg, J.W.; Zain, J.; Dunleavy, K.; Humerickhouse, R.; Hayslip, J.; Pesko, J.; Wilson, W.H. Safety and efficacy of navitoclax, a BCL-2 and BCL-XL inhibitor, in patients with relapsed or refractory lymphoid malignancies: Results from a phase 2a study. Leuk. Lymphoma 2021, 62, 810–818. [Google Scholar] [CrossRef]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Drago, J.Z.; Chandarlapaty, S.; Jhaveri, K. Targeting Apoptosis: A New Paradigm for the Treatment of Estrogen Receptor—Positive Breast Cancer. Cancer Discov. 2019, 9, 323–325. [Google Scholar] [CrossRef]
- Lindeman, G.J.; Bardia, A.; Bowen, R.; Flechais, A.; Lei, G.; Hogea, A.; Mobasher, M.; Rafii, S. Randomized phase II trial of venetoclax + fulvestrant versus fulvestrant in estrogen receptor+, HER2– locally advanced or metastatic breast cancer following recurrence or progression during or after a CDK4/6 inhibitor: VERONICA. J. Clin. Oncol. 2019, 37 (Suppl. 15), TPS1108. [Google Scholar] [CrossRef]
- Williams, M.M.; Lee, L.; Werfel, T.; Joly, M.M.M.; Hicks, D.J.; Rahman, B.; Elion, D.; McKernan, C.; Sanchez, V.; Estrada, M.V.; et al. Intrinsic apoptotic pathway activation increases response to anti-estrogens in luminal breast cancers article. Cell Death Dis. 2018, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Strasser, A.; Cory, S.; Adams, J.M. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 2011, 30, 3667–3683. [Google Scholar] [CrossRef]
- Marusyk, A.; Tabassum, D.P.; Janiszewska, M.; Place, A.E.; Trinh, A.; Rozhok, A.I.; Pyne, S.; Guerriero, J.L.; Shu, S.; Ekram, M.; et al. Spatial proximity to fibroblasts impacts molecular features and therapeutic sensitivity of breast cancer cells influencing clinical outcomes. Cancer Res. 2016, 76, 6495–6506. [Google Scholar] [CrossRef] [PubMed]
- Sansone, P.; Berishaj, M.; Rajasekhar, V.K.; Ceccarelli, C.; Chang, Q.; Strillacci, A.; Savini, C.; Shapiro, L.; Bowman, R.L.; Mastroleo, C.; et al. Evolution of cancer stem-like cells in endocrine-resistant metastatic breast cancers is mediated by stromal microvesicles. Cancer Res. 2017, 77, 1927–1941. [Google Scholar] [CrossRef] [PubMed]
- Louault, K.; Bonneaud, T.L.; Séveno, C.; Gomez-Bougie, P.; Nguyen, F.; Gautier, F.; Bourgeois, N.; Loussouarn, D.; Kerdraon, O.; Barillé-Nion, S.; et al. Interactions between cancer-associated fibroblasts and tumor cells promote MCL-1 dependency in estrogen receptor-positive breast cancers. Oncogene 2019, 38, 3261–3273. [Google Scholar] [CrossRef]
- Wang, H.; Guo, M.; Wei, H.; Chen, Y. Targeting MCL-1 in cancer: Current status and perspectives. J. Hematol. Oncol. 2021, 14, 67. [Google Scholar] [CrossRef]
- Townsend, P.A.; Kozhevnikova, M.V.; Cexus, O.N.F.; Zamyatnin, A.A.; Soond, S.M. BH3-mimetics: Recent developments in cancer therapy. J. Exp. Clin. Cancer Res. 2021, 40, 355. [Google Scholar] [CrossRef]
- Thomas, R.L.; Roberts, D.J.; Kubli, D.A.; Lee, Y.; Quinsay, M.N.; Owens, J.B.; Fischer, K.M.; Sussman, M.A.; Miyamoto, S.; Gustafsson, Å.B. Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev. 2013, 27, 1365–1377. [Google Scholar] [CrossRef]
- Wang, X.; Bathina, M.; Lynch, J.; Koss, B.; Calabrese, C.; Frase, S.; Schuetz, J.D.; Rehg, J.E.; Opferman, J.T. Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes Dev. 2013, 27, 1351–1364. [Google Scholar] [CrossRef]
- Kehr, S.; Vogler, M. It’s time to die: BH3 mimetics in solid tumors. Biochim. Biophys. Acta-Mol. Cell Res. 2021, 1868, 118987. [Google Scholar] [CrossRef]
- Kehr, S.; Haydn, T.; Bierbrauer, A.; Irmer, B.; Vogler, M.; Fulda, S. Targeting BCL-2 proteins in pediatric cancer: Dual inhibition of BCL-XL and MCL-1 leads to rapid induction of intrinsic apoptosis. Cancer Lett. 2020, 482, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Carter, R.J.; Milani, M.; Butterworth, M.; Alotibi, A.; Harper, N.; Yedida, G.; Greaves, G.; Al-Zebeeby, A.; Jorgensen, A.L.; Schache, A.G.; et al. Exploring the potential of BH3 mimetic therapy in squamous cell carcinoma of the head and neck. Cell Death Dis. 2019, 10, 912. [Google Scholar] [CrossRef] [PubMed]
- Willis, S.N.; Chen, L.; Dewson, G.; Wei, A.; Naik, E.; Fletcher, J.I.; Adams, J.M.; Huang, D.C.S. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005, 19, 1294–1305. [Google Scholar] [CrossRef] [PubMed]
- Gillissen, B.; Essmann, F.; Graupner, V.; Stärck, L.; Radetzki, S.; Dörken, B.; Schulze-Osthoff, K.; Daniel, P.T. Induction of cell death by the BH3-only Bcl-2 homolog Nbk/Bik is mediated by an entirely Bax-dependent mitochondrial pathway. EMBO J. 2003, 22, 3580–3590. [Google Scholar] [CrossRef]
- Viedma-Rodriguez, R.; Baiza-Gutman, L.A.; García-Carranća, A.; Moreno-Fierros, L.; Salamanca-Gómez, F.; Arenas-Aranda, D. Suppression of the death gene BIK is a critical factor for resistance to tamoxifen in MCF-7 breast cancer cells. Int. J. Oncol. 2013, 43, 1777–1786. [Google Scholar] [CrossRef]
- Pandya, V.; Githaka, J.M.; Patel, N.; Veldhoen, R.; Hugh, J.; Damaraju, S.; McMullen, T.; Mackey, J.; Goping, I.S. BIK drives an aggressive breast cancer phenotype through sublethal apoptosis and predicts poor prognosis of ER-positive breast cancer. Cell Death Dis. 2020, 11, 448. [Google Scholar] [CrossRef]
- Ichim, G.; Tait, S.W.G. A fate worse than death: Apoptosis as an oncogenic process. Nat. Rev. Cancer 2016, 16, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Chen, J. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef]
- Choudhuri, T.; Pal, S.; Agwarwal, M.L.; Das, T.; Sa, G. Curcumin induces apoptosis in human breast cancer cells through p53-dependent Bax induction. FEBS Lett. 2002, 512, 334–340. [Google Scholar] [CrossRef]
- Le Pen, J.; Laurent, M.; Sarosiek, K.; Vuillier, C.; Gautier, F.; Montessuit, S.; Martinou, J.C.; Letai, A.; Braun, F.; Juin, P.P. Constitutive p53 heightens mitochondrial apoptotic priming and favors cell death induction by BH3 mimetic inhibitors of BCL-xL. Cell Death Dis. 2016, 7, e2083. [Google Scholar] [CrossRef]
- Hur, J.; Bell, D.W.; Dean, K.L.; Coser, K.R.; Hilario, P.C.; Okimoto, R.A.; Tobey, E.M.; Smith, S.L.; Isselbacher, K.J.; Shioda, T. Regulation of expression of BIK proapoptotic protein in human breast cancer cells: p53-dependent induction of BIK mRNA by fulvestrant and proteasomal degradation of BIK protein. Cancer Res. 2006, 66, 10153–10161. [Google Scholar] [CrossRef]
- Bailey, S.T.; Shin, H.; Westerling, T.; Liu, X.S.; Brown, M. Estrogen receptor prevents p53-dependent apoptosis in breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 18060–18065. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BH3 Mimetic | Target | Breast Cancer Model | Effects | Reference |
|---|---|---|---|---|
| ABT-263 (Navitoclax) | Bcl-2, Bcl-xL, Bcl-w | MCF-7 cell line | Synergistic activity with camptothecin, docetaxel, etoposide, rapamycin | Chen et al. [131] |
| ABT-737 | Bcl-2, Bcl-xL, Bcl-w | ER+ PDX models, MCF-7 cells | Improved tumor and cell response to tamoxifen and PI3K/mTOR inhibitor | Vaillant et al. [114] |
| ABT-737 | Bcl-2, Bcl-xL, Bcl-w | MCF-7 cell line | Sensitization of resistant cells to paclitaxel | Kutuk and Letai [134] |
| ABT-737 | Bcl-2, Bcl-xL, Bcl-w | MCF-7 cells and ER+ primary breast tumor cells | Synergistic activity with the γ-secretase inhibitor GSIXII in apoptosis induction | Seveno et al. [135] |
| ABT-737 | Bcl-2, Bcl-xL, Bcl-w | T47D | Increased activity of PI3K/mTOR inhibitor | Muranen et al. [133] |
| ABT-737 | Bcl-2, Bcl-xL, Bcl-w | PDX model | Sensitization to docetaxel-mediated cell death | Oakes et al. [130] |
| ABT-199 (Venetoclax) | Bcl-2 | ER+ PDX models, MCF-7 cells | Improved tumor and cell response to tamoxifen and PI3K/mTOR inhibitor | Vaillant et al. [114] |
| ABT-199 (Venetoclax) | Bcl-2 | ER+, Bcl-2-expressing metastatic breast cancers | Phase I clinical trial of venetoclax in combination with tamoxifen; ORR 45% and CBR 75% | Lok et al. [136] |
| ABT-199 (Venetoclax) | Bcl-2 | ER+, locally advanced/metastatic breast cancer | VERONICA: A randomized, phase II study of second-/third-line venetoclax + fulvestrant); No increase in CBR | Lindeman et al. [137] |
| VU661013 | Mcl-1 | MCF-7, T47D, HCC1428, MCF-7 xenografts | Increases apoptosis induction in cells and reduces tumor volume in combination with with ABT-263 | Williams et al. [138] |
| A-1210477 | Mcl-1 | ER+ breast cancer cell lines | Synergistic antiproliferative activity with navitoclax | Xiao et al. [125] |
| S63845 | Mcl-1 | BT474 cell line | Increased cytotoxic activity of lapatinib | Kotschy et al. [139] |
| S63845 | Mcl-1 | MMTV-PyMT xenografts | Reduction of tumor growth | Campbell et al. [140] |
| S63845 | Mcl-1 | MDA-MB-415, T47D cell lines | Synergistic pro-apoptotic activity with Bcl-xL inhibitor | Alcon et al. [141] |
| A-1331852 | Bcl-xL | MDA-MB-415, T47D cell lines | Synergistic pro-apoptotic activity with Mcl-1 inhibitor and AKT inhibitor (Ipatasertib) | Alcon et al. [141] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kawiak, A.; Kostecka, A. Regulation of Bcl-2 Family Proteins in Estrogen Receptor-Positive Breast Cancer and Their Implications in Endocrine Therapy. Cancers 2022, 14, 279. https://doi.org/10.3390/cancers14020279
Kawiak A, Kostecka A. Regulation of Bcl-2 Family Proteins in Estrogen Receptor-Positive Breast Cancer and Their Implications in Endocrine Therapy. Cancers. 2022; 14(2):279. https://doi.org/10.3390/cancers14020279
Chicago/Turabian StyleKawiak, Anna, and Anna Kostecka. 2022. "Regulation of Bcl-2 Family Proteins in Estrogen Receptor-Positive Breast Cancer and Their Implications in Endocrine Therapy" Cancers 14, no. 2: 279. https://doi.org/10.3390/cancers14020279
APA StyleKawiak, A., & Kostecka, A. (2022). Regulation of Bcl-2 Family Proteins in Estrogen Receptor-Positive Breast Cancer and Their Implications in Endocrine Therapy. Cancers, 14(2), 279. https://doi.org/10.3390/cancers14020279