
Glucose Limitation Sensitizes Cancer Cells to Selenite-Induced Cytotoxicity via SLC7A11-Mediated Redox Collapse


Abstract
:Simple Summary
Abstract
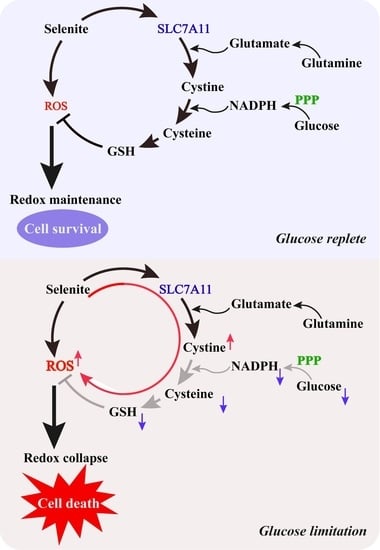
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture and Treatments
2.3. Crystal Violet Staining
2.4. Cell Death Evaluation
2.5. Determination of Cell ROS
2.6. GSH, NADP+, NADPH and Cysteine Measurement
2.7. Cystine Measurement by LC-MS
2.8. LPO, LIP Measurement
2.9. RNA Interference
2.10. Western Blotting
2.11. Xenograft Tumor Models
2.12. Statistical Analysis
3. Results
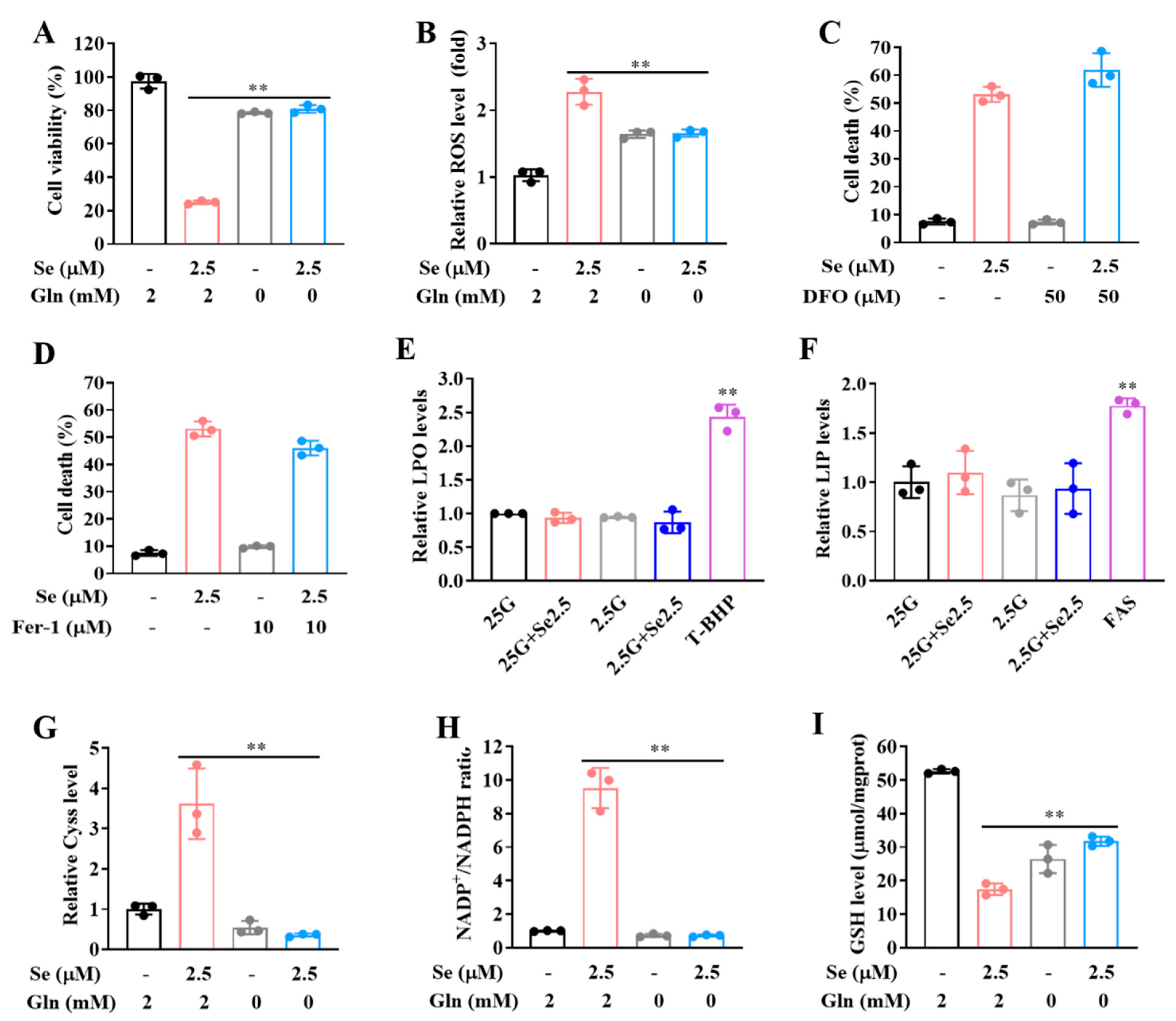
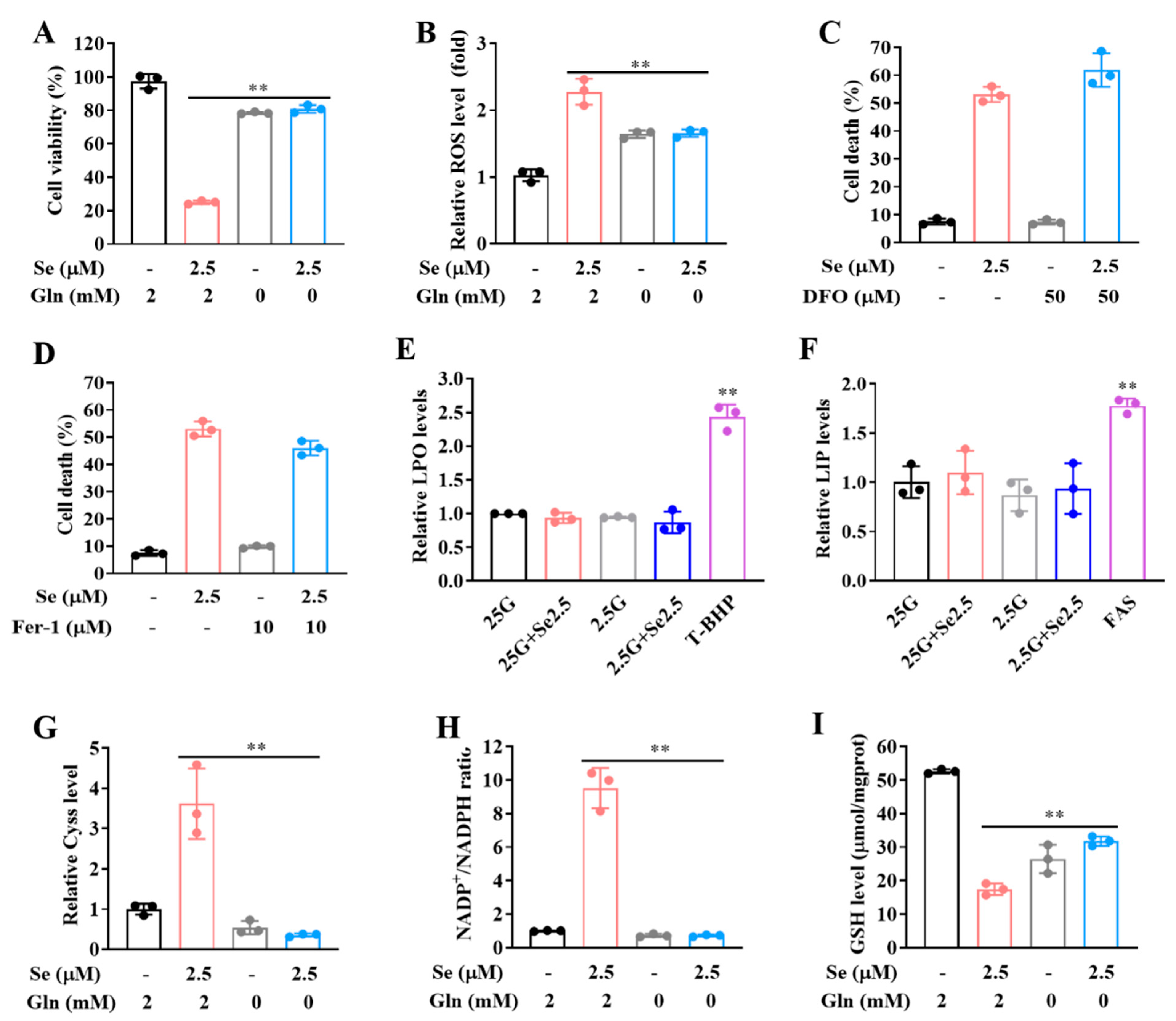
3.1. Glucose Limitation Sensitizes Cancer Cells to Selenite-Mediated Cytotoxic Effect
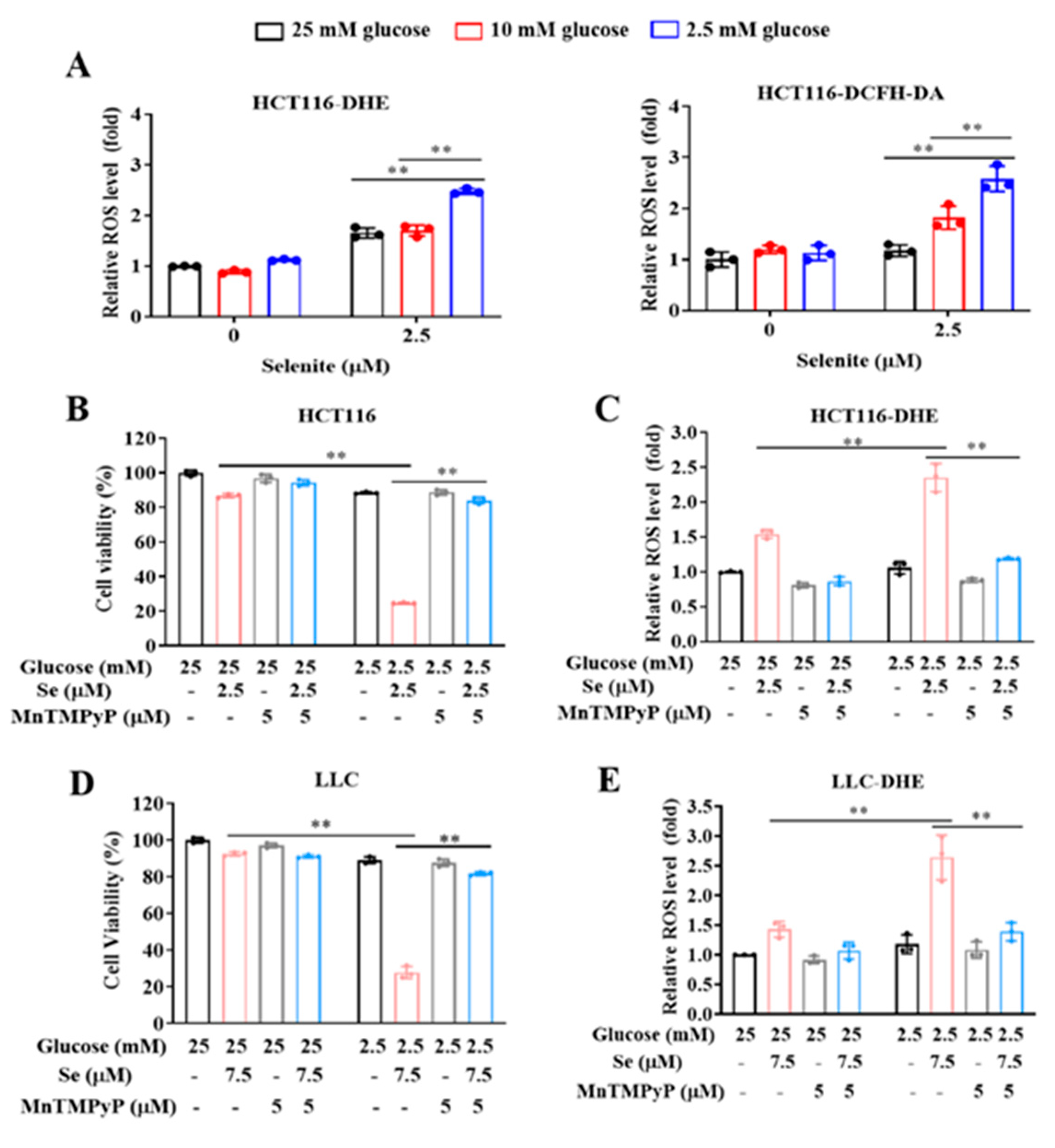
3.2. The Sensitization Effect of Glucose Limitation on Selenite Is Attributed to Elevated ROS Generation
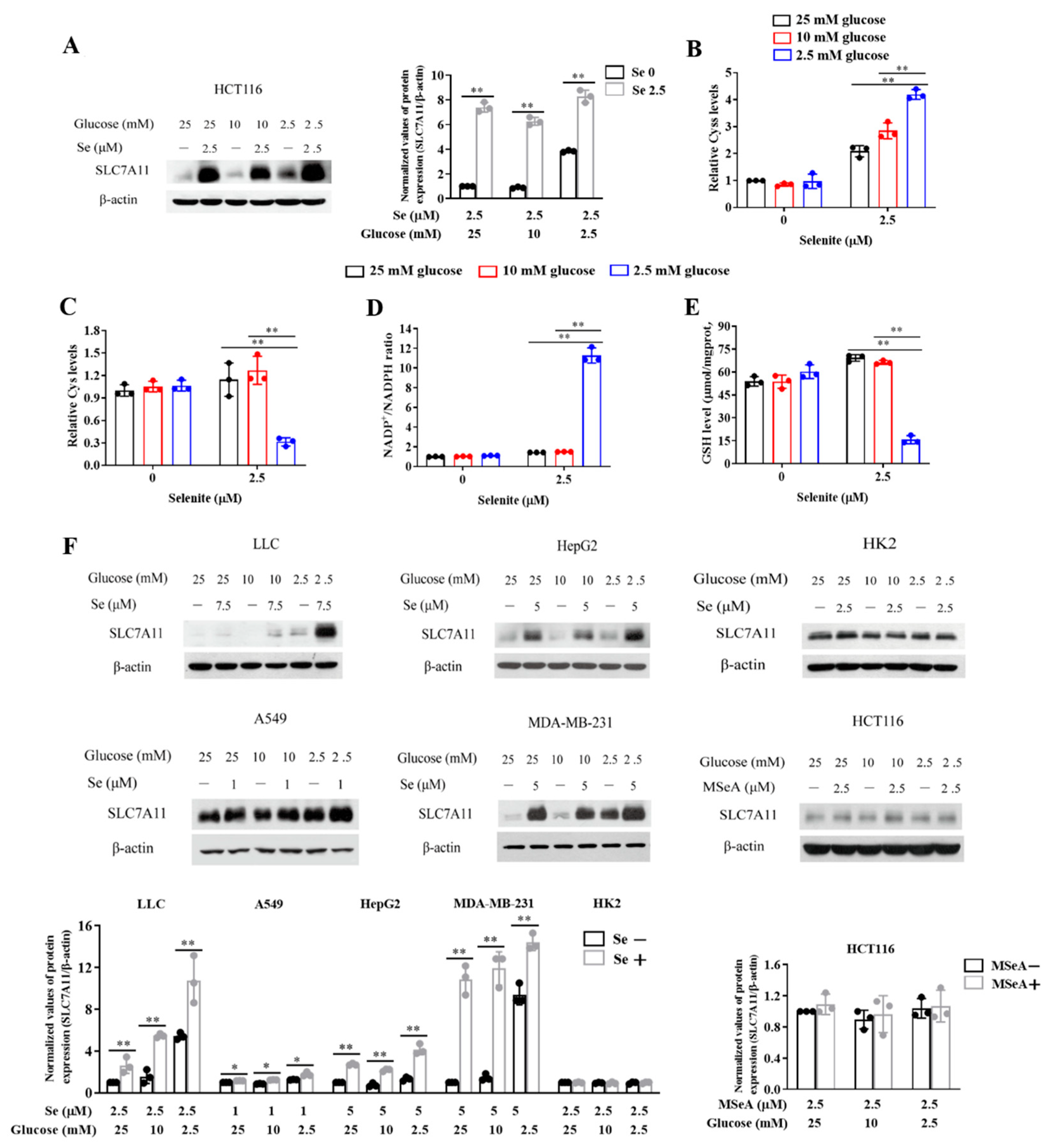
3.3. Expression of SLC7A11 Is Up-Regulated by Selenite, Accompanied by Cystine Accumulation, Cysteine Reduction and NADPH Depletion in the Context of Glucose Deprivation
3.4. SLC7A11-Mediated Cystine Accumulation and NADPH Depletion Contribute to the Elevated ROS Generation and Enhanced Cytotoxicity Induction by Combination of Selenite and Glucose Deprivation
3.5. Selenite/Glucose Deprivation-Induced Cytotoxicity Is Independent of Ferroptosis
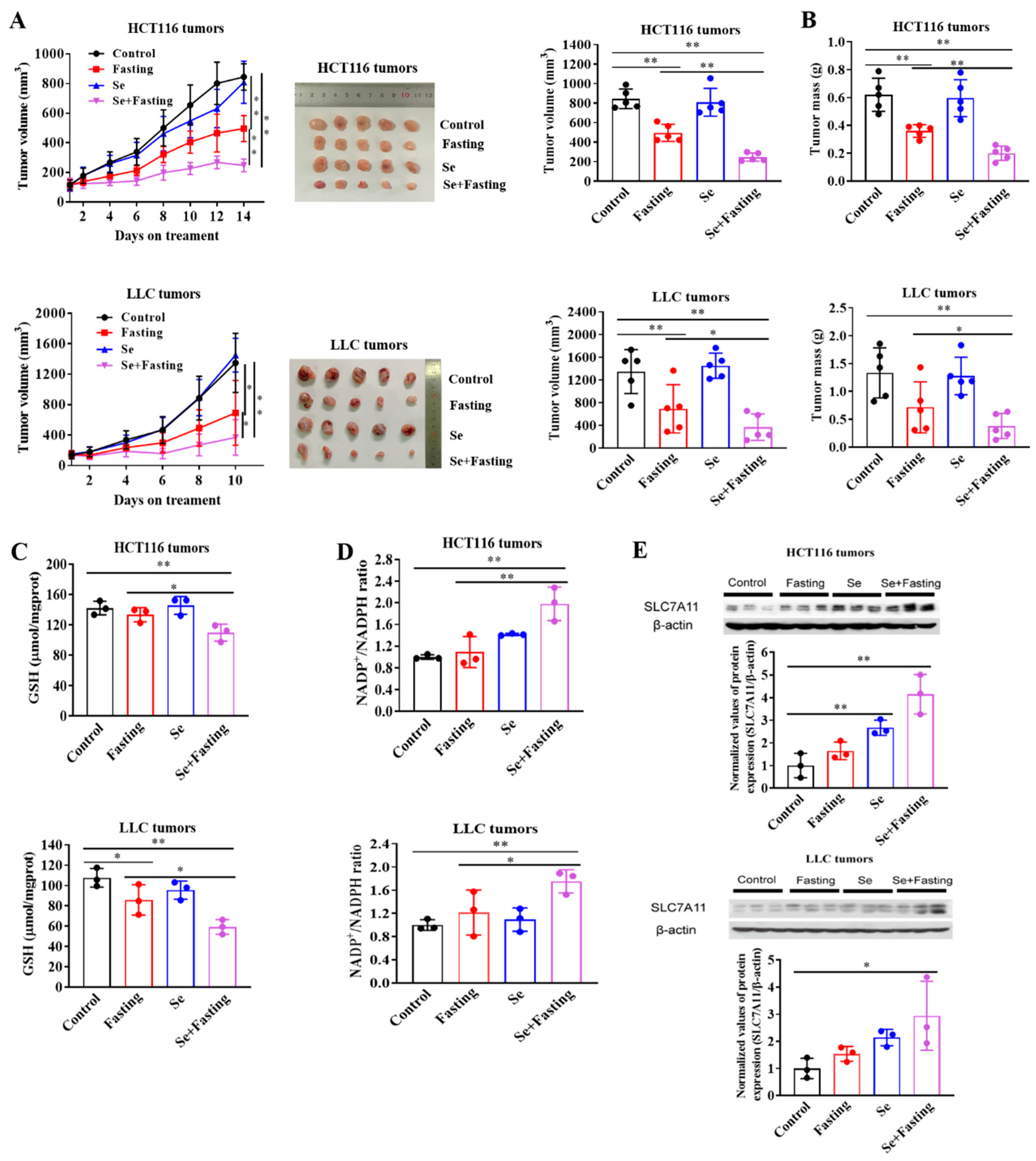
3.6. Fasting Improves Therapeutic Efficacy of Selenite In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Elgendy, M.; Cirò, M.; Hosseini, A.; Weiszmann, J.; Mazzarella, L.; Ferrari, E.; Cazzoli, R.; Curigliano, G.; De Censi, A.; Bonanni, B.; et al. Combination of hypoglycemia and metformin impairs tumor metabolic plasticity and growth by modulating the PP2A-GSK3Β-MCL-1 axis. Cancer Cell 2019, 35, 798–815. [Google Scholar] [CrossRef]
- Kim, S.W.; Cha, M.; Lee, S.-K.; Song, B.; Jin, X.; Lee, J.M.; Park, J.H.; Lee, J.D. Curcumin treatment in combination with glucose restriction inhibits intracellular alkalinization and tumor growth in hepatoma cells. Int. J. Mol. Sci. 2019, 20, 2375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Tano, M.; Raucci, F.; Vernieri, C.; Caffa, I.; Buono, R.; Fanti, M.; Brandhorst, S.; Curigliano, G.; Nencioni, A.; de Braud, F.; et al. Synergistic effect of fasting-mimicking diet and vitamin C against KRAS mutated cancers. Nat. Commun. 2020, 11, 2332–2342. [Google Scholar] [CrossRef]
- Tang, X.; Li, G.; Shi, L.; Su, F.; Qian, M.; Liu, Z.; Meng, Y.; Sun, S.; Li, J.; Liu, B. Combined intermittent fasting and ERK inhibition enhance the anti-tumor effects of chemotherapy via the GSK3beta-SIRT7 axis. Nat. Commun. 2021, 12, 5058–5075. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, B.; Mohd Omar, M.F.; Soong, R. The Warburg effect and drug resistance. Br. J. Pharmacol. 2016, 173, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Birsoy, K.; Possemato, R.; Lorbeer, F.K.; Bayraktar, E.C.; Thiru, P.; Yucel, B.; Tim Wang, T.; Chen, W.W.; Clish, C.B.; Sabatini, D.M. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 2014, 508, 108–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, G.; Martella, R.; Ravera, S.; Petretto, A.; Marimpietri, D.; Emionite, L.; Capitanio, S.; Bottone, G.; Orengo, A.; Marini, C.; et al. Fasting chemosensitizes tumor cells by affecting their metabolism. Cancer Res. 2014, 74, 3374. [Google Scholar]
- Mølck, C.; Ryall, J.; Failla, L.M.; Coates, J.L.; Pascussi, J.-M.; Heath, J.K.; Stewart, G.; Hollande, F. The A2b adenosine receptor antagonist PSB-603 promotes oxidative phosphorylation and ROS production in colorectal cancer cells via adenosine receptor independent mechanism. Cancer Lett. 2016, 383, 135–143. [Google Scholar] [CrossRef]
- Liu, X.; Olszewski, K.; Zhang, Y.; Lim, E.W.; Shi, J.; Zhang, X.; Zhang, J.; Lee, H.; Koppula, P.; Lei, G.; et al. Cystine transporter regulation of pentose phosphate pathway dependency and disulfide stress exposes a targetable metabolic vulnerability in cancer. Nat. Cell Biol. 2020, 22, 476–486. [Google Scholar] [CrossRef]
- Olm, E.; Fernandes, A.P.; Hebert, C.; Rundlöf, A.; Larsen, E.H.; Danielsson, O.; Björnstedt, M. Extracellular thiol-assisted selenium uptake dependent on the xc- cystine transporter explains the cancer-specific cytotoxicity of selenite. Proc. Natl. Acad. Sci. USA 2009, 106, 11400–11405. [Google Scholar] [CrossRef] [Green Version]
- Carlisle, A.E.; Lee, N.; Matthew-Onabanjo, A.N.; Spears, M.E.; Park, S.J.; Youkana, D.; Doschi, M.B.; Peppers, A.; Li, R.; Joseph, A.B.; et al. Selenium detoxification is required for cancer-cell survival. Nat. Metab. 2020, 2, 603–611. [Google Scholar] [CrossRef]
- Koppula, P.; Zhuang, L.; Gan, B. Cystine transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 2021, 12, 599–620. [Google Scholar] [CrossRef]
- Bekeschus, S.; Eisenmann, S.; Sagwal, K.S.; Bodnar, Y.; Moritz, J.; Poschkamp, B.; Stoffels, I.; Emmert, S.; Madesh, M.; Weltmann, K.-D.; et al. xCT (SLC7A11) expression confers intrinsic resistance to physical plasma treatment in tumor cells. Redox Biol. 30 2020, 30, 101423–101432. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Zhao, C.; Hu, H.; Yin, S. Food sources of selenium and its relationship with chronic diseases. Nutrients 2021, 13, 1739. [Google Scholar] [CrossRef] [PubMed]
- Cavalieri, R.R.; Scott, K.G.; Sairenji, E. Selenite (75Se) as a tumor-localizing agent in man. J. Nucl. Med. 1966, 7, 197–208. [Google Scholar]
- Wu, X.; Zhao, G.; He, Y.; Wang, W.; Yang, C.S.; Zhang, J. Pharmacological mechanisms of the anticancer action of sodium selenite against peritoneal cancer in mice. Pharmacol. Res. 2019, 147, 104360–104369. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; An, J.; Guo, J.; Wu, Y.; Yang, L.; Dai, J.; Gong, K.; Miao, S.; Xi, S.; Du, J. Sodium selenite attenuates lung adenocarcinoma progression by repressing SOX2-mediated stemness. Cancer Chemother. Pharmaco.l 2018, 81, 885–895. [Google Scholar] [CrossRef]
- Zhang, T.; Zhao, G.; Zhu, X.; Jiang, K.; Wu, H.; Deng, G.; Qui, C. Sodium selenite induces apoptosis via ROS-mediated NF-κB signaling and activation of the Bax caspase-9-caspase-3 axis in 4T1 cells. J. Cell Physiol. 2019, 234, 2511–2522. [Google Scholar] [CrossRef]
- Husbeck, B.; Peehl, D.M.; Knox, S.J. Redox modulation of human prostate carcinoma cells by selenite increases radiation-induced cell killing. Free Radic. Biol. Med. 2005, 38, 50–57. [Google Scholar] [CrossRef]
- Brodin, O.; Eksborg, S.; Wallenberg, M.; Asker-Hagelberg, C.; Larsen, E.H.; Mohlkert, D.; Lenneby-Helleday, C.; Jacobsson, H.; Linder, S.; Misra, S.; et al. Pharmacokinetics and toxicity of sodium selenite in the treatment of patients with carcinoma in a phase I clinical trial: The SECAR study. Nutrients 2015, 7, 4978–4994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frenkel, G.D.; Caffrey, P.B. Prevention of carboplatin-induced resistance in human ovarian tumor xenografts by selenite. Anticancer Res. 2013, 33, 4249–4254. [Google Scholar]
- Zhao, R.; Xiang, N.; Domann, F.E.; Zhong, W. Expression of p53 enhances selenite-induced superoxide production and apoptosis in human prostate cancer cells. Cancer Res. 2006, 66, 2296–2304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, M.; Ye, L.; Yin, S.; Lu, X.; Liu, X.; Lu, S.; Cui, J.; Fan, L.; Kaplowitz, N.; Hu, H. Glycycoumarin protects mice against acetaminophen-induced liver injury predominantly via activating sustained autophagy. Br. J. Pharmacol. 2018, 175, 3747–3757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Yang, X.; Zhang, Z.; Xu, H. Both calcium and ROS as common signals mediate NaSeO3-induced apoptosis in SW480 human colonic carcinoma cells. J. Inorg. Biochem. 2003, 97, 221–230. [Google Scholar] [CrossRef]
- Kim, E.H.; Sohn, S.; Kwon, H.J.; Kim, S.U.; Kim, M.; Lee, S.; Choi, K.S. Sodium selenite induces superoxide-mediated mitochondrial damage and subsequent. Cancer Res. 2007, 67, 6314–6324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, W.; Oberley, T.D. Redox-mediated effects of selenium on apoptosis and cell cycle in the LNCaP human prostate cancer cell line. Cancer Res. 2001, 61, 7071–7078. [Google Scholar]
- Zhang, D.; Li, J.; Wang, F.; Hu, J.; Wang, S.; Sun, Y. 2-Deoxy-D-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett. 2014, 355, 176–183. [Google Scholar] [CrossRef]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of mitochondria in ferroptosis. Mol. Cell 2019, 73, 354–363. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Wu, D.; Duan, J.; Xiao, H.; Zhou, Y.; Zhao, L.; Feng, Y. NRF2 regulates the sensitivity of human NSCLC cells to cystine deprivation-induced ferroptosis via FOCAD-FAK signaling pathway. Reddox Biol. 2020, 37, 101702–101714. [Google Scholar] [CrossRef]
- Kang, Y.P.; Mockabee-Macias, A.; Jiang, C.; Falzone, A.; Prieto-Farigua, N.; Stone, E.; Harris, I.S.; De Nicola, G.M. Non-canonical Glutamate-Cysteine Ligase Activity Protects against Ferroptosis. Cell Metab. 2021, 33, 174–189. [Google Scholar] [CrossRef]
- Gao, M.; Monian, P.; Jiang, X. Metabolism and iron signaling in ferroptotic cell death. Oncotarget 2015, 6, 35145–35146. [Google Scholar] [CrossRef]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and transferrin regulate ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, T.L.; Kiersgaard, M.K.; Mikkelsen, L.F.; Sørensen, D.B. Fasting of male mice—Effects of time point of initiation and duration on clinical chemistry parameters and animal welfare. Lab. Anim. UK 2019, 53, 587–597. [Google Scholar] [CrossRef]
- Bonfim, N.E.S.M.; Baranoski, A.; Mantovani, M.S. Cytotoxicity of sodium selenite in HaCaT cells induces cell death and alters the mRNA expression of PUMA, ATR, and mTOR genes. J. Trace Elem. Med. Biol. 2020, 62, 126605–126613. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, Y.; Zhang, H.; Liu, J. Safety assessment and comparison of sodium selenite and bioselenium obtained from yeast in mice. Biomed. Res. Int. 2017, 2017, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, M.; Wang, Y.Z.; Gout, P.W. The x(c)- cystine/glutamate antiporter: A potential target for therapy of cancer and other diseases. J. Cell Physiol. 2008, 215, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Koppula, P.; Zhang, Y.; Zhuang, L.; Gan, B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun. 2018, 38, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhang, Y.; Zhuang, L.; Olszewski, K.; Gan, B. NADPH debt drives redox bankruptcy: SLC7A11/xCT-mediated cystine uptake as a double-edge sword in cellular redox regulation. Genes Dis. 2021, 6, 731–745. [Google Scholar] [CrossRef]
- Deponte, M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. BBA Gen. Subj. 2013, 1830, 3217–3266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.K.M.; Delaidelli, A.; Minaker, S.W.; Zhang, H.; Colovic, M.; Yang, H.; Negri, G.L.; von Karstedt, S.; Lockwood, W.W.; Schaffer, P. Cystine/glutamate antiporter xCT (SLC7A11) facilitates oncogenic RAS transformation by preserving intracellular redox balance. Proc. Natl. Acad. Sci. USA 2019, 166, 9433–9442. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Wung, C.; Chen, M.; Chen, C.; Yin, P.; Yeh, T.-S.; Chang, Y.-L.; Chou, Y.-C.; Hung, H.H.; Lee, H.-C. Activated integrated stress response induced by salubrinal promotes cisplatin resistance in human gastric cancer cells via enhanced xCT expression and glutathione biosynthesis. Int. J. Mol. Sci. 2018, 19, 3389. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, H.; Okabe, H.; Beppu, T.; Chikamoto, A.; Hayashi, H.; Imai, K.; Nakagawa, S.; Ishimoto, T.; Miyake, K. Cystine/glutamic acid transporter is a novel marker for predicting poor survival in patients with hepatocellulcar carcinoma. Oncol. Rep. 2013, 29, 685–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Yee, D. IGF-1 regulates redox status in breast cancer by activating the amino acid transport molecule xC-. Cancer Res. 2014, 74, 2295–2305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Allouch, A.; Paoletti, A.; Leteur, C.; Mirjolet, C.; Martins, I.; Voisin, L.; Law, F.; Dakhli, H.; Mintet, E. NOX2-dependent ATM kinase activation dictates pro-inflammatory macrophage phenotype and improves effectiveness to radiation therapy. Cell Death Differ. 2017, 24, 1632–1644. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Min, K.H.; Lee, H.J.; Hwang, Y.; Lee, S.C. Fenton-like reaction performing mineralized nanocarriers as oxidative stress amplifying anticancer agents. J. Ind. Eng. Chem. 2019, 80, 829–837. [Google Scholar] [CrossRef]
- Hu, H.; Jiang, C.; Schuster, T.; Li, G.; Daniel, P.T.; Lu, J. Inorganic selenium sensitizes prostate cancer cells to TRAIL-induced apoptosis through superoxide/p53/Bax-mediated activation of mitochondrial pathway. Mol. Cancer Ther. 2006, 5, 1873–1882. [Google Scholar] [CrossRef] [Green Version]
- Klein, E.A.; Thompson, I.M., Jr.; Tangen, C.M.; Crowley, J.J.; Lucia, M.S.; Goodman, P.J.; Minasian, L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M.; et al. Vitamin E and the risk of prostate cancer: The selenium and vitamin E cancer prevention trial (SELECT). JAMA J. Am. Med. Assoc. 2011, 306, 1549–1556. [Google Scholar] [CrossRef]
- Li, G.; Lee, H.; Wang, Z.; Hu, H.; Liao, J.D.; Watts, J.C.; Combs, G.F., Jr.; Lü, J. Superior in vivo inhibitory efficacy of methylseleninic acid against human prostate cancer over selenomethionine or selenite. Carcinogenesis 2008, 29, 1005–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallenberg, M.; Misra, S.; Björnstedt, M. Selenium cytotoxicity in cancer. Basic Clin. Pharmacol. 2014, 114, 377–386. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, H.; Zhang, H.; Cao, L.; Cui, J.; Ma, X.; Zhao, C.; Yin, S.; Hu, H. Glucose Limitation Sensitizes Cancer Cells to Selenite-Induced Cytotoxicity via SLC7A11-Mediated Redox Collapse. Cancers 2022, 14, 345. https://doi.org/10.3390/cancers14020345
Chen H, Zhang H, Cao L, Cui J, Ma X, Zhao C, Yin S, Hu H. Glucose Limitation Sensitizes Cancer Cells to Selenite-Induced Cytotoxicity via SLC7A11-Mediated Redox Collapse. Cancers. 2022; 14(2):345. https://doi.org/10.3390/cancers14020345
Chicago/Turabian StyleChen, Hui, Han Zhang, Lixing Cao, Jinling Cui, Xuan Ma, Chong Zhao, Shutao Yin, and Hongbo Hu. 2022. "Glucose Limitation Sensitizes Cancer Cells to Selenite-Induced Cytotoxicity via SLC7A11-Mediated Redox Collapse" Cancers 14, no. 2: 345. https://doi.org/10.3390/cancers14020345