Simple Summary
Ovarian cancer (OC) is the second most common gynecologic malignancy and the most common cause of death among women with gynecologic cancer. Despite significant improvements having been made over the past decades, OC remains one of the most challenging malignancies to treat. Targeted therapies, such as PARPi, have emerged as one of the most interesting treatments for OC, particularly in women with BRCA1 or BRCA2 mutations. or those with a dysfunctional homologous recombination repair pathway. The purpose of our study is to address the role of NGS-targeted resequencing in the clinical routine of OC, focusing not only on BRCA1/2 but also on the homologous recombination repair genetic profile.
Abstract
Background: Pathogenic variants in homologous recombination repair (HRR) genes other than BRCA1/2 have been associated with a high risk of ovarian cancer (OC). In current clinical practice, genetic testing is generally limited to BRCA1/2. Herein, we investigated the mutational status of both BRCA1/2 and 5 HRR genes in 69 unselected OC, evaluating the advantage of multigene panel testing in everyday clinical practice. Methods: We analyzed 69 epithelial OC samples using an NGS custom multigene panel of the 5 HRR pathways genes, beyond the genetic screening routine of BRCA1/2 testing. Results: Overall, 19 pathogenic variants (27.5%) were detected. The majority (21.7%) of patients displayed a deleterious mutation in BRCA1/2, whereas 5.8% harbored a pathogenic variant in one of the HRR genes. Additionally, there were 14 (20.3%) uncertain significant variants (VUS). The assessment of germline mutational status showed that a small number of variants (five) were not detected in the corresponding blood sample. Notably, we detected one BRIP1 and four BRCA1/2 deleterious variants in the low-grade serous and endometrioid histology OC, respectively. Conclusion: We demonstrate that using a multigene panel beyond BRCA1/2 improves the diagnostic yield in OC testing, and it could produce clinically relevant results.
1. Introduction
In 2020, it was estimated that 21,750 new cases of ovarian cancer would have been diagnosed, and 13,940 women would have died from this serious disease [1]. According to the NIH (National Cancer Institute), Ovarian Cancer (OC) represents 1.2% of all new cancer cases in the US. It is a relatively rare form, and is the fifth leading cause of all cancer deaths in females and the first among gynecological cancers. OCs are diagnosed prevalently in fertile women, mainly between 40 and 50 years of age. OC is described as the “silent killer” because few signs or symptoms allow an early diagnosis: more than 70% of OCs are diagnosed when they have already progressed to stage III or IV. According to the latest data reported by NIH, the relative survival rate after 5 years is 48.6% [2].
Epithelial Ovarian Carcinomas (EOC) is the most common type of OC. EOC is a heterogeneous group with different morphological and biological features. Currently, five different sub-categories have been identified: High-Grade Serous Carcinomas (HGSC) (70%) or Low-Grade Serous Carcinomas (LGSC) (<5%), Endometrioid (10%), Mucinous (3%), and Clear Cell (10%) [3]. Risk factors for the development of OC account for a combination of environmental and genetic components and are strictly related to hormonal levels, in particular those involved in ovulation. In fact, an increased risk has been observed in postmenopausal women using hormonal replacement therapy for longer than 10 years. The number of at-term pregnancies and the use of oral contraceptives have been shown to lower the risk [4]. Most EOCs are sporadic, while in 15–25% of cases, there is evidence of a familial or inherited component [5]. Germline mutations in BRCA1 and BRCA2 (high penetrance genes) account for 20–25% of all HGSC [6]. Germline mutations in any of the MisMatch repair (MMR) genes (responsible for Lynch syndrome) have been found to be less common in OC, being present in 0.5–2% of unselected OC cases [7,8]. Several reports also demonstrated the existence of other genes at medium and low penetrance (e.g., BRIP1, RAD51C, RAD51D, BARD1, and PALB2), which have been associated with both Breast Cancer (BC) and OC risk [7,9]. These genes encode for proteins with a fundamental role in the homologous recombination (HR) mechanism (together with BRCA1 and BRCA2) by repairing DNA double-strand breaks (DSB). HR is a high-fidelity mechanism that repairs DSB using the intact sister chromatid as template, minimizing the loss of genetic information. Genes such as BARD1, BRIP1, PALB2, RAD51C, and RAD51D are all involved in the HR DNA repair mechanism interacting with both BRCA1 and BRCA2. Briefly, BRCA1 binds the site of DSB where it coordinates the repair mechanisms downstream [10]. BARD1 and BRIP1 both directly interact with BRCA1 mediating its function [11]. PALB2 dislodge BRCA2 onto DNA breaks, thus recruiting the RAD51 complex to stabilize the single strand DNA filament [12]. RAD51C and RAD51D are two RAD51 paralogs, part of the BCDX2 complex, which regulates the RAD51 filament formation [13]. Deficiency in HR process (HRD)—due to loss of function mutation in the aforementioned genes—causes the activation of alternative repair methods (e.g., non-homologous end joining). However, these methods are rather error-prone (small DNA deletions or insertions, copy number variants, or large chromosomal rearrangements) resulting in genomic instability and triggering tumorigenesis.
Patients showing a HRD profile would benefit from therapy with PARP inhibitors (PARPi) [14]. PARPi (e.g., olaparib, niraparib, and rucaparib) act throughout a mechanism of synthetic lethality preventing the repair of single strand breaks, thus leading to cancer cells apoptosis. Olaparib is approved in the management of patients affected by advanced OC as a first-line maintenance therapy, providing a substantial progression-free survival (PFS) benefit (as demonstrated in the phase III SOLO1 clinical trial) [15,16]. Other PARPi, such as Niraparib and Rucaparib, have also been approved in the United States and Europe as maintenance therapy regardless the HRD status, mostly in patients with platinum-sensitive recurrent OC [17,18,19].
Here, we report a molecular characterization of a cohort of 69 patients from Apulia (in the south-east of Italy) with primary EOC. In this study, 69 EOC samples were analyzed by next-generation sequencing (NGS) using a multigene panel including five HR pathways genes (BARD1, BRIP1, PALB2, RAD51C, and RAD51D), which represent the most frequently mutated genes (besides BRCA1/2) in OC [20].
The tumor samples were tested using an Oncomine™ BRCA Research Assay Thermo Fisher, Waltham, MA, USA, Devyser’s CE-IVD, and an Ion AmpliSeq Custom Panel (Thermo Fisher, Waltham, MA, USA) designed with five genes related to HR repair (BARD1, BRIP1, RAD51D, RAD51C, and PALB2). This study aimed to (1) carry out a clinical-molecular screening of 69 patients, to verify the correlation between type of mutation and clinical status of patients; (2) to define the mutation profiles of HR genes; and (3) evaluate the usefulness of multigene panel in terms of prevention protocols and therapeutic approaches.
2. Results
2.1. Cohort Characteristics
All the clinical and demographic characteristics of our cohort have been summarized in Table 1. The median age of OC onset was 61 years (range from 31 to 88). As far as concerns the histology, most cases were classified as serous (45 out of 69), with a prevalence of high-grade serous type (38 out of 45). Among the other 24 OC cases several histological types have been identified, including endometrioid (10), clear cell (5), and mucinous (1).

Table 1.
Clinical and demographic characteristics of our cohort.
2.2. Overall Mutation Spectrum
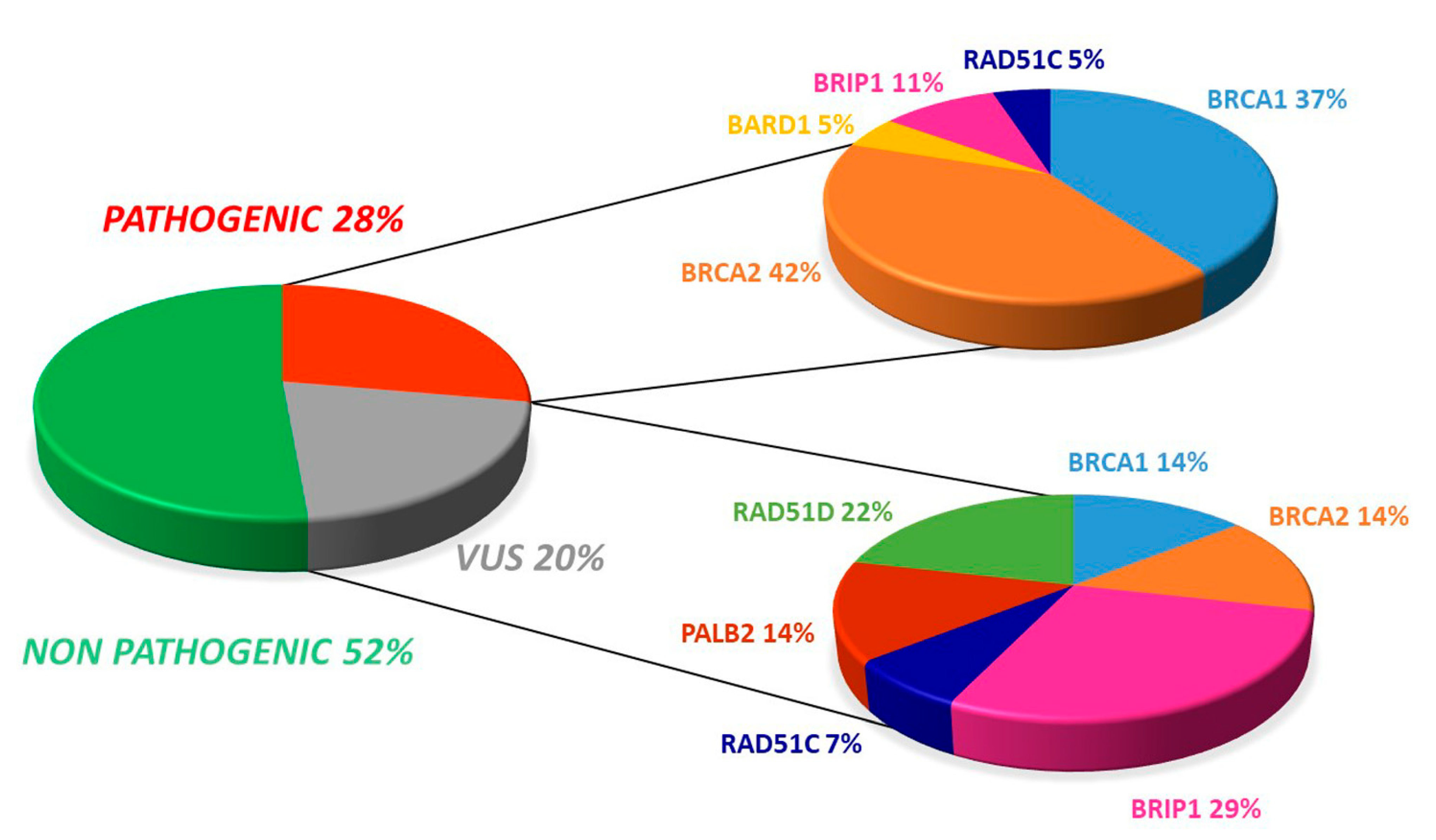
The somatic testing of our samples showed the presence of 19 pathogenic variants (27.5%). The distribution of mutations revealed that most (21.7%) of the 69 patients displayed a deleterious mutation in either BRCA1 or BRCA2, whereas 5.8% harbored a pathogenic variant in one of the HR pathway genes.
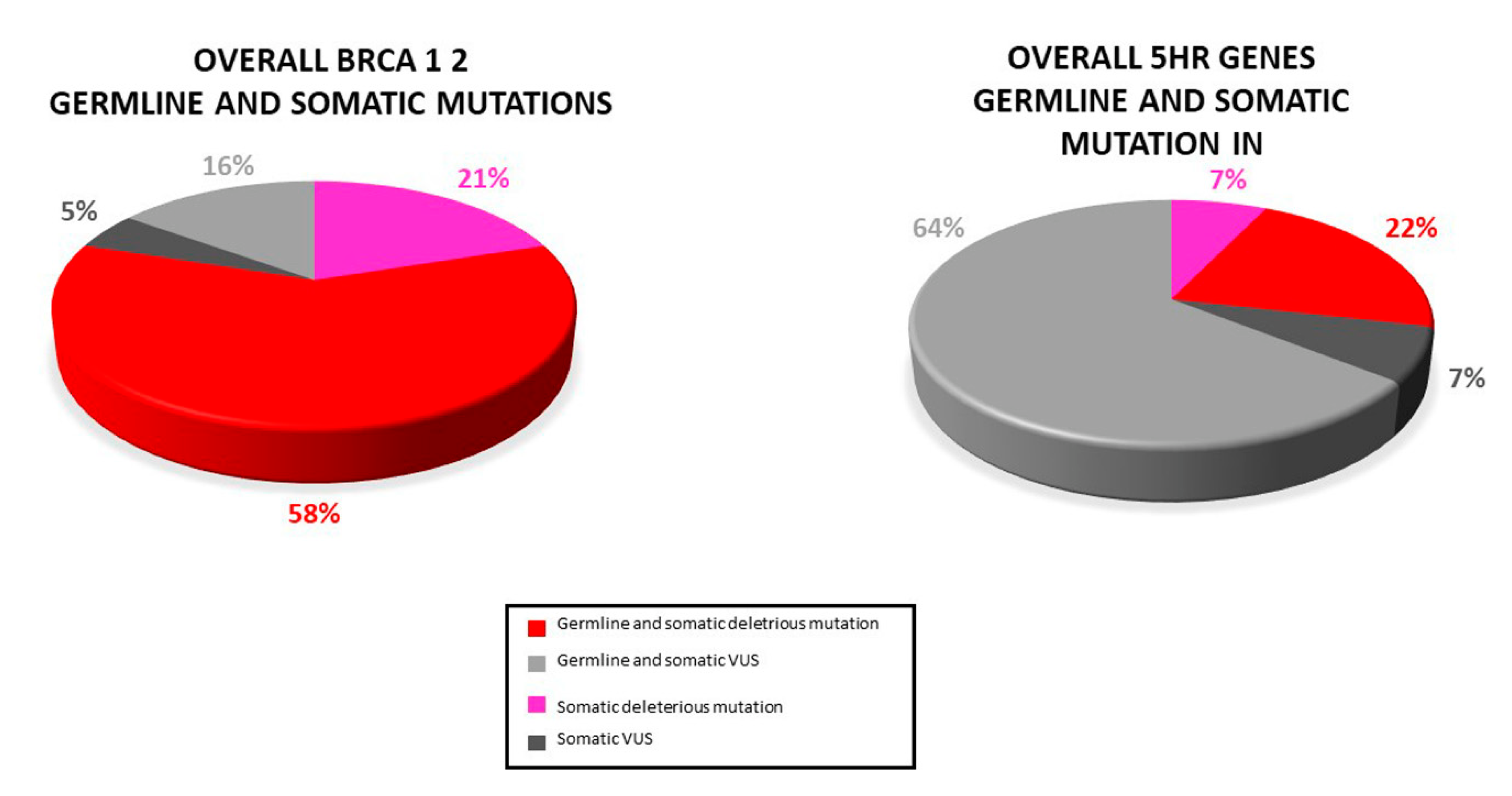
Additionally, there were 14 (20.3%) uncertain significant variants (VUS), with four occurring in BRCA1/2 while ten involved at least one HR gene (Figure 1).
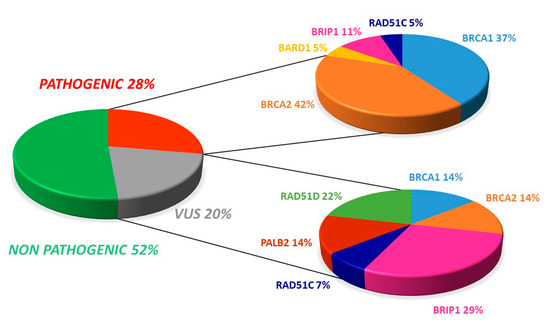
Figure 1.
Overall distribution of deleterious and uncertain significance variants in our cohort.
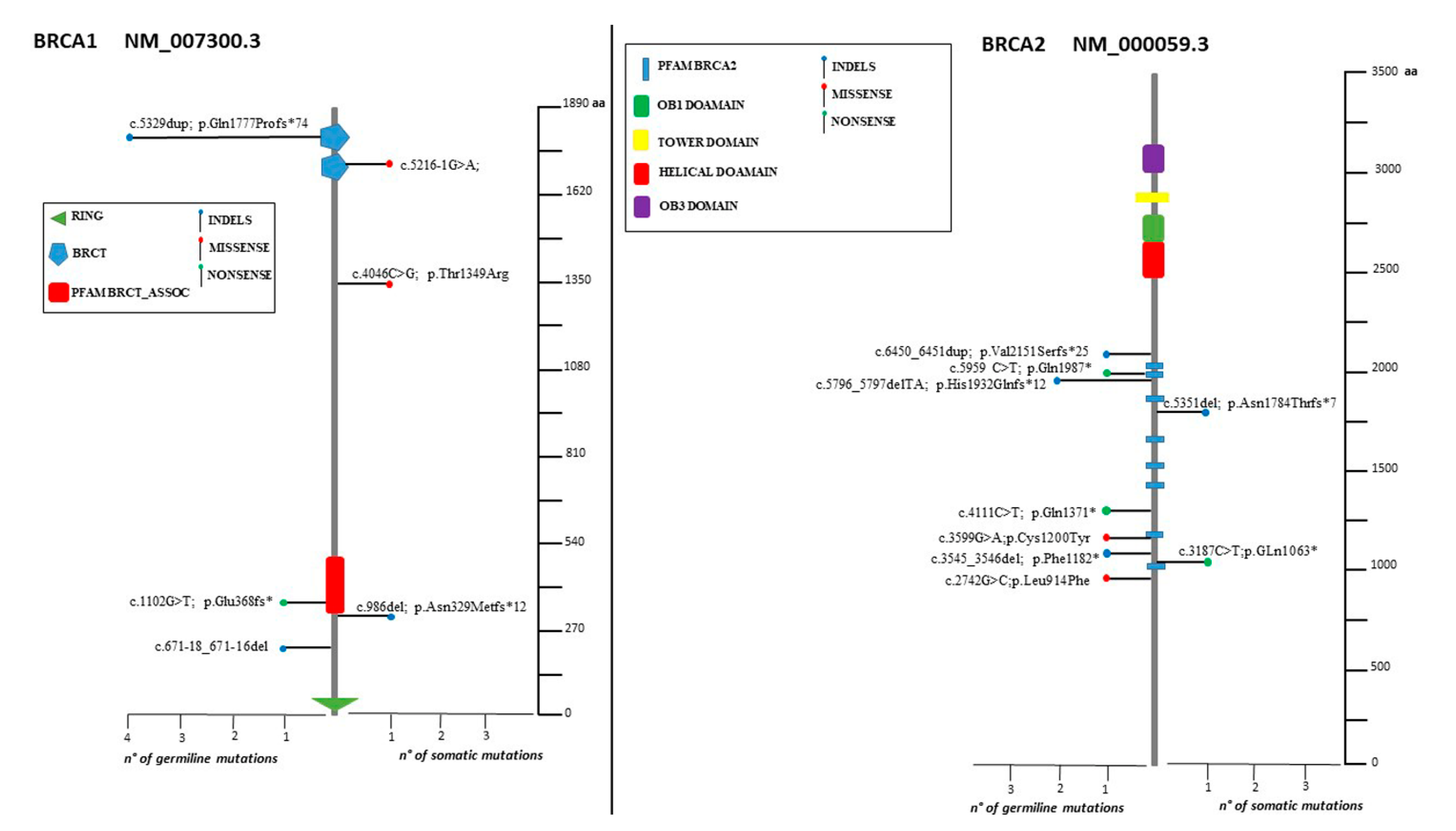
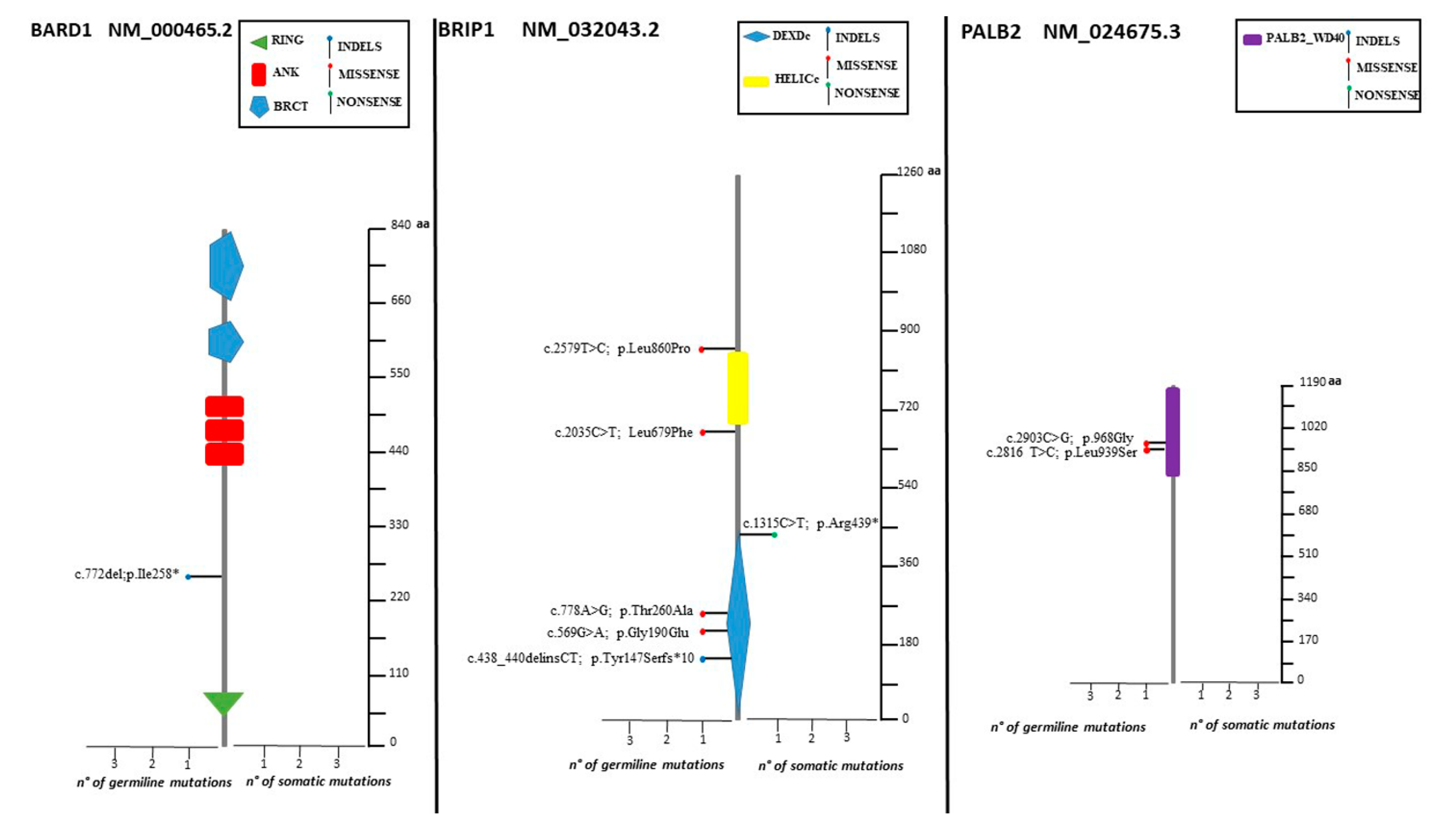
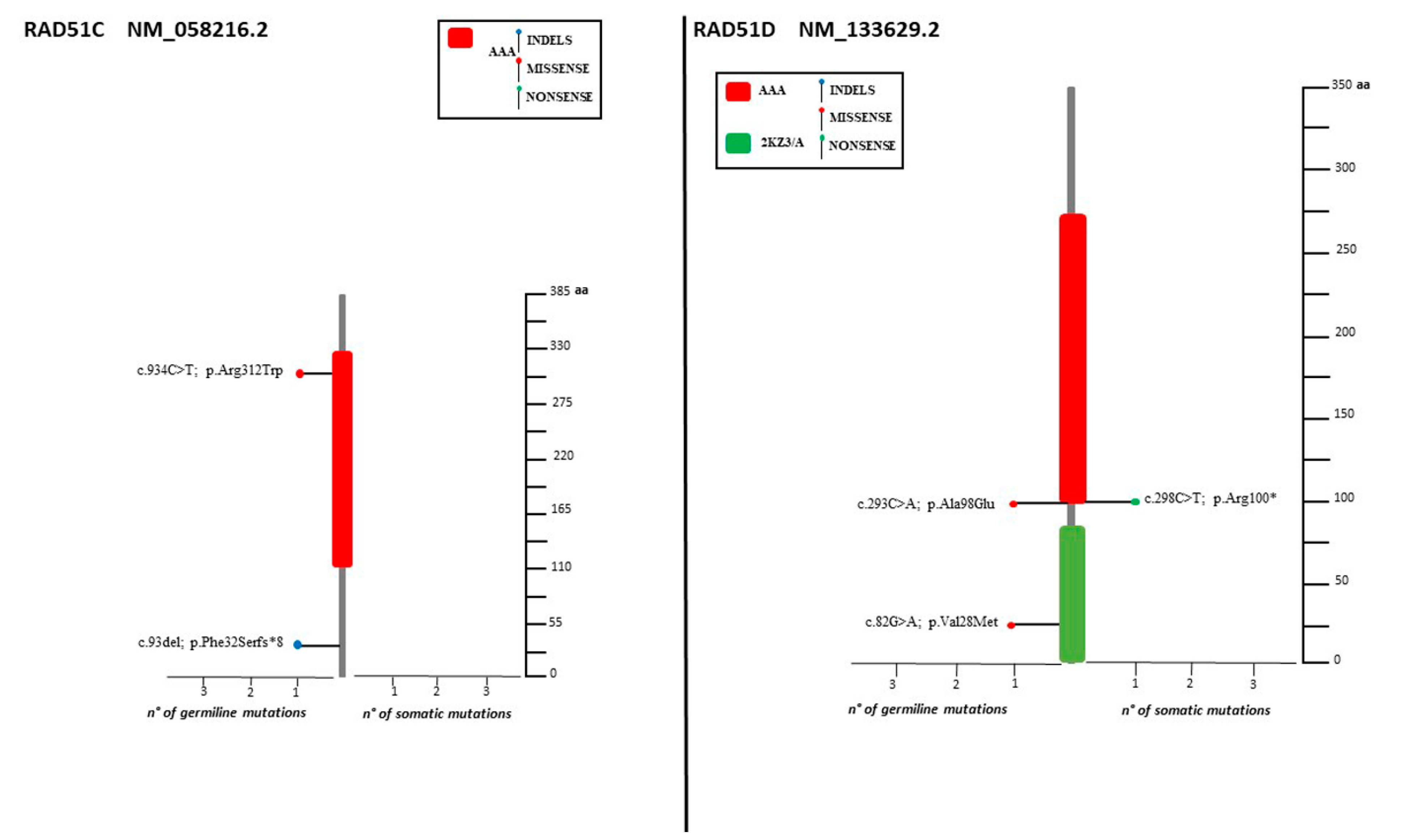
The spectrum of identified mutations includes 19 single-nucleotide variants (6 nonsense, 12 missense, and 1 splice variant) and 14 indels (8 deletions, 5 duplications, and 1 indel) (Figure 2, Figure 3 and Figure 4).
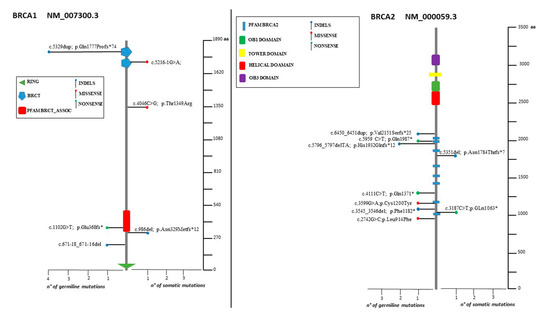
Figure 2.
Lolliplots showing germline and somatic variant spectrum (both pathogenic and VUS) throughout the whole protein sequences of BRCA1 and BRCA2. The vertical black scale bar represents the length (amino acids) of the protein sequence. The horizontal bar represents the number of patients affected by the specific mutation. Each lolliplot represents a variant identified in this study. The blue lolliplot identifies a shorts indels mutation (deletion or duplication). The red lolliplot indicate a missense variant, while the green ones a nonsense mutation.
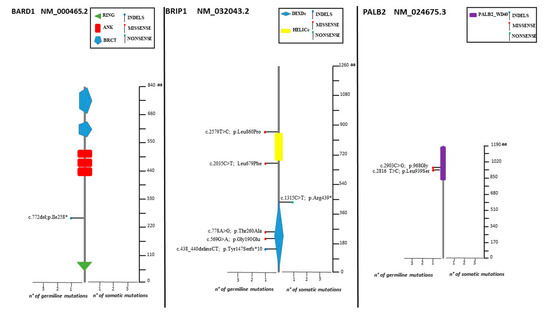
Figure 3.
Lolliplots showing germline and somatic variant spectrum (both pathogenic and VUS) throughout the whole protein sequences of BARD1, BRIP1, and PALB2. The vertical black scale bar represents the length (amino acids) of the protein sequence. The horizontal bar represents the number of patients affected by the specific mutation. Each lolliplot represents a variant identified in this study. The blue lolliplot identifies a shorts indels mutation (deletion or duplication). The red lolliplot indicate a missense variant, while the green ones a nonsense mutation.
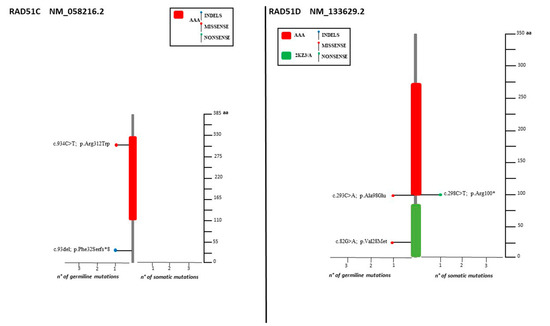
Figure 4.
Lolliplots showing germline and somatic variant spectrum (both pathogenic and VUS) throughout the whole protein sequences of RAD51C and RAD51D. The vertical black scale bar represents the length (amino acids) of the protein sequence. The horizontal bar represents the number of patients affected by the specific mutation. Each lolliplot represents a variant identified in this study. The blue lolliplot identifies a shorts indels mutation (deletion or duplication). The red lolliplot indicate a missense variant, while the green ones a nonsense mutation.
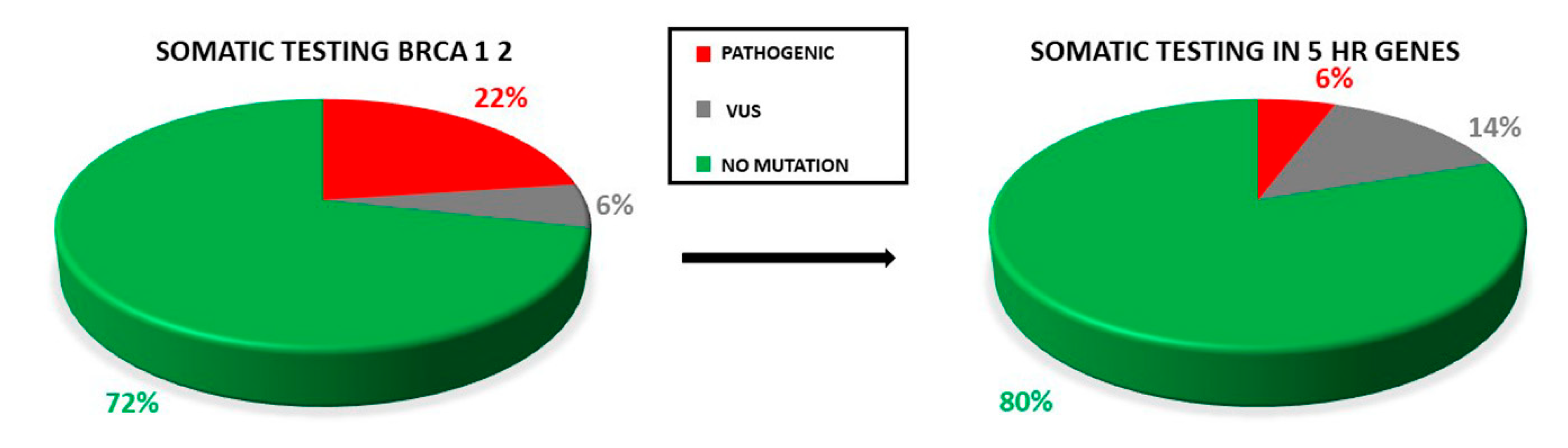
Only two samples displayed the co-occurrence of two variants: two VUS (RAD51D: c.82G>A; RAD51D: c.293C>A) in one case, one VUS and one pathogenic variant (BRCA1: 671-18_671-16del, BARD1: c.772delA) in the second case. Retesting all the samples for the HR genes panel increases the yield of VUS detected (Figure 5).
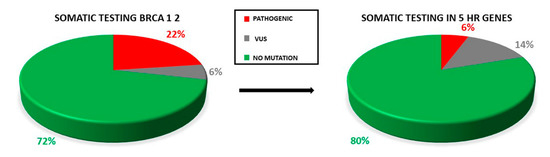
Figure 5.
Proportion of pathogenic variants and VUS in testing only BRCA1/2 genes and retesting all the OC samples for the five HR genes. The rate of VUS detection is increased.
2.3. Distribution of Mutation in BRCA1/2
Of the 19 pathogenic variants, seven (36.8%) occurred in BRCA1 and eight involved BRCA2 (42.1%). Regarding VUS, both BRCA1 and BRCA2 displayed two variants (14.3%, Figure 1 and Figure 2).
Two deleterious mutations were recurrent (BRCA2: c.5796_5797del, BRCA1: c.5329dup) in two and four individuals, respectively (Figure 2).
2.4. Distribution of Mutation in Non BRCA Genes
2.5. Germline Mutation Status
Based on PB samples availability, the germline occurrence of variants was evaluated. A total of 24 (34.8%) individuals presented both germline and somatic mutations: 11 (15.9%) deleterious BRCA1/2 mutations, 3 (4.3%) HR genes pathogenic mutation, 3 (4.3%) BRCA1/2 VUS, and 9 (13%) HR genes VUS (Figure 6).
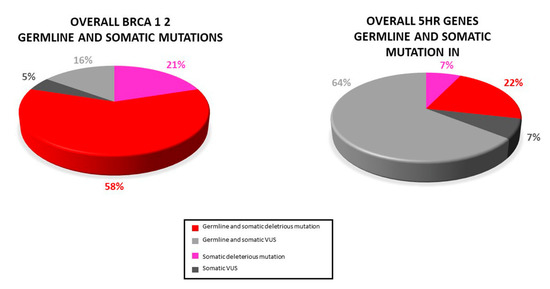
Figure 6.
Distribution of somatic and germline variant detected in our study: Most somatic variants (pathogenic and VUS) were also found in the paired blood sample.
Our analysis revealed that the majority (78.8%) of the somatic variants detected was also found in PB. However, three deleterious mutations (BRCA1: c.986del, BRCA2: C.3187C>T, BRCA2: c.5351del) as well as two VUS (BRCA1: c.4046 C>G, RAD51D: c.298C>T) were not present in PB. Because of the lack of the corresponding PB sample, for two somatic variants (BRCA1: c.5216-1G>A, BRIP1: c.1315C>T) the germline testing was not possible.
2.6. Mutations and Histology
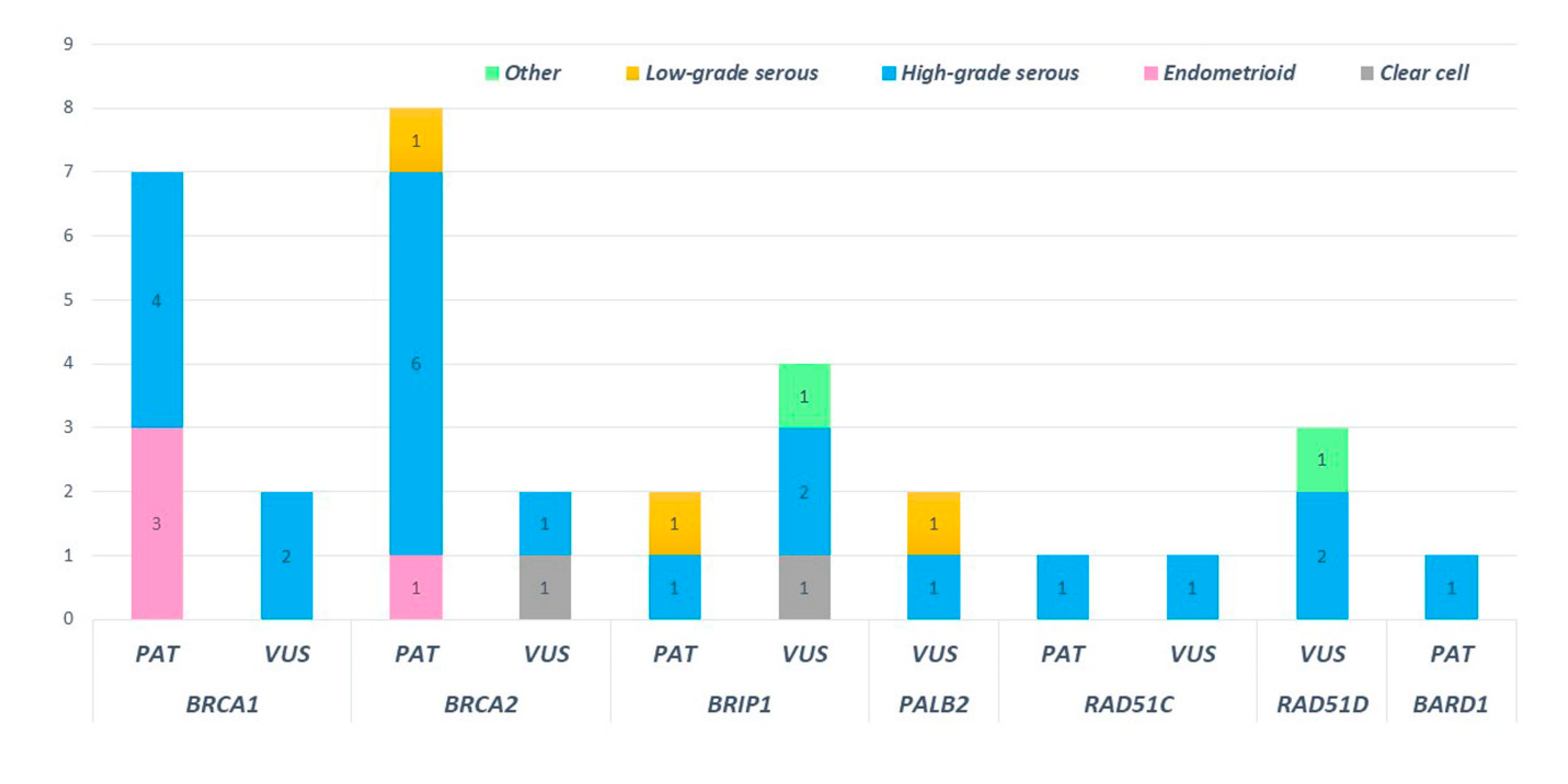
High-grade serous histotypes displayed a heterogeneous spectrum of variants with a prevalence of BRCA1/2 mutations. Eighteen high-grade serous tumors out of thirty-eight (47.4%) were not affected by mutation in any tested genes. Three out of four deleterious mutations in HR genes were found in subjects with high-grade serous histology. Of note, a pathogenic mutation in BRIP1 was detected in low-grade serous histology (BRIP1: c.438_440delinsCT). Moreover, ten deleterious variants in BRCA1 and BRCA2 were detected in patients showing the high-grade serous histotype. Among non-serous histology tumors, four endometrioid cancers were mutated, revealing three deleterious mutations in BRCA1 and one in BRCA2 (Figure 7).
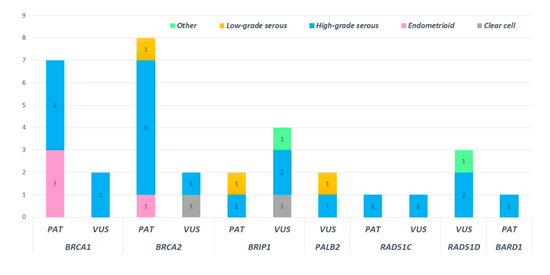
Figure 7.
Distribution of pathogenic and uncertain significance variants according to the ovarian cancer histology of our cohort.
2.7. BRCA1 and BRCA2 Copy Number Variant
In 67 patients of our cohort, for whom PB sample was available, MLPA analysis of BRCA1/2 was carried out. None revealed a CNV alteration.
3. Discussion
Several pieces of evidence have emerged regarding variations in HR genes other than BRCA1/2 (e.g., BRIP1, RAD51C, and RAD51D) associated with a high risk of OC [21,22]. These new findings might lead to the implementation of novel therapeutic procedures, such as PARPi. In this scenario, NGS is rapidly developing as a powerful tool that allows the analysis of multiple genes simultaneously and providing both prognostic and predictive value for OC. The importance of genetic testing in clinical practice is constantly increasing; although, it is generally limited to the BRCA1/2 screening. Herein, we investigated the mutational status of both BRCA1/2 and 5 HR genes (BRIP1, RAD51C, RAD51D, PALB2, and BARD1) in 69 unselected OC, with the aim of evaluating the advantages of multigene panel testing in the everyday clinical practice.
Overall, our pathogenic variants’ frequency of 21.7% in BRCA1/2 (10.1% and 11.6% for BRCA1 and BRCA2, respectively) is within the range of other published studies on OC, ranging from 3% to 15% [21,23,24,25].
As far as concerns non-BRCA genes, we detected a deleterious mutation in 5.8% of our cohort patients (2.9% in BRIP1, 1.5% each in BARD1 and RAD51C), which is widely consistent with previous literature [21,23,24]. Pathogenic variants in BRIP1, BARD1, and RAD51C are all acknowledged as being associated with an increased risk of OC [22,26,27].
Hence, identifying deleterious variants in these latter genes might have a clinical impact resulting in a change in the patients’ management (in terms of either therapeutic approach or testing relatives) [28,29,30]. In BRCA1 and BRCA2, multiple mutation cluster regions have been observed and are associated with relatively different BC and OC risks [31]. In our cohort, five out of seven BRCA1 pathogenic mutations are located from c.5216 to c.5563, which has been described as a region associated with higher BC risk [10]. Deleterious mutation of BRCA2 were detected in the coding sequence region comprised between c.3187 and c.6450, which is consistent with the higher OC risk compared to BC risk associated to mutations in this cluster [10]. To our knowledge, no significance differences have been reported so far in the assessment of OC risk vs. BC risk in correlation with the distribution of the pathogenic variants along BRIP1, RAD51C, and BARD1 coding sequences. Nevertheless, truncating variants in BARD1, BRIP1, and RAD51C are associated with a moderate risk of female BC, generally accounting for less than 1% of Breast Cancers [32].
The National Comprehensive Cancer Network (NCCN) guidelines report on mutations in BRIP1 and RAD51C as being a cause for potential increase in breast cancer risk (specifically for triple negative breast cancers), but there is insufficient evidence for risk management [33].
Two recurrent variants were detected (BRCA1: c.5329dup and BRCA2: c.5796_5797del). Both have already been reported as recurrently found in a large cohort of individuals, particularly in Italian cohorts [34,35,36,37,38].
Of note, the multigene panel approach provides a higher yield of VUS in non BRCA genes (5.8% of VUS in BRCA1/2 compared to 14.5% in HR genes). This result was not unexpected since the rate of the “hard to interpret” variants increase with the introduction of a multigene panel [39]. Consequently, a prioritization strategy is useful to select variants with the highest probability of being associated with OC risk.
The assessment of germline mutational status showed that a minority of variants (three pathogenic mutations in BRCA1/2 and two VUS in BRCA1 and RAD51D) were not detected in the corresponding blood sample. Therefore, most of our mutated cases revealed a hereditary predisposition to OC. To date [40,41], 13–18% of OC is associated with germline mutations in BRCA1/2, whereas an additional 4% with inherited mutations in other susceptibility genes [41], which are both in concordance with our overall germline deleterious mutation frequency (17.4% for BRCA1/2 and 4.3% for HR genes). As a matter of fact, simultaneous germline and somatic analysis are needed in clinical practice to guide the OC patients’ care.
Concerning the distribution of variants among the different histotypes, 10 high-grade serous subtypes (26.3%) out of 38 individuals revealed a BRCA1/2 pathogenic mutation, and three carried a pathogenic mutation in one of the HR genes. These results further confirm previous literature reports [6,42,43,44]. Remarkably, four (40%) endometrioid cancers out of ten in our cohort were found to harbor three deleterious mutations in BRCA1 and 1 in BRCA2. This finding differs from other published research, which refer to a lower BRCA1/2 mutation rate in the endometrioid histology [6,43]. Of note, in two patients affected by low serous OC, two pathogenic mutations in BRIP1 (c.438_440delinsCT) and in BRCA2 (c.6450dup), respectively, were disclosed. Generally, activating somatic mutations in mitogen-activated protein kinase pathway genes, most commonly in KRAS, BRAF, and NRAS, are common in LGSC. Such genetic alterations promote tumorigenesis through constitutive activation of MAPK/ERK pathway. Considering this evidence, therapies based on MEK and BRAF inhibitors use have shown efficacy and improved clinical outcomes [45]. Deleterious germline mutations in BRIP1 have been rarely reported as being associated with low-grade serous histology [43,46]. Weber-Lassalle et al. [47] have previously reported the occurrence of BRIP1 deleterious variants in tumors with low-grade histology. Similarly, BRCA2 pathogenic mutation are unusual in LGCS. Previous work reported BRCA1/2 deleterious mutations as very rare in the low-grade histology [48,49]. Having identified pathogenic mutations in genes such as BRCA2 and BRIP1, albeit in a small percentage of cases, the possibility for these patients to access therapies that were previously precluded opens up. Furthermore, the BRCA2 mutated-LGCS patient presented also a somatic KRAS mutation (G12A). This result further confirms that dysregulation of the MAPK/ERK pathway is important in tumors with low-grade histology.
The majority (55%) of individuals in our cohort had neither pathogenic mutations nor deletions or duplications. We speculate that other mechanisms could be possibly involved in the OC onset in our cohort. For instance, a large body of research points out to the role of BRCA methylation in causing BRCA dysfunction in OC [50]. Thus, future studies should include the assessment of BRCA methylation status to evaluate the reliability of this new molecular entity in the clinical management. Similarly, somatic and germline CNV evaluation for genes other than BRCA1/2 should be performed to ascertain frequency and involvement of large duplications/deletions in OC, demanding for the implementation of a more sophisticated NGS system and bioinformatic pipeline.
Furthermore, mutations in MMR genes should also be investigated, specifically in tumors with clear cell and endometrioid histology.
Clinically, the relatively short follow-up times in our study, lower than the average survival of women affected by EOC (5 years), did not achieve a statistically significant result concerning the PFS in patients with mutations versus patients without mutations. However, the concurrent diagnosis of high HRD and EOC is associated with an increased PFS. Therefore, the HRD score can be translated as a valid prognostic score particularly useful in a precision medicine scenario [19,51]. The synthetic lethality provoked by PARPi is based on HRD (due to BRCA mutations and the BRCAness phenotype), even if the clinical response to PARPi depends on time and chemosensitivity. This variability can be explained throughout PARPi resistance due to tumoral restoration of HRR functions, thus evading the combination of the genetical and iatrogenic “two hits” (HRD-PARPi). In other words, the HRD status is dynamic, and therefore is crucial the early introduction of PARPi in the EOC treatment timeline. In fact, in the SOLO-1 trial, more than 50% of BRCA 1/2 mutated women with advanced EOC who gained a complete response after frontline chemotherapy, experienced at least 5 years free from EOC’s relapses. Interestingly, clinical response rates and survival outcomes of the PFS are similar in cases of somatic or germline mutation of BRCA 1/2. Notably, in case of platinum sensitivity, we can predict a positive response to PARPi. However, considering all the HRD panels, there is an evident trend of higher PFS in crescent order: HRD-negative, HRD-positive, and BRCA1/2-mutant subgroups [52].
4. Materials and Methods
4.1. Patient Recruitment
Sixty-nine patients with a primary diagnosis of malignant epithelial OC were referred to the UOC of Obstetrics and Gynecology at the Università Degli Studi di Bari, from September 2018 to October 2020. Neither age at diagnosis nor tumor histotype was used as selection criteria. Clinicopathological characteristics of the patients were collected to analyze the distribution of identified mutations in subgroups of patients. Fresh cancer tissues were collected from all 69 patients. Peripheral blood (PB) samples were withdrawn (when available) to eventually assess both the presence of a corresponding germline mutation after mutation identification in the tumor sample and to verify the presence of germline copy number variation (CNV) in BRCA1/2 genes in patients who tested negative for somatic analysis for BRCA 1/2 genes. In case of a positive genetic test for a germline mutation, post-test genetic counseling was offered to all patients to discuss the risk for their relatives. Written informed consent was obtained from patients to perform the genetic tests for diagnostics and research purposes on peripheral blood and tissue biopsy according to the local ethic committee’s policy (approval code study: 5517 N0017671|26/02/2018, Policlinico of Bari, Italy).
4.2. Next-Generation Sequencing
Genomic DNA was isolated from PB and biopsy samples using the QIAamp Mini Kit (Qiagen, Hilden, Germany), according to the manufacturer’s instructions, and quantified on a BioSpectrometer Plus (Eppendorf, Hamburg, Germany) and Qubit ds DNA HS Assay Kit on Qubit 2.0 Fluorimeter (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. The BRCA1/2 analysis was performed using Oncomine™ BRCA Research Assay (ThermoFisher, Carlsbad, CA, USA) for 60 individuals.
An Ion AmpliSeq Custom Panel was designed online using Ion AmpliSeq Designer v.7.06 (http://www.ampliseq.com/ accessed on 27 March 2019) (Thermo Fisher, Waltham, MA, USA) to analyze the CDSs (+/− 25 bp of intronic flanking regions) of five HR genes (BRIP1, RAD51C, RAD51D, BARD1, and PALB2). The final custom panel was composed of 197 amplicons divided into three primer pools for a total of 19.01 kb of target size DNA and coverage of 98.78%. Library preparation was performed using the Ion AmpliSeqTM Library Kit Plus (Life Technologies, Carlsbad, CA, USA) following the manufacturer’s instructions. The equimolar concentration of each library was used to prepare templates for clonal amplification. Emulsion PCR was carried out on Ion OneTouch 2 Instrument (Thermo Fisher Scientific, Carlsbad, CA, USA) using the Ion PGMTM Hi-Q View OT2 Kit (Thermo Fisher Scientific, Carlsbad, CA, USA). Templates were enriched using Ion OneTouch ES (Enrichment System) (Thermo Fisher Scientific, Carlsbad, CA, USA), loaded on a 316v2/318v2 chip, and sequenced on an Ion Torrent Personal Genome Machine (Life Technologies, Carlsbad, CA, USA) using Ion PGM Hi-Q View Sequencing Kit (Thermo Fisher Scientific, Carlsbad, CA, USA). Nine samples were tested for BRCA1/2 by the Devyser’s CE-IVD library prep kit (Devyser, Hägersten, Sweden). Sequencing run was performed on an Illumina Miseq instrument (Illumina, San Diego, CA, USA) following the manufacturer’s instructions using a standard flow cell (Illumina, San Diego, CA, USA) and V2 300 cycle cartridge (Illumina, San Diego, CA, USA). To validate variants detected with NGS and to confirm the corresponding presence in PB samples, direct sequencing was performed using the BigDye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems, Waltham, MA, USA) on SeqStudio Genetic Analyzer (Applied Biosystems, Waltham, MA, USA) following the manufacturer’s instructions.
4.3. Variant Analysis and Data Interpretation
For the Oncomine BRCA panel, PGM sequencing produced mean reads per patient of about 600,000 with the Mean Read Length of 108 bp. The average base coverage depth was about 3626×. The mean percentage of regions of interest (ROI), covered at least by 100×, was 100.00% with a Uniformity of base coverage of 99.98%. For the custom panel including 5 HR genes, mean reads per patient are about 550,000 with a mean read length of 115 bp. The average base coverage depth was about 3000×. The mean percentage of regions of interest (ROI), covered at least by 100×, was 98.71%, with uniformity of base coverage of 98.48%. Data analysis was performed using Torrent Suite version 5.0.4 and Ion Reporter version 5.14.1.2 (Thermo Fisher Scientific, Carlsbad, CA, USA). Alamut Visual 2.13-0 (Sophia Genetics, Lausanne, Switzerland) was used to visually check the sequencing data and the corresponding BAM file. For the remaining nine samples tested by the Devyser’s CE-IVD kit for BRCA1/2, a mean of 2,245,135 reads with a mean read length of 147 bp per sample was obtained. The average coverage depth was about 11,019, with uniformity of base coverage of 100%. Data analysis was performed by MiSeq™ Control Software 3.1 (Illumina, San Diego, CA, USA). FASTQ files were analyzed with Amplicon Suite software version 3.1 (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions. Variants were analyzed Online databases, including dbSNP (database the single nucleotide polymorphism database), 1000 Genomes, ClinVar, EXAC (exome aggregation consortium), COSMIC (catalog of somatic mutations in cancer), ESP (exome sequencing project), and ENIGMA (Evidence-based Network for the Interpretation of Germline Mutant Alleles, for BRCA 1 2 variant annotation). Pathogenicity prediction programs (e.g., PolyPhen2, SIFT, Mutation Taster) and splice prediction programs were used to check out variants not formerly described.
4.4. MLPA
To assess the presence of germline Copy Number Variation (CNV) in BRCA1/2 genes, MLPA (Multiplex ligation-dependent probe amplification MRC Holland® (Amsterdam, The Netherlands)), BRCA1 P002 D1, and BRCA2 P045 D1 were performed on blood samples (when available) of patients who tested negative for somatic analysis for BRCA1/2 genes. Data analyses were performed with Coffalyser.Net-MRC Holland software® (Amsterdam, The Netherlands).
4.5. KRAS and BRAF Mutational Analysis
Mutational analysis of the BRAF and KRAS genes was performed on OC samples by real-time polymerase chain reaction (Therascreen, Qiagen, Hilden, Germany). Tumor specimens were screened for hot-spot mutations in codon 600 of the BRAF gene and in codons 12 and 13 of the KRAS gene.
5. Conclusions
Using a multigene panel testing in patients affected by OC, rather than testing BRCA1/2 alone, could generate clinically relevant results. Further research is warranted to verify the clinical implications to develop a standard clinical multigene panel and narrow down the number of candidates VUS detected.
Author Contributions
Conceptualization, N.R., A.T. and D.C.L.; investigation, A.T., D.C.L., R.D.N., G.C. (Giulia Chiarello), F.A., S.P., S.M., A.P., M.I., R.B., A.G., C.R. and A.D.L.; resources, G.C. (Gennaro Cormio), V.L., M.M. and E.C.; supervision, N.R., G.C. (Gennaro Cormio), V.L., M.M. and E.C.; visualization, A.T., D.C.L., R.D.N., G.C. (Giulia Chiarello), F.A., S.P., S.M., A.P., M.I., R.B., C.R. and A.D.L.; writing—original draft, A.T., D.C.L., N.R., G.C. (Gennaro Cormio) and R.D.N.; writing—review and editing, A.T., D.C.L., N.R., G.C. (Gennaro Cormio), A.S. and C.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded partially by AstraZeneca, grant number #ESR-16-12553-D0810C00108” (Apulia Network Ovary-ANOVA: molecular characterization of the ovarian cancers in Apulian population).
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the local Ethics Committee (approval code 5517—prot n 0017671|26/02/2018).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
We thank the Apulian association ACTO (Alleanza Contro Il Tumore Ovarico) and its founder, Adele Leone.
Conflicts of Interest
The authors declare no conflict of interest. The sponsors had no role in the design, execution, interpretation, or writing of the study.
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute Ovarian Cancer-Cancer Stat Facts. Available online: https://seer.cancer.gov/statfacts/html/ovary.html (accessed on 9 December 2021).
- Prat, J.; D’Angelo, E.; Espinosa, I. Ovarian carcinomas: At least five different diseases with distinct histological features and molecular genetics. Hum. Pathol. 2018, 80, 11–27. [Google Scholar] [CrossRef] [PubMed]
- Mørch, L.S.; Løkkegaard, E.; Andreasen, A.H.; Krüger-Kjær, S.; Lidegaard, Ø. Hormone Therapy and Ovarian Cancer. JAMA 2009, 302, 298–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lheureux, S.; Msc, M.B.; Oza, A.M. Epithelial ovarian cancer: Evolution of management in the era of precision medicine. CA Cancer J. Clin. 2019, 69, 280–304. [Google Scholar] [CrossRef] [Green Version]
- Manchana, T.; Phoolcharoen, N.; Tantbirojn, P. BRCA mutation in high grade epithelial ovarian cancers. Gynecol. Oncol. Rep. 2019, 29, 102–105. [Google Scholar] [CrossRef]
- Walsh, T.; Casadei, S.; Lee, M.K.; Pennil, C.C.; Nord, A.; Thornton, A.M.; Roeb, W.; Agnew, K.J.; Stray, S.M.; Wickramanayake, A.; et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 18032–18037. [Google Scholar] [CrossRef] [Green Version]
- Malander, S.; Rambech, E.; Kristoffersson, U.; Halvarsson, B.; Ridderheim, M.; Borg, A.; Nilbert, M. The contribution of the hereditary nonpolyposis colorectal cancer syndrome to the development of ovarian cancer. Gynecol. Oncol. 2006, 101, 238–243. [Google Scholar] [CrossRef]
- Easton, D.F.; Pharoah, P.D.; Antoniou, A.C.; Tischkowitz, M.; Tavtigian, S.V.; Nathanson, K.L.; Devilee, P.; Meindl, A.; Couch, F.J.; Southey, M.; et al. Gene-Panel Sequencing and the Prediction of Breast-Cancer Risk. N. Engl. J. Med. 2015, 372, 2243–2257. [Google Scholar] [CrossRef] [Green Version]
- Creeden, J.F.; Nanavaty, N.S.; Einloth, K.R.; Gillman, C.E.; Stanbery, L.; Hamouda, D.M.; Dworkin, L.; Nemunaitis, J. Homologous recombination proficiency in ovarian and breast cancer patients. BMC Cancer 2021, 21, 1154. [Google Scholar] [CrossRef]
- Toh, M.R.; Ngeow, J. Homologous Recombination Deficiency: Cancer Predispositions and Treatment Implications. Oncologist 2021, 26, 1526–1537. [Google Scholar] [CrossRef]
- Haunschild, C.E.; Tewari, K.S. The current landscape of molecular profiling in the treatment of epithelial ovarian cancer. Gynecol. Oncol. 2021, 160, 333–345. [Google Scholar] [CrossRef]
- Rein, H.L.; Bernstein, K.A.; Baldock, R.A. RAD51 paralog function in replicative DNA damage and tolerance. Curr. Opin. Genet. Dev. 2021, 71, 86–91. [Google Scholar] [CrossRef]
- Hodgson, D.R.; Dougherty, B.A.; Lai, Z.; Fielding, A.; Grinsted, L.; Spencer, S.; O’Connor, M.J.; Ho, T.W.; Robertson, J.D.; Lanchbury, J.S.; et al. Candidate biomarkers of PARP inhibitor sensitivity in ovarian cancer beyond the BRCA genes. Br. J. Cancer 2018, 119, 1401–1409. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961, Erratum in Lancet 2017, 390, 1948. [Google Scholar] [CrossRef] [Green Version]
- González-Martín, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef] [Green Version]
- Norquist, B.M.; Harrell, M.I.; Brady, M.F.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Bernards, S.S.; Casadei, S.; Yi, Q.; Burger, R.A.; et al. Inherited Mutations in Women With Ovarian Carcinoma. JAMA Oncol. 2016, 2, 482–490. [Google Scholar] [CrossRef]
- Boussios, S.; Mikropoulos, C.; Samartzis, E.; Karihtala, P.; Moschetta, M.; Sheriff, M.; Karathanasi, A.; Sadauskaite, A.; Rassy, E.; Pavlidis, N. Wise Management of Ovarian Cancer: On the Cutting Edge. J. Pers. Med. 2020, 10, 41. [Google Scholar] [CrossRef]
- Suszynska, M.; Ratajska, M.; Kozlowski, P. BRIP1, RAD51C, and RAD51D mutations are associated with high susceptibility to ovarian cancer: Mutation prevalence and precise risk estimates based on a pooled analysis of ~30,000 cases. J. Ovarian Res. 2020, 13, 50. [Google Scholar] [CrossRef]
- Wallbillich, J.J.; Morris, R.T.; Ali-Fehmi, R. Comparing mutation frequencies for homologous recombination genes in uterine serous and high-grade serous ovarian carcinomas: A case for homologous recombination deficiency testing in uterine serous carcinoma. Gynecol. Oncol. 2020, 159, 381–386. [Google Scholar] [CrossRef]
- Yadav, V.; Yadav, G.; Vashisht, M.; Shyam, R. Molecular biomarkers for early detection and prevention of ovarian cancer—A gateway for good prognosis: A narrative review. Int. J. Prev. Med. 2020, 11, 135. [Google Scholar] [CrossRef]
- Amin, N.; Chaabouni, N.; George, A. Genetic testing for epithelial ovarian cancer. Best Pract. Res. Clin. Obstet. Gynaecol. 2020, 65, 125–138. [Google Scholar] [CrossRef]
- Lerner-Ellis, J.; Sopik, V.; Wong, A.; Lázaro, C.; Narod, S.A.; Charames, G.S. Retesting of women who are negative for a BRCA1 and BRCA2 mutation using a 20-gene panel. J. Med. Genet. 2019, 57, 380–384. [Google Scholar] [CrossRef]
- Alenezi, W.M.; Fierheller, C.T.; Recio, N.; Tonin, P.N. Literature Review of BARD1 as a Cancer Predisposing Gene with a Focus on Breast and Ovarian Cancers. Genes 2020, 11, 856. [Google Scholar] [CrossRef]
- Lord, C.; Ashworth, A. BRCAness revisited. Nat. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Matsumoto, K.; Nishimura, M.; Onoe, T.; Sakai, H.; Urakawa, Y.; Onda, T.; Yaegashi, N. PARP inhibitors for BRCA wild type ovarian cancer; gene alterations, homologous recombination deficiency and combination therapy. Jpn. J. Clin. Oncol. 2019, 49, 703–707. [Google Scholar] [CrossRef]
- Li, H.; Liu, Z.-Y.; Wu, N.; Chen, Y.-C.; Cheng, Q.; Wang, J. PARP inhibitor resistance: The underlying mechanisms and clinical implications. Mol. Cancer 2020, 19, 1–16. [Google Scholar] [CrossRef]
- Rebbeck, T.R.; Mitra, N.; Wan, F.; Sinilnikova, O.M.; Healey, S.; McGuffog, L.; Mazoyer, S.; Chenevix-Trench, G.; Easton, D.F.; Antoniou, A.C.; et al. Association of Type and Location of BRCA1 and BRCA2 Mutations with Risk of Breast and Ovarian Cancer. JAMA J. Am. Med. Assoc. 2015, 313, 1347–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiovitz, S.; Korde, L.A. Genetics of breast cancer: A topic in evolution. Ann. Oncol. 2015, 26, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Daly, M.B.; Pal, T.; Berry, M.P.; Buys, S.S.; Dickson, P.; Domchek, S.M.; Elkhanany, A.; Friedman, S.; Goggins, M.; Htton, M.L.L.; et al. Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic. NCCN Guidel. 2021, 19, 77–102. [Google Scholar] [CrossRef] [PubMed]
- Figlioli, G.; De Nicolo, A.; Catucci, I.; Manoukian, S.; Peissel, B.; Azzollini, J.; Beltrami, B.; Bonanni, B.; Calvello, M.; Bondavalli, D.; et al. Analysis of Italian BRCA1/2 Pathogenic Variants Identifies a Private Spectrum in the Population from the Bergamo Province in Northern Italy. Cancers 2021, 13, 532. [Google Scholar] [CrossRef] [PubMed]
- Coppa, A.; Buffone, A.; Capalbo, C.; Nicolussi, A.; D’Inzeo, S.; Belardinilli, F.; Colicchia, V.; Petroni, M.; Granato, T.; Midulla, C.; et al. Novel and recurrent BRCA2 mutations in Italian breast/ovarian cancer families widen the ovarian cancer cluster region boundaries to exons 13 and 14. Breast Cancer Res. Treat. 2014, 148, 629–635. [Google Scholar] [CrossRef]
- Santonocito, C.; Rizza, R.; Paris, I.; De Marchis, L.; Paolillo, C.; Tiberi, G.; Scambia, G.; Capoluongo, E. Spectrum of Germline BRCA1 and BRCA2 Variants Identified in 2351 Ovarian and Breast Cancer Patients Referring to a Reference Cancer Hospital of Rome. Cancers 2020, 12, 1286. [Google Scholar] [CrossRef]
- Kluz, T.; Jasiewicz, A.; Marczyk, E.; Jach, R.; Jakubowska, A.; Lubiński, J.; Narod, S.A.; Gronwald, J. Frequency of BRCA1 and BRCA2 causative founder variants in ovarian cancer patients in South-East Poland. Hered. Cancer Clin. Pract. 2018, 16, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Cotrim, D.P.; Ribeiro, A.R.G.; Paixão, D.; Soares, D.C.D.Q.; Jbili, R.; Pandolfi, N.C.; Cezana, C.; Mauro, C.D.C.; Mantoan, H.; Bovolim, G.; et al. Prevalence of BRCA1 and BRCA2 pathogenic and likely pathogenic variants in non-selected ovarian carcinoma patients in Brazil. BMC Cancer 2019, 19, 4. [Google Scholar] [CrossRef] [Green Version]
- Toss, A.; Tomasello, C.; Razzaboni, E.; Contu, G.; Grandi, G.; Cagnacci, A.; Schilder, R.J.; Cortesi, L. Hereditary Ovarian Cancer: Not OnlyBRCA1 and 2 Genes. BioMed Res. Int. 2015, 2015, 341723. [Google Scholar] [CrossRef] [Green Version]
- Moschetta, M.; George, A.; Kaye, S.B.; Banerjee, S. BRCA somatic mutations and epigenetic BRCA modifications in serous ovarian cancer. Ann. Oncol. 2016, 27, 1449–1455. [Google Scholar] [CrossRef]
- Jorge, S.; McFaddin, A.S.; Doll, K.M.; Pennington, K.P.; Norquist, B.M.; Bennett, R.L.; Pritchard, C.C.; Swisher, E.M. Simultaneous germline and somatic sequencing in ovarian carcinoma: Mutation rate and impact on clinical decision-making. Gynecol. Oncol. 2020, 156, 517–522. [Google Scholar] [CrossRef]
- Lertkhachonsuk, A.-A.; Suprasert, P.; Manchana, T.; Kittisiam, T.; Kantathavorn, N.; Chansoon, T.; Khunamornpong, S.; Pohthipornthawat, N.; Tangjitgamol, S.; Luasiripanthu, T.; et al. Prevalence of Tissue BRCA Gene Mutation in Ovarian, Fallopian Tube, and Primary Peritoneal Cancers: A Multi-Institutional Study. Asian Pac. J. Cancer Prev. 2020, 21, 2381–2388. [Google Scholar] [CrossRef]
- Pavanello, M.; Chan, I.H.Y.; Ariff, A.; Pharoah, P.D.P.; Gayther, S.A.; Ramus, S.J. Rare Germline Genetic Variants and the Risks of Epithelial Ovarian Cancer. Cancers 2020, 12, 3046. [Google Scholar] [CrossRef]
- Loizzi, V.; Cicinelli, E.; Santamaria, F.; Murgia, F.; Minicucci, V.; Resta, L.; Resta, N.; Natalicchio, M.I.; Ranieri, G.; Cormio, G. BRCAmut and “founder effect”: A prospective study in a single academic institution. Oncotarget 2018, 9, 22353–22358. [Google Scholar] [CrossRef] [Green Version]
- Moujaber, T.; Balleine, R.L.; Gao, B.; Madsen, I.; Harnett, P.R.; DeFazio, A. New therapeutic opportunities for women with low-grade serous ovarian cancer. Endocr.-Relat. Cancer 2022, 29, R1–R16. [Google Scholar] [CrossRef]
- Ramus, S.J.; Song, H.; Dicks, E.; Tyrer, J.P.; Rosenthal, A.N.; Intermaggio, M.P.; Fraser, L.; Gentry-Maharaj, A.; Hayward, J.; Philpott, S.; et al. Germline Mutations in the BRIP1, BARD1, PALB2, and NBN Genes in Women with Ovarian Cancer. JNCI J. Natl. Cancer Inst. 2015, 107, djv214. [Google Scholar] [CrossRef]
- Weber-Lassalle, N.; Hauke, J.; Ramser, J.; Richters, L.; Groß, E.; Blümcke, B.; Gehrig, A.; Kahlert, A.-K.; Müller, C.R.; Hackmann, K.; et al. BRIP1 loss-of-function mutations confer high risk for familial ovarian cancer, but not familial breast cancer. Breast Cancer Res. 2018, 20, 1–6. [Google Scholar] [CrossRef]
- Kaldawy, A.; Segev, Y.; Lavie, O.; Auslender, R.; Sopik, V.; Narod, S.A. Low-grade serous ovarian cancer: A review. Gynecol. Oncol. 2016, 143, 433–438. [Google Scholar] [CrossRef]
- Vineyard, M.A.; Daniels, M.S.; Urbauer, D.L.; Deavers, M.T.; Sun, C.C.; Boerwinkle, E.; Bodurka, D.C.; Gershenson, D.M.; Crawford, J.; Lu, K.H. Is low-grade serous ovarian cancer part of the tumor spectrum of Hereditary Breast and Ovarian Cancer? Gynecol. Oncol. 2011, 120, 229–232. [Google Scholar] [CrossRef]
- Kalachand, R.D.; O’Riain, C.; Toomey, S.; Carr, A.; Timms, K.M.; O’Toole, S.; Madden, S.; Bates, M.; O’Leary, J.J.; Gleeson, N.; et al. Prevalence of tumor BRCA1 and BRCA2 dysfunction in unselected patients with ovarian cancer. Obstet. Gynecol. Sci. 2020, 63, 643–654. [Google Scholar] [CrossRef]
- Coleman, R.L.; Fleming, G.F.; Brady, M.F.; Swisher, E.M.; Steffensen, K.D.; Friedlander, M.; Okamoto, A.; Moore, K.N.; Efrat Ben-Baruch, N.; Werner, T.L.; et al. Veliparib with First-Line Chemotherapy and as Maintenance Therapy in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2403–2415. [Google Scholar] [CrossRef]
- Ngoi, N.Y.L.; Tan, D.S.P. The role of homologous recombination deficiency testing in ovarian cancer and its clinical implications: Do we need it? ESMO Open 2021, 6, 100144. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).