
A Small Molecule–Drug Conjugate (SMDC) Consisting of a Modified Camptothecin Payload Linked to an αVß3 Binder for the Treatment of Multiple Cancer Types

 , ,
, ,
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Compounds
2.3. Caco-2 and P-gp-Expressing LLC-PK1 Cell Permeability Assays
2.4. Cytotoxicity of the VIP126 and SN38 Payloads in NCI-H1975 Parental and Transporter-Expressing Mutant Cells
2.5. In Vitro Proliferation Assay
2.6. Tumor Homing of the αVβ3 Binder in 786-O RCC Tumor-Bearing Mice
2.7. Pharmacokinetics of VIP236 and VIP126 in Plasma and Tumor
2.8. In Vivo Antitumor Efficacy of VIP236
2.9. Statistical Analyses
3. Results
3.1. The αVβ3 Integrin-Targeting Moiety Mediates Tumor Homing
3.2. The Modified CPT Payload VIP126 Shows an Improved In Vitro Profile
3.3. In Vitro Cytotoxic Activity of VIP236 against Tumor Cell Lines Is NE-Dependent
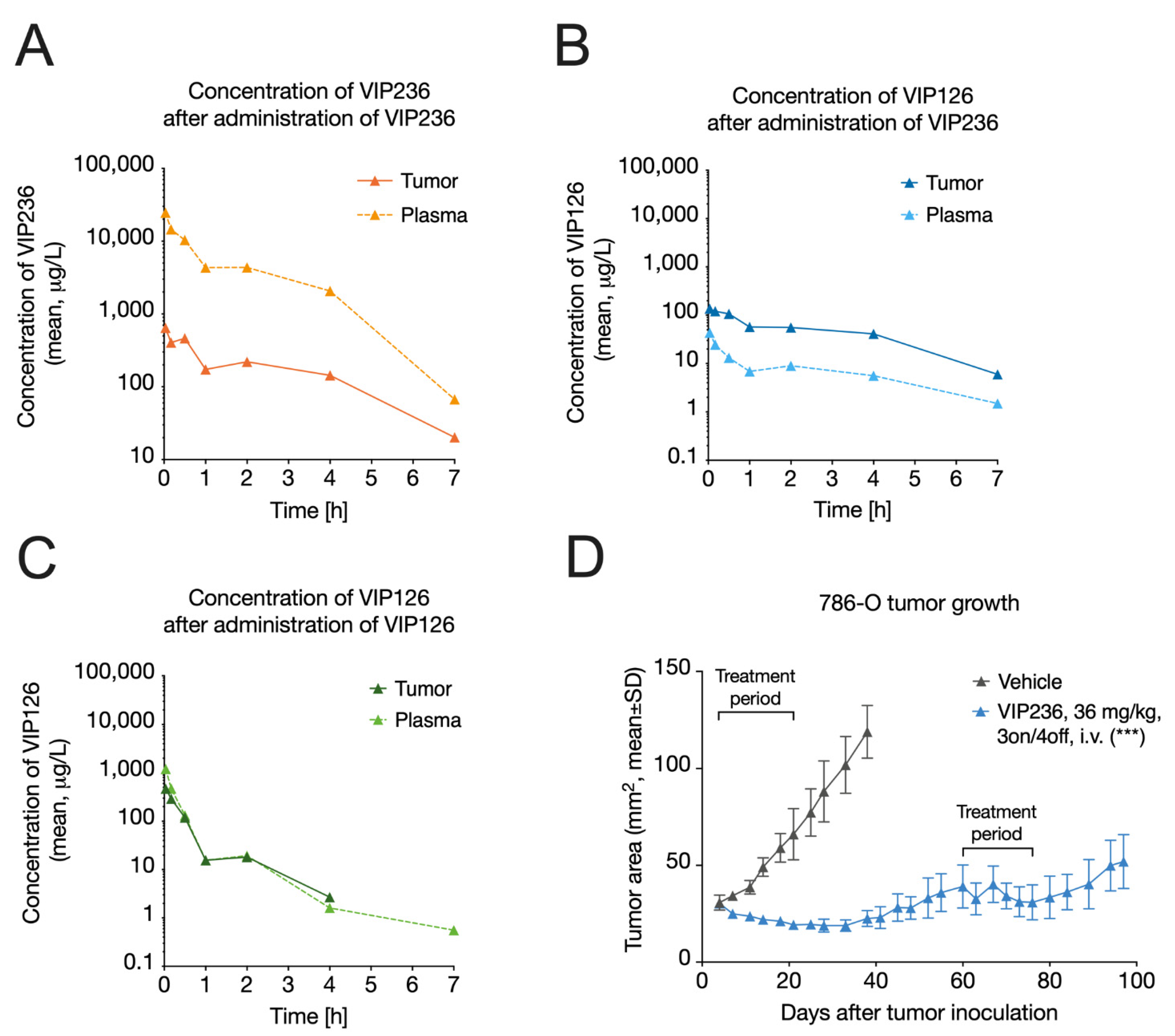
3.4. VIP236 Has Low Clearance and Results in Reduced Systemic Exposure of the Payload
3.5. VIP236 Results in a Higher Tumor-to-Plasma Ratio of the VIP126 Payload and Leads to Anti-Tumor Efficacy In Vivo
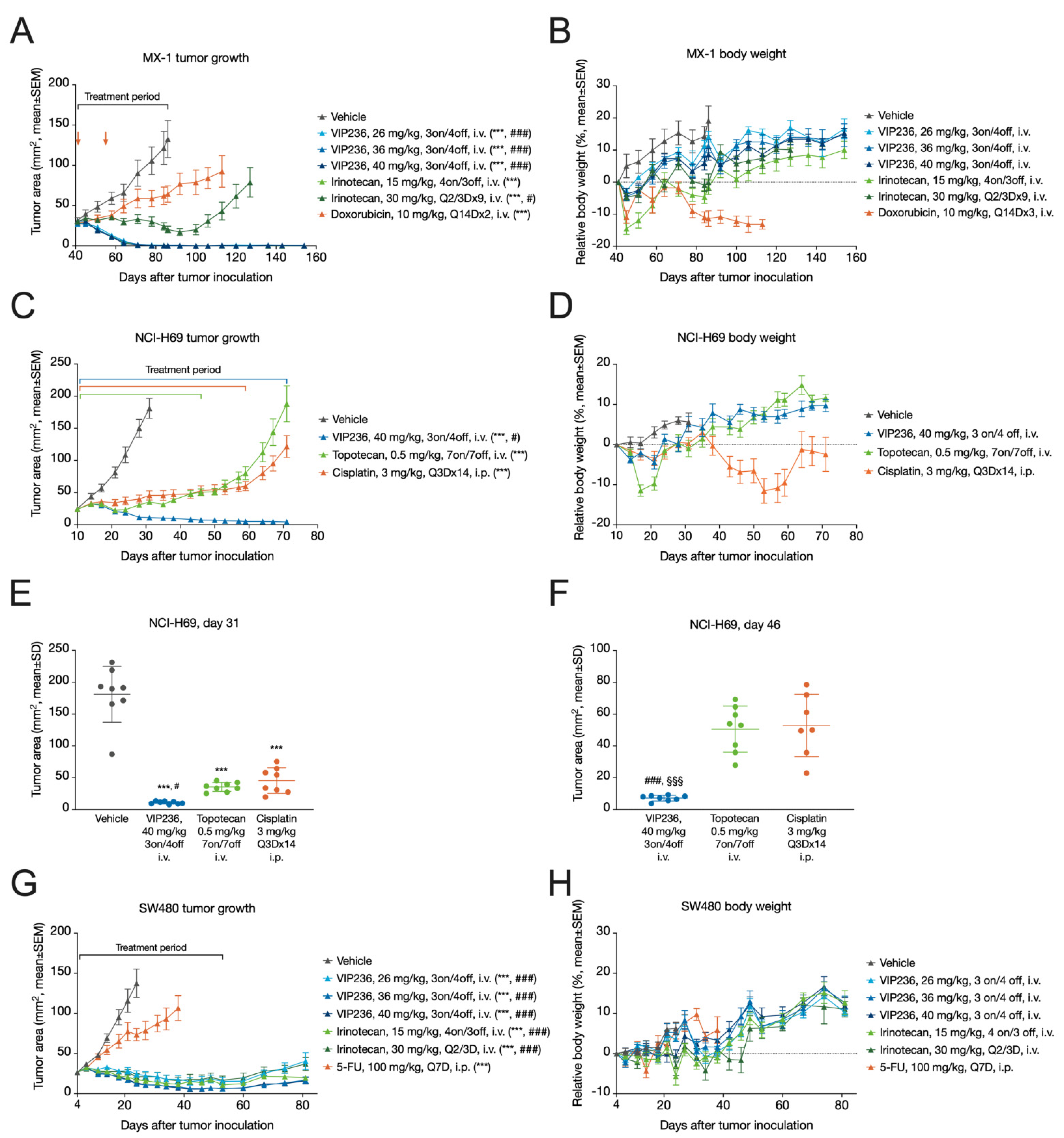
3.6. VIP236 Shows High Antitumor Efficacy and Good Tolerability In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable linkers in antibody-drug conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef]
- Chari, R.V.; Miller, M.L.; Widdison, W.C. Antibody-drug conjugates: An emerging concept in cancer therapy. Angew. Chem. Int. Ed. Engl. 2014, 53, 3796–3827. [Google Scholar] [CrossRef]
- Joubert, N.; Beck, A.; Dumontet, C.; Denevault-Sabourin, C. Antibody-Drug Conjugates: The Last Decade. Pharmaceuticals 2020, 13, 245. [Google Scholar] [CrossRef] [PubMed]
- Katz, J.; Janik, J.E.; Younes, A. Brentuximab Vedotin (SGN-35). Clin. Cancer Res. 2011, 17, 6428–6436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senter, P.D.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.M.; Chari, R.V. Ado-trastuzumab Emtansine (T-DM1): An antibody-drug conjugate (ADC) for HER2-positive breast cancer. J. Med. Chem. 2014, 57, 6949–6964. [Google Scholar] [CrossRef]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.Y.; Dieras, V.; Guardino, E.; et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantarjian, H.M.; DeAngelo, D.J.; Stelljes, M.; Martinelli, G.; Liedtke, M.; Stock, W.; Gokbuget, N.; O’Brien, S.; Wang, K.; Wang, T.; et al. Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2016, 375, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Amadori, S.; Suciu, S.; Selleslag, D.; Aversa, F.; Gaidano, G.; Musso, M.; Annino, L.; Venditti, A.; Voso, M.T.; Mazzone, C.; et al. Gemtuzumab Ozogamicin versus Best Supportive Care in Older Patients with Newly Diagnosed Acute Myeloid Leukemia Unsuitable for Intensive Chemotherapy: Results of the Randomized Phase III EORTC-GIMEMA AML-19 Trial. J. Clin. Oncol. 2016, 34, 972–979. [Google Scholar] [CrossRef]
- Bross, P.F.; Beitz, J.; Chen, G.; Chen, X.H.; Duffy, E.; Kieffer, L.; Roy, S.; Sridhara, R.; Rahman, A.; Williams, G.; et al. Approval summary: Gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin. Cancer Res. 2001, 7, 1490–1496. [Google Scholar]
- Morschhauser, F.; Flinn, I.W.; Advani, R.; Sehn, L.H.; Diefenbach, C.; Kolibaba, K.; Press, O.W.; Salles, G.; Tilly, H.; Chen, A.I.; et al. Polatuzumab vedotin or pinatuzumab vedotin plus rituximab in patients with relapsed or refractory non-Hodgkin lymphoma: Final results from a phase 2 randomised study (ROMULUS). Lancet Haematol. 2019, 6, e254–e265. [Google Scholar] [CrossRef]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N. Engl. J. Med. 2020, 382, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.E.; O’Donnell, P.H.; Balar, A.V.; McGregor, B.A.; Heath, E.I.; Yu, E.Y.; Galsky, M.D.; Hahn, N.M.; Gartner, E.M.; Pinelli, J.M.; et al. Pivotal Trial of Enfortumab Vedotin in Urothelial Carcinoma after Platinum and Anti-Programmed Death 1/Programmed Death Ligand 1 Therapy. J. Clin. Oncol. 2019, 37, 2592–2600. [Google Scholar] [CrossRef]
- Rugo, H.S.; Bardia, A.; Tolaney, S.M.; Arteaga, C.; Cortes, J.; Sohn, J.; Marme, F.; Hong, Q.; Delaney, R.J.; Hafeez, A.; et al. TROPiCS-02: A Phase III study investigating sacituzumab govitecan in the treatment of HR+/HER2− metastatic breast cancer. Future Oncol. 2020, 16, 705–715. [Google Scholar] [CrossRef] [Green Version]
- Caimi, P.F.; Ai, W.; Alderuccio, J.P.; Ardeshna, K.M.; Hamadani, M.; Hess, B.; Kahl, B.S.; Radford, J.; Solh, M.; Stathis, A.; et al. Loncastuximab tesirine in relapsed or refractory diffuse large B-cell lymphoma (LOTIS-2): A multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2021, 22, 790–800. [Google Scholar] [CrossRef]
- Jiang, J.; Li, S.; Shan, X.; Wang, L.; Ma, J.; Huang, M.; Dong, L.; Chen, F. Preclinical safety profile of disitamab vedotina novel anti-HER2 antibody conjugated with MMAE. Toxicol. Lett. 2020, 324, 30–37. [Google Scholar] [CrossRef]
- de Bono, J.S.; Concin, N.; Hong, D.S.; Thistlethwaite, F.C.; Machiels, J.P.; Arkenau, H.T.; Plummer, R.; Jones, R.H.; Nielsen, D.; Windfeld, K.; et al. Tisotumab vedotin in patients with advanced or metastatic solid tumours (InnovaTV 201): A first-in-human, multicentre, phase 1–2 trial. Lancet Oncol. 2019, 20, 383–393. [Google Scholar] [CrossRef]
- Krall, N.; Scheuermann, J.; Neri, D. Small targeted cytotoxics: Current state and promises from DNA-encoded chemical libraries. Angew. Chem. Int. Ed. Engl. 2013, 52, 1384–1402. [Google Scholar] [CrossRef]
- Kumar, A.; Mastren, T.; Wang, B.; Hsieh, J.T.; Hao, G.; Sun, X. Design of a Small-Molecule Drug Conjugate for Prostate Cancer Targeted Theranostics. Bioconjug. Chem. 2016, 27, 1681–1689. [Google Scholar] [CrossRef] [PubMed]
- Seattle Genetics, Inc. Adcetris (Brentuximab Vedotin) [Package Insert]. U.S. Food and Drug Administration Website. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/125388_s056s078lbl.pdf (accessed on 11 October 2021).
- Genentech, Inc. Kadcyla (Ado-Trastuzumab Emtansine) [Package Insert]. U.S. Food and Drug Administration Website. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/125427s105lbl.pdf (accessed on 11 October 2021).
- Wyeth Pharmaceuticals Inc. Besponsa (Inotuzumab Ozogamicin) [Package Insert]. U.S. Food and Drug Administration Website. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761040s000lbl.pdf (accessed on 11 October 2021).
- Wyeth Pharmaceuticals Inc. Mylotarg (Gemtuzumab Ozogamicin) [Package Insert]. U.S. Food and Drug Administration Website. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761060lbl.pdf (accessed on 11 October 2021).
- Astellas Pharma US, Inc. Padcev (Enfortumab Vedotin-Ejfv) [Package Insert]. U.S. Food and Drug Administration Website. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761137s000lbl.pdf (accessed on 11 October 2021).
- Daiichi Sankyo, Inc. Enhertu (Fam-Trastuzumab Deruxtecan-Nxki) [Package Insert]. U.S. Food and Drug Administration Website. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761139s011lbl.pdf (accessed on 11 October 2021).
- Immunomedics, Inc. Trodelvy (Sacituzumab Govitecan-Hziy) [Package Insert]. U.S. Food and Drug Administration Website. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761115s000lbl.pdf (accessed on 11 October 2021).
- GlaxoSmithKline Intellectual Property Development Ltd. Blenrep (Belantamab Mafodotin-Blmf) [Package Insert]. U.S. Food and Drug Administration Website. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761158s000lbl.pdf (accessed on 11 October 2021).
- Seagen Inc. Tivdak (Tisotumab Vedotin-Tftv) [Package Insert]. U.S. Food and Drug Administration Website. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761208s000lbl.pdf (accessed on 11 October 2021).
- Zhuang, C.; Guan, X.; Ma, H.; Cong, H.; Zhang, W.; Miao, Z. Small molecule-drug conjugates: A novel strategy for cancer-targeted treatment. Eur. J. Med. Chem. 2019, 163, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Cazzamalli, S.; Dal Corso, A.; Widmayer, F.; Neri, D. Chemically Defined Antibody- and Small Molecule-Drug Conjugates for in vivo Tumor Targeting Applications: A Comparative Analysis. J. Am. Chem. Soc. 2018, 140, 1617–1621. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Jiang, T.; Li, Q.; Ling, X. Camptothecin (CPT) and its derivatives are known to target topoisomerase I (Top1) as their mechanism of action: Did we miss something in CPT analogue molecular targets for treating human disease such as cancer? Am. J. Cancer Res. 2017, 7, 2350–2394. [Google Scholar]
- Lerchen, H.-G.; von dem Bruch, K. Synthesis of 20-O-linked 20(S)-Camptothecin Glycoconjugates: Impact of the Side Chain of the Ester-linked Amino Acid on Epimerization during the Acylation Reaction and on Hydrolytic Stability of the Final Glycoconjugates. J. Prakt. Chem. 2000, 342, 753–760. [Google Scholar] [CrossRef]
- Lerchen, H.G.; Baumgarten, J.; von dem Bruch, K.; Lehmann, T.E.; Sperzel, M.; Kempka, G.; Fiebig, H.H. Design and optimization of 20-O-linked camptothecin glycoconjugates as anticancer agents. J. Med. Chem. 2001, 44, 4186–4195. [Google Scholar] [CrossRef] [PubMed]
- Kciuk, M.; Marciniak, B.; Kontek, R. Irinotecan-Still an Important Player in Cancer Chemotherapy: A Comprehensive Overview. Int. J. Mol. Sci. 2020, 21, 4919. [Google Scholar] [CrossRef] [PubMed]
- Danen, E.H.; Ten Berge, P.J.; Van Muijen, G.N.; Van’t Hof-Grootenboer, A.E.; Brocker, E.B.; Ruiter, D.J. Emergence of alpha 5 beta 1 fibronectin- and alpha v beta 3 vitronectin-receptor expression in melanocytic tumour progression. Histopathology 1994, 24, 249–256. [Google Scholar] [CrossRef]
- Felding-Habermann, B.; O’Toole, T.E.; Smith, J.W.; Fransvea, E.; Ruggeri, Z.M.; Ginsberg, M.H.; Hughes, P.E.; Pampori, N.; Shattil, S.J.; Saven, A.; et al. Integrin activation controls metastasis in human breast cancer. Proc. Natl. Acad. Sci. USA 2001, 98, 1853–1858. [Google Scholar] [CrossRef] [Green Version]
- Gruber, G.; Hess, J.; Stiefel, C.; Aebersold, D.M.; Zimmer, Y.; Greiner, R.H.; Studer, U.; Altermatt, H.J.; Hlushchuk, R.; Djonov, V. Correlation between the tumoral expression of beta3-integrin and outcome in cervical cancer patients who had undergone radiotherapy. Br. J. Cancer 2005, 92, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Hosotani, R.; Kawaguchi, M.; Masui, T.; Koshiba, T.; Ida, J.; Fujimoto, K.; Wada, M.; Doi, R.; Imamura, M. Expression of integrin alphaVbeta3 in pancreatic carcinoma: Relation to MMP-2 activation and lymph node metastasis. Pancreas 2002, 25, e30–e35. [Google Scholar] [CrossRef]
- Landen, C.N.; Kim, T.J.; Lin, Y.G.; Merritt, W.M.; Kamat, A.A.; Han, L.Y.; Spannuth, W.A.; Nick, A.M.; Jennnings, N.B.; Kinch, M.S.; et al. Tumor-selective response to antibody-mediated targeting of alphavbeta3 integrin in ovarian cancer. Neoplasia 2008, 10, 1259–1267. [Google Scholar] [CrossRef] [Green Version]
- McCabe, N.P.; De, S.; Vasanji, A.; Brainard, J.; Byzova, T.V. Prostate cancer specific integrin alphavbeta3 modulates bone metastatic growth and tissue remodeling. Oncogene 2007, 26, 6238–6243. [Google Scholar] [CrossRef] [Green Version]
- Nip, J.; Shibata, H.; Loskutoff, D.J.; Cheresh, D.A.; Brodt, P. Human melanoma cells derived from lymphatic metastases use integrin alpha v beta 3 to adhere to lymph node vitronectin. J. Clin. Investig. 1992, 90, 1406–1413. [Google Scholar] [CrossRef] [Green Version]
- Brooks, P.C.; Montgomery, A.M.; Rosenfeld, M.; Reisfeld, R.A.; Hu, T.; Klier, G.; Cheresh, D.A. Integrin alpha v beta 3 antagonists promote tumor regression by inducing apoptosis of angiogenic blood vessels. Cell 1994, 79, 1157–1164. [Google Scholar] [CrossRef]
- Alday-Parejo, B.; Stupp, R.; Ruegg, C. Are Integrins Still Practicable Targets for Anti-Cancer Therapy? Cancers 2019, 11, 978. [Google Scholar] [CrossRef] [Green Version]
- Argraves, W.S.; Pytela, R.; Suzuki, S.; Millan, J.L.; Pierschbacher, M.D.; Ruoslahti, E. cDNA sequences from the alpha subunit of the fibronectin receptor predict a transmembrane domain and a short cytoplasmic peptide. J. Biol. Chem. 1986, 261, 12922–12924. [Google Scholar] [CrossRef]
- Pierschbacher, M.D.; Ruoslahti, E. Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature 1984, 309, 30–33. [Google Scholar] [CrossRef]
- Pytela, R.; Pierschbacher, M.D.; Ruoslahti, E. A 125/115-kDa cell surface receptor specific for vitronectin interacts with the arginine-glycine-aspartic acid adhesion sequence derived from fibronectin. Proc. Natl. Acad. Sci. USA 1985, 82, 5766–5770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, S.; Argraves, W.S.; Pytela, R.; Arai, H.; Krusius, T.; Pierschbacher, M.D.; Ruoslahti, E. cDNA and amino acid sequences of the cell adhesion protein receptor recognizing vitronectin reveal a transmembrane domain and homologies with other adhesion protein receptors. Proc. Natl. Acad. Sci. USA 1986, 83, 8614–8618. [Google Scholar] [CrossRef] [Green Version]
- Murphy, E.A.; Majeti, B.K.; Barnes, L.A.; Makale, M.; Weis, S.M.; Lutu-Fuga, K.; Wrasidlo, W.; Cheresh, D.A. Nanoparticle-mediated drug delivery to tumor vasculature suppresses metastasis. Proc. Natl. Acad. Sci. USA 2008, 105, 9343–9348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugahara, K.N.; Teesalu, T.; Karmali, P.P.; Kotamraju, V.R.; Agemy, L.; Girard, O.M.; Hanahan, D.; Mattrey, R.F.; Ruoslahti, E. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell 2009, 16, 510–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Bajjuri, K.M.; Liu, C.; Sinha, S.C. Targeting cell surface alpha(v)beta(3) integrin increases therapeutic efficacies of a legumain protease-activated auristatin prodrug. Mol. Pharm. 2012, 9, 168–175. [Google Scholar] [CrossRef] [Green Version]
- Hood, J.D.; Bednarski, M.; Frausto, R.; Guccione, S.; Reisfeld, R.A.; Xiang, R.; Cheresh, D.A. Tumor regression by targeted gene delivery to the neovasculature. Science 2002, 296, 2404–2407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiffelers, R.M.; Ansari, A.; Xu, J.; Zhou, Q.; Tang, Q.; Storm, G.; Molema, G.; Lu, P.Y.; Scaria, P.V.; Woodle, M.C. Cancer siRNA therapy by tumor selective delivery with ligand-targeted sterically stabilized nanoparticle. Nucleic Acids Res. 2004, 32, e149. [Google Scholar] [CrossRef]
- Raposo Moreira Dias, A.; Pina, A.; Dean, A.; Lerchen, H.G.; Caruso, M.; Gasparri, F.; Fraietta, I.; Troiani, S.; Arosio, D.; Belvisi, L.; et al. Neutrophil Elastase Promotes Linker Cleavage and Paclitaxel Release from an Integrin-Targeted Conjugate. Chemistry 2019, 25, 1696–1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, T.; Takahashi, S.; Mizumoto, T.; Harao, M.; Akizuki, M.; Takasugi, M.; Fukutomi, T.; Yamashita, J. Neutrophil elastase and cancer. Surg. Oncol. 2006, 15, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Troutman, M.D.; Thakker, D.R. Novel experimental parameters to quantify the modulation of absorptive and secretory transport of compounds by P-glycoprotein in cell culture models of intestinal epithelium. Pharm. Res. 2003, 20, 1210–1224. [Google Scholar] [CrossRef]
- Schinkel, A.H.; Wagenaar, E.; van Deemter, L.; Mol, C.A.; Borst, P. Absence of the mdr1a P-Glycoprotein in mice affects tissue distribution and pharmacokinetics of dexamethasone, digoxin, and cyclosporin A. J. Clin. Investig. 1995, 96, 1698–1705. [Google Scholar] [CrossRef] [Green Version]
- Schwab, D.; Fischer, H.; Tabatabaei, A.; Poli, S.; Huwyler, J. Comparison of in vitro P-glycoprotein screening assays: Recommendations for their use in drug discovery. J. Med. Chem. 2003, 46, 1716–1725. [Google Scholar] [CrossRef]
- Gibaldi, M.; Perrier, D. Pharmacokinetics; Marcel Dekker: New York, NY, USA, 1982. [Google Scholar]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Seelig, A. P-Glycoprotein: One Mechanism, Many Tasks and the Consequences for Pharmacotherapy of Cancers. Front. Oncol. 2020, 10, 576559. [Google Scholar] [CrossRef]
- Bailly, C. Irinotecan: 25 years of cancer treatment. Pharmacol. Res. 2019, 148, 104398. [Google Scholar] [CrossRef] [PubMed]
- Davies, B.; Morris, T. Physiological parameters in laboratory animals and humans. Pharm. Res. 1993, 10, 1093–1095. [Google Scholar] [CrossRef]
- Liu, L.F.; Desai, S.D.; Li, T.K.; Mao, Y.; Sun, M.; Sim, S.P. Mechanism of action of camptothecin. Ann. N. Y. Acad. Sci. 2000, 922, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.H.; Yu, A.M. ABC transporters in multidrug resistance and pharmacokinetics, and strategies for drug development. Curr. Pharm. Des. 2014, 20, 793–807. [Google Scholar] [CrossRef]
- Brooks, P.C.; Stromblad, S.; Klemke, R.; Visscher, D.; Sarkar, F.H.; Cheresh, D.A. Antiintegrin alpha v beta 3 blocks human breast cancer growth and angiogenesis in human skin. J. Clin. Investig. 1995, 96, 1815–1822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, S.Y.; Shin, J.; Kim, J.H.; Kang, M.S.; Yoo, H.Y.; Kim, H.H.; Um, S.H.; Kim, S.H. Overexpression of integrin alphav correlates with poor prognosis in colorectal cancer. J. Clin. Pathol. 2014, 67, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Vonlaufen, A.; Wiedle, G.; Borisch, B.; Birrer, S.; Luder, P.; Imhof, B.A. Integrin alpha(v)beta(3) expression in colon carcinoma correlates with survival. Mod. Pathol. 2001, 14, 1126–1132. [Google Scholar] [CrossRef]
- Hieken, T.J.; Farolan, M.; Ronan, S.G.; Shilkaitis, A.; Wild, L.; Das Gupta, T.K. Beta3 integrin expression in melanoma predicts subsequent metastasis. J. Surg. Res. 1996, 63, 169–173. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, F.; Chen, X. Integrin alpha(v)beta(3)-Targeted Cancer Therapy. Drug Dev. Res. 2008, 69, 329–339. [Google Scholar] [CrossRef] [Green Version]
- Delbaldo, C.; Raymond, E.; Vera, K.; Hammershaimb, L.; Kaucic, K.; Lozahic, S.; Marty, M.; Faivre, S. Phase I and pharmacokinetic study of etaracizumab (Abegrin), a humanized monoclonal antibody against alphavbeta3 integrin receptor, in patients with advanced solid tumors. Investig. New Drugs 2008, 26, 35–43. [Google Scholar] [CrossRef]
- Gutheil, J.C.; Campbell, T.N.; Pierce, P.R.; Watkins, J.D.; Huse, W.D.; Bodkin, D.J.; Cheresh, D.A. Targeted antiangiogenic therapy for cancer using Vitaxin: A humanized monoclonal antibody to the integrin alphavbeta3. Clin. Cancer Res. 2000, 6, 3056–3061. [Google Scholar] [PubMed]
- Mitjans, F.; Sander, D.; Adan, J.; Sutter, A.; Martinez, J.M.; Jaggle, C.S.; Moyano, J.M.; Kreysch, H.G.; Piulats, J.; Goodman, S.L. An anti-alpha v-integrin antibody that blocks integrin function inhibits the development of a human melanoma in nude mice. J. Cell Sci. 1995, 108, 2825–2838. [Google Scholar] [CrossRef]
- O’Day, S.; Pavlick, A.; Loquai, C.; Lawson, D.; Gutzmer, R.; Richards, J.; Schadendorf, D.; Thompson, J.A.; Gonzalez, R.; Trefzer, U.; et al. A randomised, phase II study of intetumumab, an anti-alphav-integrin mAb, alone and with dacarbazine in stage IV melanoma. Br. J. Cancer 2011, 105, 346–352. [Google Scholar] [CrossRef] [Green Version]
- Bauerle, T.; Komljenovic, D.; Merz, M.; Berger, M.R.; Goodman, S.L.; Semmler, W. Cilengitide inhibits progression of experimental breast cancer bone metastases as imaged noninvasively using VCT, MRI and DCE-MRI in a longitudinal in vivo study. Int. J. Cancer 2011, 128, 2453–2462. [Google Scholar] [CrossRef]
- Bretschi, M.; Cheng, C.; Witt, H.; Dimitrakopoulou-Strauss, A.; Strauss, L.G.; Semmler, W.; Bauerle, T. Cilengitide affects tumor compartment, vascularization and microenvironment in experimental bone metastases as shown by longitudinal (1)(8)F-FDG PET and gene expression analysis. J. Cancer Res. Clin. Oncol. 2013, 139, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Mas-Moruno, C.; Rechenmacher, F.; Kessler, H. Cilengitide: The first anti-angiogenic small molecule drug candidate design, synthesis and clinical evaluation. Anticancer. Agents Med. Chem. 2010, 10, 753–768. [Google Scholar] [CrossRef] [Green Version]
- Elez, E.; Kocakova, I.; Hohler, T.; Martens, U.M.; Bokemeyer, C.; Van Cutsem, E.; Melichar, B.; Smakal, M.; Csoszi, T.; Topuzov, E.; et al. Abituzumab combined with cetuximab plus irinotecan versus cetuximab plus irinotecan alone for patients with KRAS wild-type metastatic colorectal cancer: The randomised phase I/II POSEIDON trial. Ann. Oncol. 2015, 26, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, A.; Rawal, S.K.; Szkarlat, K.; Bogdanova, N.; Dirix, L.; Stenzl, A.; Welslau, M.; Wang, G.; Dawkins, F.; de Boer, C.J.; et al. A randomized, double-blind, multicenter, phase 2 study of a human monoclonal antibody to human alphanu integrins (intetumumab) in combination with docetaxel and prednisone for the first-line treatment of patients with metastatic castration-resistant prostate cancer. Ann. Oncol. 2013, 24, 329–336. [Google Scholar] [CrossRef]
- Hersey, P.; Sosman, J.; O’Day, S.; Richards, J.; Bedikian, A.; Gonzalez, R.; Sharfman, W.; Weber, R.; Logan, T.; Buzoianu, M.; et al. A randomized phase 2 study of etaracizumab, a monoclonal antibody against integrin alpha(v)beta(3), + or − dacarbazine in patients with stage IV metastatic melanoma. Cancer 2010, 116, 1526–1534. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Gorlia, T.; Erridge, S.C.; Perry, J.; Hong, Y.K.; Aldape, K.D.; Lhermitte, B.; Pietsch, T.; Grujicic, D.; et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1100–1108. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, A.R.; Hart, I.R.; Watson, A.R.; Welti, J.C.; Silva, R.G.; Robinson, S.D.; Da Violante, G.; Gourlaouen, M.; Salih, M.; Jones, M.C.; et al. Stimulation of tumor growth and angiogenesis by low concentrations of RGD-mimetic integrin inhibitors. Nat. Med. 2009, 15, 392–400. [Google Scholar] [CrossRef]
- Humphries, J.D.; Byron, A.; Humphries, M.J. Integrin ligands at a glance. J. Cell Sci. 2006, 119, 3901–3903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, D.S.; Patnaik, A.; Bendell, J.C.; Papadopoulos, K.; Infante, J.R.; Mastico, R.A.; Johnson, D.; Qin, A.; O’Leary, J.J.; Tolcher, A.W. A phase I dose-escalation study of IMGN388 in patients with solid tumors. J. Clin. Oncol. 2010, 28, 3058. [Google Scholar] [CrossRef]
- Korkmaz, B.; Moreau, T.; Gauthier, F. Neutrophil elastase, proteinase 3 and cathepsin G: Physicochemical properties, activity and physiopathological functions. Biochimie 2008, 90, 227–242. [Google Scholar] [CrossRef]
- Lerman, I.; Hammes, S.R. Neutrophil elastase in the tumor microenvironment. Steroids 2018, 133, 96–101. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Payload | Permeability A → B [nm/s] | Efflux Ratio |
|---|---|---|---|
| P-gp-expressing LLC-PK1 | VIP126 | 196 | 0.6 |
| SN38 | 10 | 16 | |
| Caco-2 | VIP126 | 171 | 1 |
| SN38 | 8 | 36 |
| Compound | IC50 [nM] | ||
|---|---|---|---|
| NCI-H1975 | NCI-H1975-P-gp | NCI-H1975-BCRP | |
| VIP126 | 19 | 34 | 27 |
| SN38 | 45 | 141 | 512 |
| Cancer Cell Line | IC50 (nM) | |||
|---|---|---|---|---|
| Cancer Type | VIP236 without NE | VIP236 with NE | VIP126 | |
| 786-O | Human renal cell carcinoma | 188 | 1.1 | 1.2 |
| HT29 | Human colorectal cancer | 245 | 8.7 | 6.8 |
| LoVo | Human colorectal cancer | 91 | 2.9 | 1.8 |
| SW480 | Human colorectal cancer | 41 | 1.2 | 1.8 |
| NCI-H292 | Human lung mucoepidermoid carcinoma | 209 | 1.8 | 1.5 |
| NCI-H69 | Human small cell lung cancer | 486 | 3.0 | 2.9 |
| 4T1 | Murine mammary carcinoma | >1000 | 59 | 49 |
| PK Parameters | VIP236 | VIP126 | |
|---|---|---|---|
| Dose | (mg/kg) | 4 | 1 |
| AUCinf | (µg·h/L) | 93,400 | 301 |
| CL | (L/h/kg) | 0.0428 | 3.33 |
| Cmax | (µg/L) | 112,000 | 1510 |
| Vss | (L/kg) | 0.0688 | 1.54 |
| t1/2 | (h) | 0.807 | 1.02 |
| Compound Dosed | VIP236 (4 mg/kg) | VIP126 (1 mg/kg) | |
|---|---|---|---|
| PK Parameters | VIP236 | VIP126 | VIP126 |
| AUCtumor (µg·h/L) | 4660 | 318 | 185 |
| AUCplasma (µg·h/L) | 93,400 | 52.1 | 301 |
| AUCtumor/AUCplasma | 0.0499 | 6.10 | 0.616 |
| Treatment | Corresponding Payload Dose (mg/kg) | Response a | Treatment/ Control Ratio b |
|---|---|---|---|
| MX-1 TNBC | |||
| VIP236, 26 mg/kg, 3 on/4 off | 6.5 | CR: 7/8, PR: 1/8 | 0.003 ***, ### |
| VIP236, 36 mg/kg, 3 on/4 off | 9 | CR: 6/8, PR: 2/8 | 0.004 ***, ### |
| VIP236, 40 mg/kg, 3 on/4 off | 10 | CR: 7/8, PR: 1/8 | 0.001 ***, ### |
| Irinotecan, 15 mg/kg, 4 on/3 off | 9.5 | CR: 4/8, PR: 4/8 | 0.002 *** |
| Irinotecan, 30 mg/kg, Q2/3Dx9 | 19 | PR: 5/7, SD: 2/7 | 0.15 ***, # |
| Doxorubicin, 10 mg/kg, Q14Dx2 | n/a | SD: 1/7, PD: 2/7 | 0.49 *** |
| NCI-H69 SCLC | |||
| VIP236, 40 mg/kg, 3 on/4 off | 10 | PR: 8/8 | 0.06 ***, # |
| Topotecan, 0.5 mg/kg, 7 on/7 off | n/a | SD: 1/8, PD: 7/8 | 0.20 *** |
| Cisplatin, 3 mg/kg, Q3Dx14 | n/a | SD: 1/8, PD: 7/8 | 0.25 *** |
| SW480 CRC | |||
| VIP236, 26 mg/kg, 3 on/4 off | 6.5 | PR: 2/8, SD: 4/8, PD: 2/8 | 0.18 ***, ### |
| VIP236, 36 mg/kg, 3 on/4 off | 9 | PR: 7/8, SD: 1/8 | 0.09 ***, ### |
| VIP236, 40 mg/kg, 3 on/4 off | 10 | PR: 8/8 | 0.10 ***, ### |
| Irinotecan, 15 mg/kg, 4 on/3 off | 9.5 | PR: 5/8, SD: 3/8 | 0.13 *** |
| Irinotecan, 30 mg/kg, Q2/3D | 19 | PR: 4/8, SD: 3/8, PD: 1/8 | 0.15 *** |
| 5-FU, 100 mg/kg, Q7D | n/a | PD: 8/8 | 0.54 *** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lerchen, H.-G.; Stelte-Ludwig, B.; Kopitz, C.; Heroult, M.; Zubov, D.; Willuda, J.; Schlange, T.; Kahnert, A.; Wong, H.; Izumi, R.; et al. A Small Molecule–Drug Conjugate (SMDC) Consisting of a Modified Camptothecin Payload Linked to an αVß3 Binder for the Treatment of Multiple Cancer Types. Cancers 2022, 14, 391. https://doi.org/10.3390/cancers14020391
Lerchen H-G, Stelte-Ludwig B, Kopitz C, Heroult M, Zubov D, Willuda J, Schlange T, Kahnert A, Wong H, Izumi R, et al. A Small Molecule–Drug Conjugate (SMDC) Consisting of a Modified Camptothecin Payload Linked to an αVß3 Binder for the Treatment of Multiple Cancer Types. Cancers. 2022; 14(2):391. https://doi.org/10.3390/cancers14020391
Chicago/Turabian StyleLerchen, Hans-Georg, Beatrix Stelte-Ludwig, Charlotte Kopitz, Melanie Heroult, Dmitry Zubov, Joerg Willuda, Thomas Schlange, Antje Kahnert, Harvey Wong, Raquel Izumi, and et al. 2022. "A Small Molecule–Drug Conjugate (SMDC) Consisting of a Modified Camptothecin Payload Linked to an αVß3 Binder for the Treatment of Multiple Cancer Types" Cancers 14, no. 2: 391. https://doi.org/10.3390/cancers14020391
APA StyleLerchen, H.-G., Stelte-Ludwig, B., Kopitz, C., Heroult, M., Zubov, D., Willuda, J., Schlange, T., Kahnert, A., Wong, H., Izumi, R., & Hamdy, A. (2022). A Small Molecule–Drug Conjugate (SMDC) Consisting of a Modified Camptothecin Payload Linked to an αVß3 Binder for the Treatment of Multiple Cancer Types. Cancers, 14(2), 391. https://doi.org/10.3390/cancers14020391