
HIF-1 and NRF2; Key Molecules for Malignant Phenotypes of Pancreatic Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Dense Fibrotic Stroma in Pancreatic Cancer
3. Hypoxia-Inducible Factor 1: A Central Machinery for Hypoxia Response Mechanism
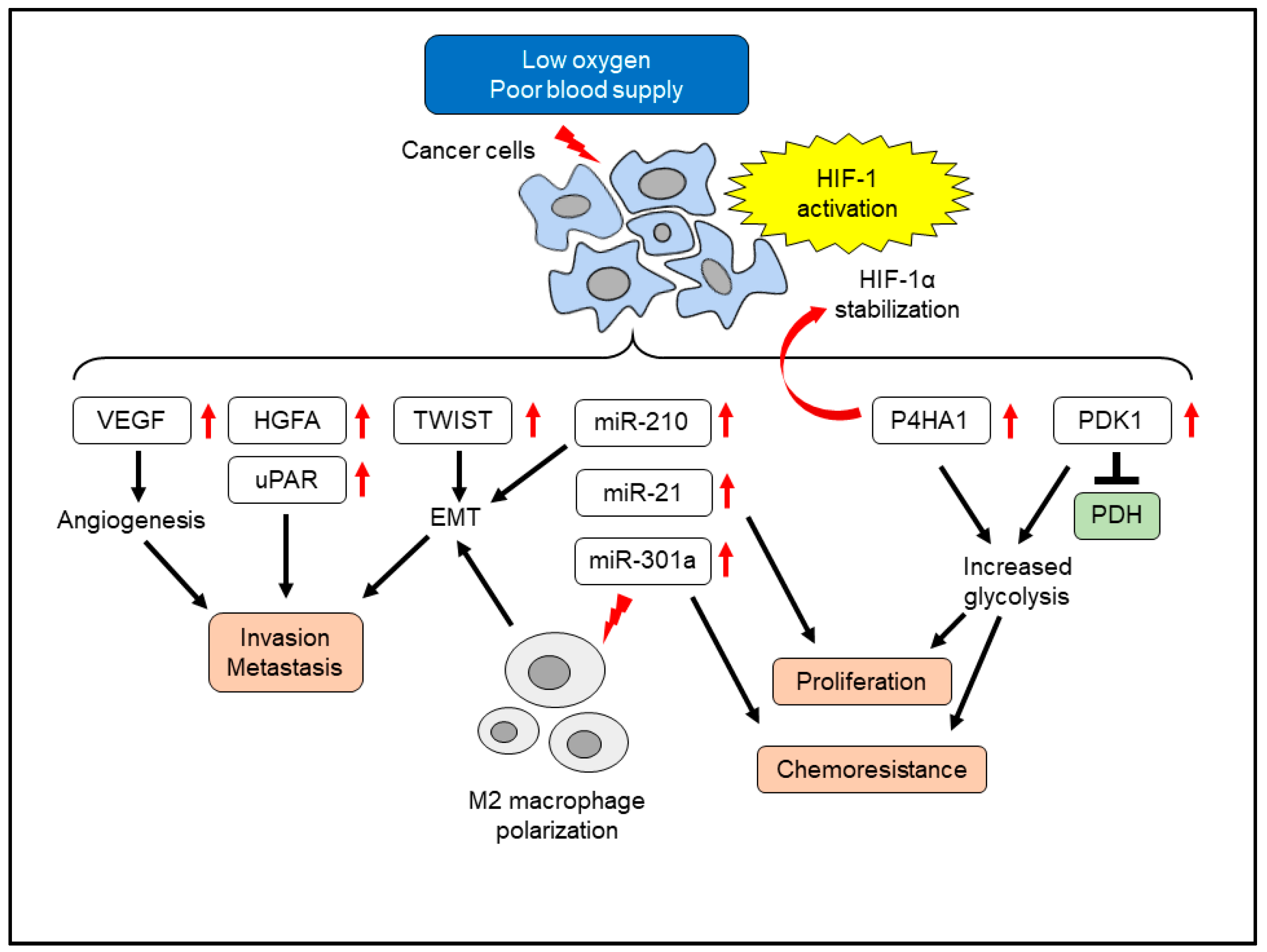
4. Effects of Hypoxia on Pancreatic Cancer Cells
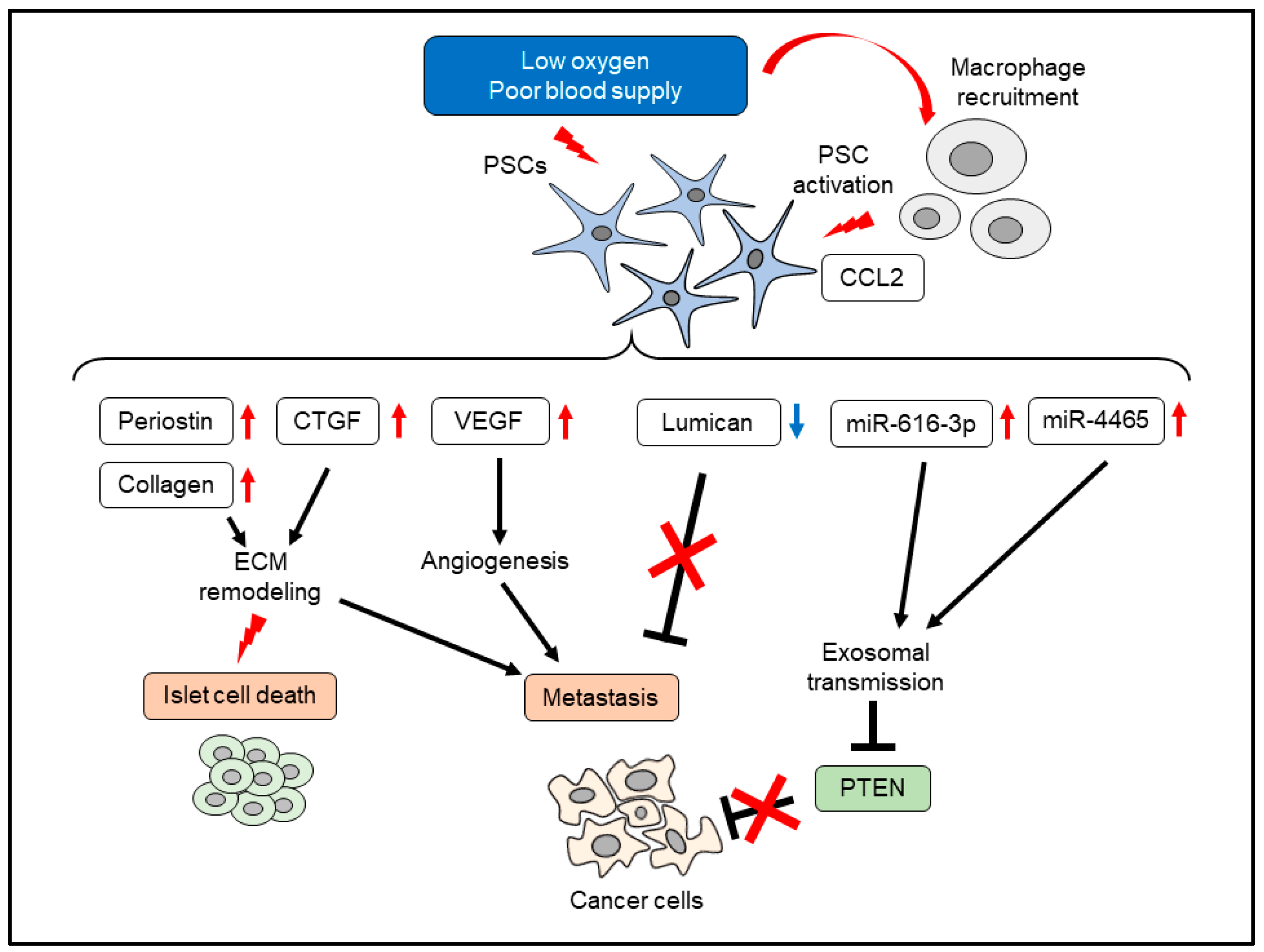
5. Effects of Hypoxia on PSCs
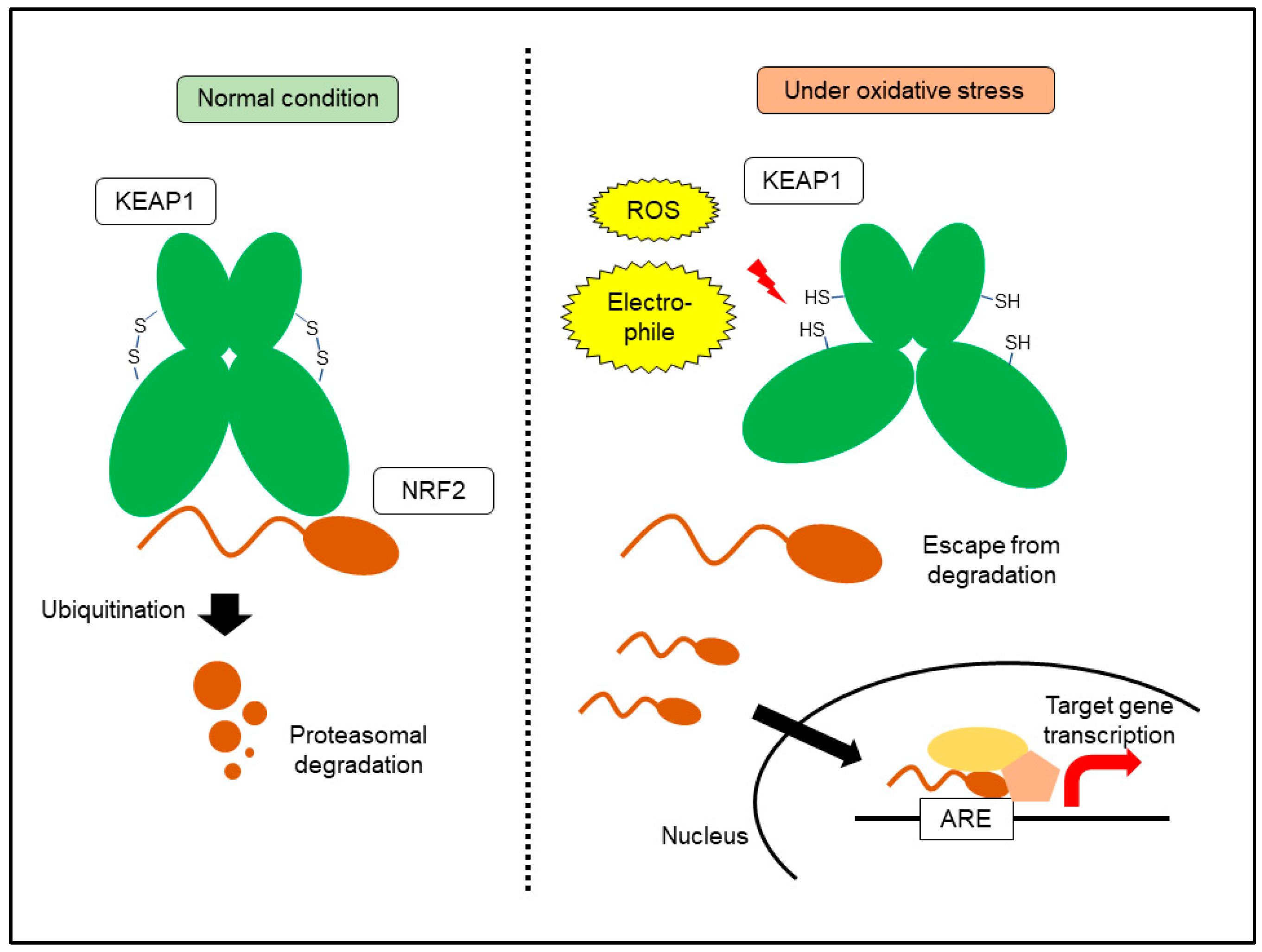
6. KEAP1-NRF2 System: A Central Machinery for Oxidative Stress Response
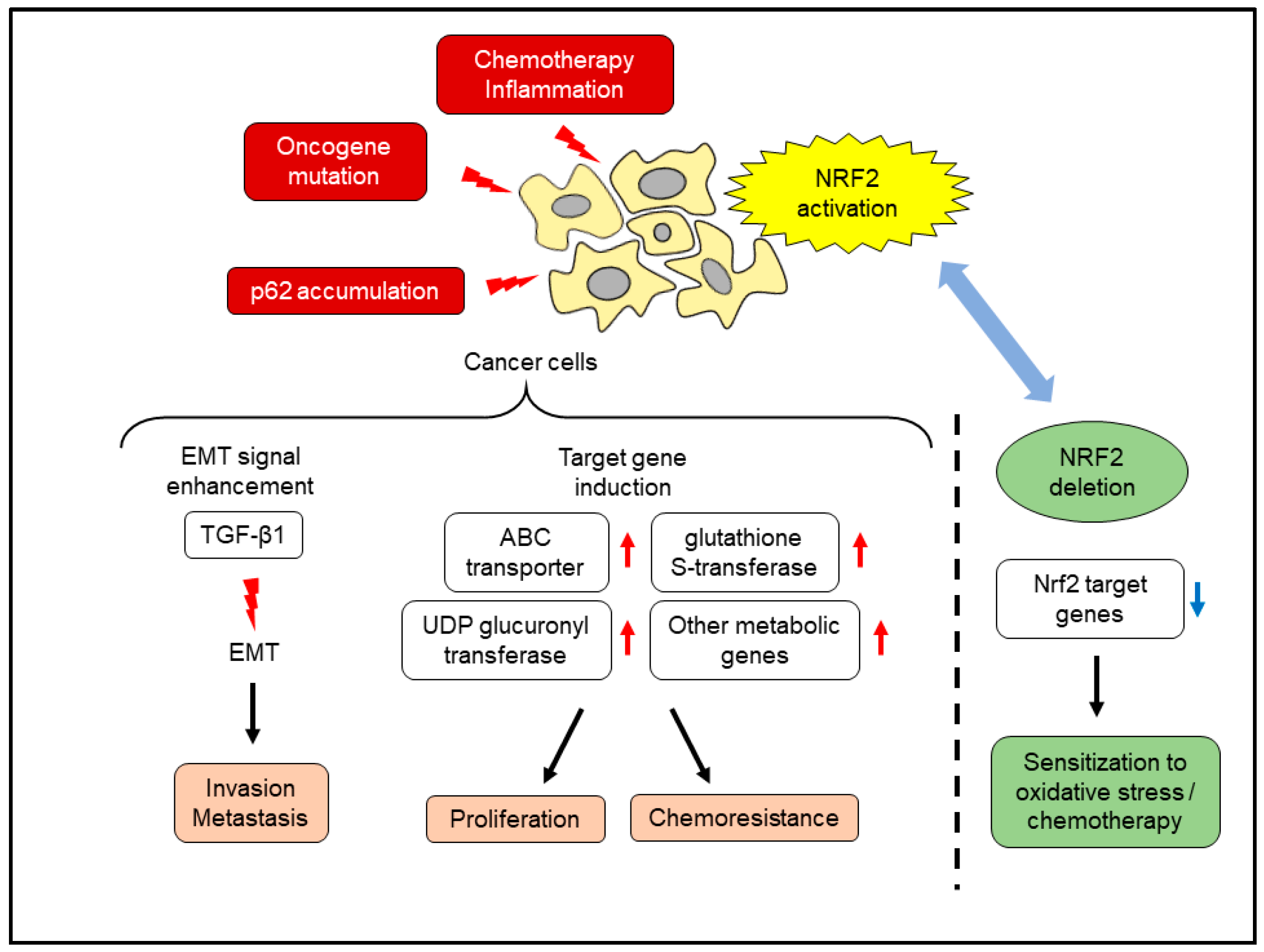
7. Effects of NRF2 Activation in Pancreatic Cancer Cells
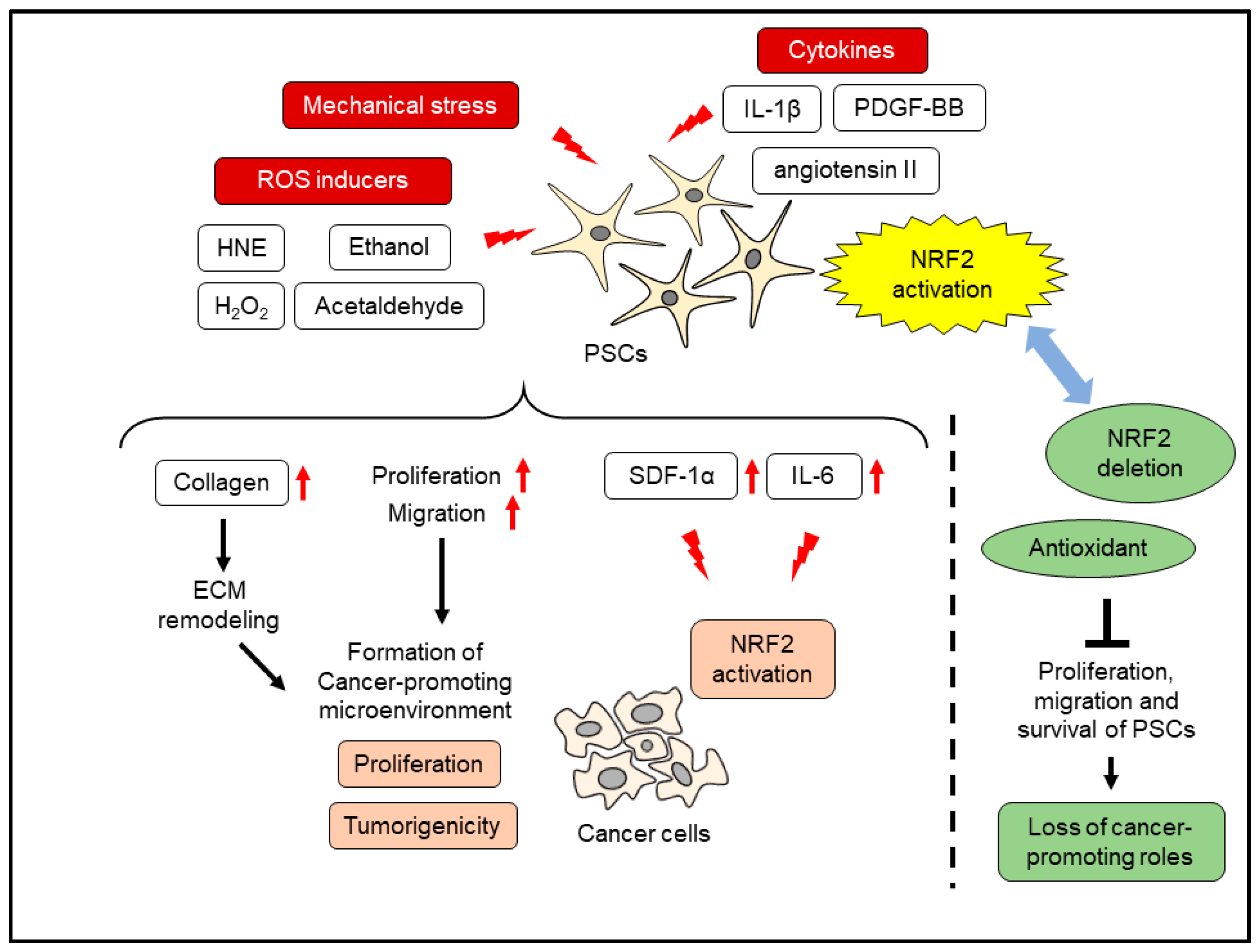
8. Oxidative Stress and PSC Activation
9. Application to Therapeutic Strategy
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Park, W.; Chawla, A.; O’Reilly, E.M. Pancreatic cancer: A review. JAMA 2021, 326, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Okusaka, T.; Furuse, J. Recent advances in chemotherapy for pancreatic cancer: Evidence from Japan and recommendations in guidelines. J. Gastroenterol. 2020, 55, 369–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yachida, S.; Jones, S.; Bozic, I.; Antal, T.; Leary, R.; Fu, B.; Kamiyama, M.; Hruban, R.H.; Eshleman, J.R.; Nowak, M.A.; et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010, 467, 1114–1117. [Google Scholar] [CrossRef] [Green Version]
- Samulitis, B.K.; Pond, K.W.; Pond, E.; Cress, A.E.; Patel, H.; Wisner, L.; Patel, C.; Dorr, R.T.; Landowski, T.H. Gemcitabine resistant pancreatic cancer cell lines acquire an invasive phenotype with collateral hypersensitivity to histone deacetylase inhibitors. Cancer Biol. Ther. 2015, 16, 43–51. [Google Scholar] [CrossRef]
- Masamune, A.; Shimosegawa, T. Pancreatic stellate cells: A dynamic player of the intercellular communication in pancreatic cancer. Clin. Res. Hepatol. Gastroenterol. 2015, 39, S98–S103. [Google Scholar] [CrossRef]
- Erkan, M.; Adler, G.; Apte, M.V.; Bachem, M.G.; Buchholz, M.; Detlefsen, S.; Esposito, I.; Friess, H.; Gress, T.M.; Habisch, H.J.; et al. StellaTUM: Current consensus and discussion on pancreatic stellate cell research. Gut 2012, 61, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Kikuta, K.; Masamune, A.; Watanabe, T.; Ariga, H.; Itoh, H.; Hamada, S.; Satoh, K.; Egawa, S.; Unno, M.; Shimosegawa, T. Pancreatic stellate cells promote epithelial-mesenchymal transition in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2010, 403, 380–384. [Google Scholar] [CrossRef]
- Hamada, S.; Masamune, A.; Takikawa, T.; Suzuki, N.; Kikuta, K.; Hirota, M.; Hamada, H.; Kobune, M.; Satoh, K.; Shimosegawa, T. Pancreatic stellate cells enhance stem cell-like phenotypes in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2012, 421, 349–354. [Google Scholar] [CrossRef]
- Hamada, S.; Masamune, A.; Yoshida, N.; Takikawa, T.; Shimosegawa, T. IL-6/STAT3 plays a regulatory role in the interaction between pancreatic stellate cells and cancer cells. Dig. Dis. Sci. 2016, 61, 1561–1571. [Google Scholar] [CrossRef]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [Green Version]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [Green Version]
- Mizutani, Y.; Kobayashi, H.; Iida, T.; Asai, N.; Masamune, A.; Hara, A.; Esaki, N.; Ushida, K.; Mii, S.; Shiraki, Y.; et al. Meflin-positive cancer-associated fibroblasts inhibit pancreatic carcinogenesis. Cancer Res. 2019, 79, 5367–5381. [Google Scholar] [CrossRef] [Green Version]
- Guillaumond, F.; Iovanna, J.L.; Vasseur, S. Pancreatic tumor cell metabolism: Focus on glycolysis and its connected metabolic pathways. Arch. Biochem. Biophys. 2014, 545, 69–73. [Google Scholar] [CrossRef]
- Carvalho, T.M.A.; Di Molfetta, D.; Greco, M.R.; Koltai, T.; Alfarouk, K.O.; Reshkin, S.J.; Cardone, R.A. Tumor microenvironment features and chemoresistance in pancreatic ductal adenocarcinoma: Insights into targeting physicochemical barriers and metabolism as therapeutic approaches. Cancers 2021, 13, 6135. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; He, C.; Hua, X.; Kan, A.; Mao, Y.; Sun, S.; Duan, F.; Wang, J.; Huang, P.; Li, S. Oxidative stress induces monocyte-to-myofibroblast transdifferentiation through p38 in pancreatic ductal adenocarcinoma. Clin. Transl. Med. 2020, 10, e41. [Google Scholar] [CrossRef]
- Corzo, C.A.; Cotter, M.J.; Cheng, P.; Cheng, F.; Kusmartsev, S.; Sotomayor, E.; Padhya, T.; McCaffrey, T.V.; McCaffrey, J.C.; Gabrilovich, D.I. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J. Immunol. 2009, 182, 5693–5701. [Google Scholar] [CrossRef] [PubMed]
- Seong, J.B.; Kim, B.; Kim, S.; Kim, M.H.; Park, Y.H.; Lee, Y.; Lee, H.J.; Hong, C.W.; Lee, D.S. Macrophage peroxiredoxin 5 deficiency promotes lung cancer progression via ROS-dependent M2-like polarization. Free Radic. Biol. Med. 2021, 176, 322–334. [Google Scholar] [CrossRef]
- Edderkaoui, M.; Hong, P.; Vaquero, E.; Lee, J.; Fischer, L.; Friess, H.; Buchler, M.; Lerch, M.; Pandol, S.J.; Gukovskaya, A. Extracellular matrix stimulates reactive oxygen species production and increases pancreatic cancer cell survival through 5-lipoxygenase and NADPH oxidase. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G1137–G1147. [Google Scholar] [CrossRef]
- Wang, Y.; Qi, H.; Liu, Y.; Duan, C.; Liu, X.; Xia, T.; Chen, D.; Piao, H.L.; Liu, H.X. The double-edged roles of ROS in cancer prevention and therapy. Theranostics 2021, 11, 4839–4857. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, J.; Zong, L.; Chen, X.; Chen, K.; Jiang, Z.; Nan, L.; Li, X.; Li, W.; Shan, T.; et al. Reactive oxygen species and targeted therapy for pancreatic cancer. Oxid. Med. Cell. Longev. 2016, 2016, 1616781. [Google Scholar] [CrossRef] [Green Version]
- Gervasoni, J.E., Jr.; Hindenburg, A.A.; Vezeridis, M.P.; Schulze, S.; Wanebo, H.J.; Mehta, S. An effective in vitro antitumor response against human pancreatic carcinoma with paclitaxel and daunorubicin by induction of both necrosis and apoptosis. Anticancer Res. 2004, 24, 2617–2626. [Google Scholar]
- Infantino, V.; Santarsiero, A.; Convertini, P.; Todisco, S.; Iacobazzi, V. Cancer cell metabolism in hypoxia: Role of HIF-1 as key regulator and therapeutic target. Int. J. Mol. Sci. 2021, 22, 5703. [Google Scholar] [CrossRef]
- Baird, L.; Yamamoto, M. The molecular mechanisms regulating the KEAP1-NRF2 pathway. Mol. Cell Biol. 2020, 40, e00099-20. [Google Scholar] [CrossRef]
- Koong, A.C.; Mehta, V.K.; Le, Q.T.; Fisher, G.A.; Terris, D.J.; Brown, J.M.; Bastidas, A.J.; Vierra, M. Pancreatic tumors show high levels of hypoxia. Int. J. Radiat. Oncol. Biol. Phys. 2000, 48, 919–922. [Google Scholar] [CrossRef]
- Morfoisse, F.; Renaud, E.; Hantelys, F.; Prats, A.C.; Garmy-Susini, B. Role of hypoxia and vascular endothelial growth factors in lymphangiogenesis. Mol. Cell Oncol. 2015, 2, e1024821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakazawa, M.S.; Keith, B.; Simon, M.C. Oxygen availability and metabolic adaptations. Nat. Rev. Cancer 2016, 16, 663–673. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Simon, M.C. Oxygen availability and metabolic reprogramming in cancer. J. Biol. Chem. 2017, 292, 16825–16832. [Google Scholar] [CrossRef] [Green Version]
- Morotti, M.; Bridges, E.; Valli, A.; Choudhry, H.; Sheldon, H.; Wigfield, S.; Gray, N.; Zois, C.E.; Grimm, F.; Jones, D.; et al. Hypoxia-induced switch in SNAT2/SLC38A2 regulation generates endocrine resistance in breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 12452–12461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latif, F.; Tory, K.; Gnarra, J.; Yao, M.; Duh, F.M.; Orcutt, M.L.; Stackhouse, T.; Kuzmin, I.; Modi, W.; Geil, L.; et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 1993, 260, 1317–1320. [Google Scholar] [CrossRef]
- Natsuizaka, M.; Ozasa, M.; Darmanin, S.; Miyamoto, M.; Kondo, S.; Kamada, S.; Shindoh, M.; Higashino, F.; Suhara, W.; Koide, H.; et al. Synergistic up-regulation of Hexokinase-2, glucose transporters and angiogenic factors in pancreatic cancer cells by glucose deprivation and hypoxia. Exp. Cell Res. 2007, 313, 3337–3348. [Google Scholar] [CrossRef] [Green Version]
- Kitajima, Y.; Ide, T.; Ohtsuka, T.; Miyazaki, K. Induction of hepatocyte growth factor activator gene expression under hypoxia activates the hepatocyte growth factor/c-Met system via hypoxia inducible factor-1 in pancreatic cancer. Cancer Sci. 2008, 99, 1341–1347. [Google Scholar] [CrossRef]
- Buchler, P.; Reber, H.A.; Tomlinson, J.S.; Hankinson, O.; Kallifatidis, G.; Friess, H.; Herr, I.; Hines, O.J. Transcriptional regulation of urokinase-type plasminogen activator receptor by hypoxia-inducible factor 1 is crucial for invasion of pancreatic and liver cancer. Neoplasia 2009, 11, 196–206. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.P.; Cao, Y.; Li, W.J.; Zhang, H.H.; Zhu, Z.M. P4HA1/HIF1alpha feedback loop drives the glycolytic and malignant phenotypes of pancreatic cancer. Biochem. Biophys. Res. Commun. 2019, 516, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golias, T.; Papandreou, I.; Sun, R.; Kumar, B.; Brown, N.V.; Swanson, B.J.; Pai, R.; Jaitin, D.; Le, Q.T.; Teknos, T.N.; et al. Hypoxic repression of pyruvate dehydrogenase activity is necessary for metabolic reprogramming and growth of model tumours. Sci. Rep. 2016, 6, 31146. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Chen, J.Z.; Zhang, J.Q.; Chen, H.X.; Yan, M.L.; Huang, L.; Tian, Y.F.; Chen, Y.L.; Wang, Y.D. Hypoxia induces TWIST-activated epithelial-mesenchymal transition and proliferation of pancreatic cancer cells in vitro and in nude mice. Cancer Lett. 2016, 383, 73–84. [Google Scholar] [CrossRef]
- Higgins, D.F.; Biju, M.P.; Akai, Y.; Wutz, A.; Johnson, R.S.; Haase, V.H. Hypoxic induction of Ctgf is directly mediated by Hif-1. Am. J. Physiol. Renal Physiol. 2004, 287, F1223–F1232. [Google Scholar] [CrossRef]
- Hong, K.H.; Yoo, S.A.; Kang, S.S.; Choi, J.J.; Kim, W.U.; Cho, C.S. Hypoxia induces expression of connective tissue growth factor in scleroderma skin fibroblasts. Clin. Exp. Immunol. 2006, 146, 362–370. [Google Scholar] [CrossRef]
- Bennewith, K.L.; Huang, X.; Ham, C.M.; Graves, E.E.; Erler, J.T.; Kambham, N.; Feazell, J.; Yang, G.P.; Koong, A.; Giaccia, A.J. The role of tumor cell-derived connective tissue growth factor (CTGF/CCN2) in pancreatic tumor growth. Cancer Res. 2009, 69, 775–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maity, G.; Ghosh, A.; Gupta, V.; Haque, I.; Sarkar, S.; Das, A.; Dhar, K.; Bhavanasi, S.; Gunewardena, S.S.; Von Hoff, D.D.; et al. CYR61/CCN1 regulates dCK and CTGF and causes gemcitabine-resistant phenotype in pancreatic ductal adenocarcinoma. Mol. Cancer Ther. 2019, 18, 788–800. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Dai, L.; Ma, Y.; Wang, J.; Liu, Z. Implications of HIF-1alpha in the tumorigenesis and progression of pancreatic cancer. Cancer Cell Int. 2020, 20, 273. [Google Scholar] [CrossRef]
- Mace, T.A.; Collins, A.L.; Wojcik, S.E.; Croce, C.M.; Lesinski, G.B.; Bloomston, M. Hypoxia induces the overexpression of microRNA-21 in pancreatic cancer cells. J. Surg. Res. 2013, 184, 855–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greco, S.; Martelli, F. MicroRNAs in hypoxia response. Antioxid. Redox Signal. 2014, 21, 1164–1166. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Cao, Y.; Sun, M.; Feng, H. Expression, regulation, and function of exosome-derived miRNAs in cancer progression and therapy. FASEB J. 2021, 35, e21916. [Google Scholar] [CrossRef]
- Greither, T.; Grochola, L.F.; Udelnow, A.; Lautenschlager, C.; Wurl, P.; Taubert, H. Elevated expression of microRNAs 155, 203, 210 and 222 in pancreatic tumors is associated with poorer survival. Int. J. Cancer 2010, 126, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Takikawa, T.; Masamune, A.; Hamada, S.; Nakano, E.; Yoshida, N.; Shimosegawa, T. miR-210 regulates the interaction between pancreatic cancer cells and stellate cells. Biochem. Biophys. Res. Commun. 2013, 437, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Giovannetti, E.; Funel, N.; Peters, G.J.; Del Chiaro, M.; Erozenci, L.A.; Vasile, E.; Leon, L.G.; Pollina, L.E.; Groen, A.; Falcone, A.; et al. MicroRNA-21 in pancreatic cancer: Correlation with clinical outcome and pharmacologic aspects underlying its role in the modulation of gemcitabine activity. Cancer Res. 2010, 70, 4528–4538. [Google Scholar] [CrossRef] [Green Version]
- Luo, G.; Xia, X.; Wang, X.; Zhang, K.; Cao, J.; Jiang, T.; Zhao, Q.; Qiu, Z. miR-301a plays a pivotal role in hypoxia-induced gemcitabine resistance in pancreatic cancer. Exp. Cell Res. 2018, 369, 120–128. [Google Scholar] [CrossRef]
- Wang, X.; Luo, G.; Zhang, K.; Cao, J.; Huang, C.; Jiang, T.; Liu, B.; Su, L.; Qiu, Z. Hypoxic tumor-derived exosomal miR-301a mediates M2 macrophage polarization via PTEN/PI3Kgamma to promote pancreatic cancer metastasis. Cancer Res. 2018, 78, 4586–4598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masamune, A.; Kikuta, K.; Watanabe, T.; Satoh, K.; Hirota, M.; Shimosegawa, T. Hypoxia stimulates pancreatic stellate cells to induce fibrosis and angiogenesis in pancreatic cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G709–G717. [Google Scholar] [CrossRef] [Green Version]
- Erkan, M.; Reiser-Erkan, C.; Michalski, C.W.; Deucker, S.; Sauliunaite, D.; Streit, S.; Esposito, I.; Friess, H.; Kleeff, J. Cancer-stellate cell interactions perpetuate the hypoxia-fibrosis cycle in pancreatic ductal adenocarcinoma. Neoplasia 2009, 11, 497–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eguchi, D.; Ikenaga, N.; Ohuchida, K.; Kozono, S.; Cui, L.; Fujiwara, K.; Fujino, M.; Ohtsuka, T.; Mizumoto, K.; Tanaka, M. Hypoxia enhances the interaction between pancreatic stellate cells and cancer cells via increased secretion of connective tissue growth factor. J. Surg. Res. 2013, 181, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Sada, M.; Ohuchida, K.; Horioka, K.; Okumura, T.; Moriyama, T.; Miyasaka, Y.; Ohtsuka, T.; Mizumoto, K.; Oda, Y.; Nakamura, M. Hypoxic stellate cells of pancreatic cancer stroma regulate extracellular matrix fiber organization and cancer cell motility. Cancer Lett. 2016, 372, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Kikuta, K.; Masamune, A.; Hamada, S.; Takikawa, T.; Nakano, E.; Shimosegawa, T. Pancreatic stellate cells reduce insulin expression and induce apoptosis in pancreatic beta-cells. Biochem. Biophys. Res. Commun. 2013, 433, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Lee, E.; Ryu, G.R.; Ko, S.H.; Ahn, Y.B.; Song, K.H. Hypoxia increases beta-cell death by activating pancreatic stellate cells within the islet. Diabetes Metab. J. 2020, 44, 919–927. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Truty, M.A.; Kang, Y.; Chopin-Laly, X.; Zhang, R.; Roife, D.; Chatterjee, D.; Lin, E.; Thomas, R.M.; Wang, H.; et al. Extracellular lumican inhibits pancreatic cancer cell growth and is associated with prolonged survival after surgery. Clin. Cancer Res. 2014, 20, 6529–6540. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Lee, Y.; Kang, Y.; Dai, B.; Perez, M.R.; Pratt, M.; Koay, E.J.; Kim, M.; Brekken, R.A.; Fleming, J.B. Hypoxia-induced autophagy of stellate cells inhibits expression and secretion of lumican into microenvironment of pancreatic ductal adenocarcinoma. Cell Death Differ. 2019, 26, 382–393. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Li, Y.; Li, Z.; Huang, C.; Yang, Y.; Lang, M.; Cao, J.; Jiang, W.; Xu, Y.; Dong, J.; et al. Hypoxia inducible factor 1 (HIF-1) recruits macrophage to activate pancreatic stellate cells in pancreatic ductal adenocarcinoma. Int. J. Mol. Sci. 2016, 17, 799. [Google Scholar] [CrossRef] [Green Version]
- Cao, W.; Zeng, Z.; He, Z.; Lei, S. Hypoxic pancreatic stellate cell-derived exosomal mirnas promote proliferation and invasion of pancreatic cancer through the PTEN/AKT pathway. Aging 2021, 13, 7120–7132. [Google Scholar] [CrossRef]
- Taguchi, K.; Yamamoto, M. The KEAP1-NRF2 system as a molecular target of cancer treatment. Cancers 2020, 13, 46. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 system: A thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, K.; Yamamoto, M. The KEAP1-NRF2 system in cancer. Front. Oncol. 2017, 7, 85. [Google Scholar] [CrossRef]
- Frank, R.; Scheffler, M.; Merkelbach-Bruse, S.; Ihle, M.A.; Kron, A.; Rauer, M.; Ueckeroth, F.; Konig, K.; Michels, S.; Fischer, R.; et al. Clinical and pathological characteristics of KEAP1- and NFE2L2-mutated non-small cell lung carcinoma (NSCLC). Clin. Cancer Res. 2018, 24, 3087–3096. [Google Scholar] [CrossRef] [Green Version]
- Kerins, M.J.; Ooi, A. A catalogue of somatic NRF2 gain-of-function mutations in cancer. Sci. Rep. 2018, 8, 12846. [Google Scholar] [CrossRef] [PubMed]
- Shibata, T.; Kokubu, A.; Saito, S.; Narisawa-Saito, M.; Sasaki, H.; Aoyagi, K.; Yoshimatsu, Y.; Tachimori, Y.; Kushima, R.; Kiyono, T.; et al. NRF2 mutation confers malignant potential and resistance to chemoradiation therapy in advanced esophageal squamous cancer. Neoplasia 2011, 13, 864–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umemura, A.; He, F.; Taniguchi, K.; Nakagawa, H.; Yamachika, S.; Font-Burgada, J.; Zhong, Z.; Subramaniam, S.; Raghunandan, S.; Duran, A.; et al. p62, upregulated during preneoplasia, induces hepatocellular carcinogenesis by maintaining survival of stressed HCC-initiating cells. Cancer Cell 2016, 29, 935–948. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Ichimura, Y.; Taguchi, K.; Suzuki, T.; Mizushima, T.; Takagi, K.; Hirose, Y.; Nagahashi, M.; Iso, T.; Fukutomi, T.; et al. p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat. Commun. 2016, 7, 12030. [Google Scholar] [CrossRef] [PubMed]
- Purohit, V.; Wang, L.; Yang, H.; Li, J.; Ney, G.M.; Gumkowski, E.R.; Vaidya, A.J.; Wang, A.; Bhardwaj, A.; Zhao, E.; et al. ATDC binds to KEAP1 to drive NRF2-mediated tumorigenesis and chemoresistance in pancreatic cancer. Genes Dev. 2021, 35, 218–233. [Google Scholar] [CrossRef]
- Ju, H.Q.; Gocho, T.; Aguilar, M.; Wu, M.; Zhuang, Z.N.; Fu, J.; Yanaga, K.; Huang, P.; Chiao, P.J. Mechanisms of overcoming intrinsic resistance to gemcitabine in pancreatic ductal adenocarcinoma through the redox modulation. Mol. Cancer Ther. 2015, 14, 788–798. [Google Scholar] [CrossRef] [Green Version]
- Arfmann-Knubel, S.; Struck, B.; Genrich, G.; Helm, O.; Sipos, B.; Sebens, S.; Schafer, H. The crosstalk between Nrf2 and TGF-beta1 in the epithelial-mesenchymal transition of pancreatic duct epithelial cells. PLoS ONE 2015, 10, e0132978. [Google Scholar]
- Todoric, J.; Antonucci, L.; Di Caro, G.; Li, N.; Wu, X.; Lytle, N.K.; Dhar, D.; Banerjee, S.; Fagman, J.B.; Browne, C.D.; et al. Stress-activated NRF2-MDM2 cascade controls neoplastic progression in pancreas. Cancer Cell 2017, 32, 824–839 e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef] [Green Version]
- Chio, I.I.C.; Jafarnejad, S.M.; Ponz-Sarvise, M.; Park, Y.; Rivera, K.; Palm, W.; Wilson, J.; Sangar, V.; Hao, Y.; Ohlund, D.; et al. NRF2 promotes tumor maintenance by modulating mRNA translation in pancreatic cancer. Cell 2016, 166, 963–976. [Google Scholar] [CrossRef] [Green Version]
- Hamada, S.; Taguchi, K.; Masamune, A.; Yamamoto, M.; Shimosegawa, T. Nrf2 promotes mutant K-ras/p53-driven pancreatic carcinogenesis. Carcinogenesis 2017, 38, 661–670. [Google Scholar] [CrossRef]
- Hamada, S.; Shimosegawa, T.; Taguchi, K.; Nabeshima, T.; Yamamoto, M.; Masamune, A. Simultaneous K-ras activation and Keap1 deletion cause atrophy of pancreatic parenchyma. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 314, G65–G74. [Google Scholar] [CrossRef]
- O’Dell, M.R.; Huang, J.L.; Whitney-Miller, C.L.; Deshpande, V.; Rothberg, P.; Grose, V.; Rossi, R.M.; Zhu, A.X.; Land, H.; Bardeesy, N.; et al. Kras(G12D) and p53 mutation cause primary intrahepatic cholangiocarcinoma. Cancer Res. 2012, 72, 1557–1567. [Google Scholar] [CrossRef] [Green Version]
- Nabeshima, T.; Hamada, S.; Taguchi, K.; Tanaka, Y.; Matsumoto, R.; Yamamoto, M.; Masamune, A. Keap1 deletion accelerates mutant K-ras/p53-driven cholangiocarcinoma. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G419–G427. [Google Scholar] [CrossRef]
- Hayashi, M.; Kuga, A.; Suzuki, M.; Panda, H.; Kitamura, H.; Motohashi, H.; Yamamoto, M. Microenvironmental activation of Nrf2 restricts the progression of Nrf2-activated malignant tumors. Cancer Res. 2020, 80, 3331–3344. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.J.; Cheng, X.D.; Zhang, J.; Zhang, W.D. Dual roles and therapeutic potential of Keap1-Nrf2 pathway in pancreatic cancer: A systematic review. Cell Commun. Signal. 2019, 17, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, B.; Cheng, L.; Jiang, Z.; Chen, K.; Zhou, C.; Sun, L.; Cao, J.; Qian, W.; Li, J.; Shan, T.; et al. Resveratrol inhibits ROS-promoted activation and glycolysis of pancreatic stellate cells via suppression of miR-21. Oxid. Med. Cell Longev. 2018, 2018, 1346958. [Google Scholar] [CrossRef]
- Masamune, A.; Watanabe, T.; Kikuta, K.; Satoh, K.; Shimosegawa, T. NADPH oxidase plays a crucial role in the activation of pancreatic stellate cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G99–G108. [Google Scholar] [CrossRef] [PubMed]
- Masamune, A.; Kikuta, K.; Satoh, M.; Satoh, A.; Shimosegawa, T. Alcohol activates activator protein-1 and mitogen-activated protein kinases in rat pancreatic stellate cells. J. Pharmacol. Exp. Ther. 2002, 302, 36–42. [Google Scholar] [CrossRef] [Green Version]
- Ryu, G.R.; Lee, E.; Chun, H.J.; Yoon, K.H.; Ko, S.H.; Ahn, Y.B.; Song, K.H. Oxidative stress plays a role in high glucose-induced activation of pancreatic stellate cells. Biochem. Biophys. Res. Commun. 2013, 439, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.H.; Lin, L.; Zhu, X.Y.; Wen, T.; Hu, D.M.; Dong, Y.; Li, L.Y.; Wang, S.F. Protective effects of edaravone on experimental chronic pancreatitis induced by dibutyltin dichloride in rats. Pancreatology 2013, 13, 125–132. [Google Scholar] [CrossRef]
- Wu, Y.S.; Looi, C.Y.; Subramaniam, K.S.; Masamune, A.; Chung, I. Soluble factors from stellate cells induce pancreatic cancer cell proliferation via Nrf2-activated metabolic reprogramming and ROS detoxification. Oncotarget 2016, 7, 36719–36732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Hamada, S.; Matsumoto, R.; Taguchi, K.; Yamamoto, M.; Masamune, A. Nrf2 expression in pancreatic stellate cells promotes progression of cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 321, G378–G388. [Google Scholar] [CrossRef]
- Schwartz, D.L.; Bankson, J.A.; Lemos, R., Jr.; Lai, S.Y.; Thittai, A.K.; He, Y.; Hostetter, G.; Demeure, M.J.; Von Hoff, D.D.; Powis, G. Radiosensitization and stromal imaging response correlates for the HIF-1 inhibitor PX-478 given with or without chemotherapy in pancreatic cancer. Mol. Cancer Ther. 2010, 9, 2057–2067. [Google Scholar] [CrossRef] [Green Version]
- Miyake, K.; Nishioka, M.; Imura, S.; Batmunkh, E.; Uto, Y.; Nagasawa, H.; Hori, H.; Shimada, M. The novel hypoxic cytotoxin, TX-2098 has antitumor effect in pancreatic cancer; possible mechanism through inhibiting VEGF and hypoxia inducible factor-1alpha targeted gene expression. Exp. Cell Res. 2012, 318, 1554–1563. [Google Scholar] [CrossRef]
- Picozzi, V.; Alseidi, A.; Winter, J.; Pishvaian, M.; Mody, K.; Glaspy, J.; Larson, T.; Matrana, M.; Carney, M.; Porter, S.; et al. Gemcitabine/nab-paclitaxel with pamrevlumab: A novel drug combination and trial design for the treatment of locally advanced pancreatic cancer. ESMO Open 2020, 5, e000668. [Google Scholar] [CrossRef]
- Neesse, A.; Frese, K.K.; Bapiro, T.E.; Nakagawa, T.; Sternlicht, M.D.; Seeley, T.W.; Pilarsky, C.; Jodrell, D.I.; Spong, S.M.; Tuveson, D.A. CTGF antagonism with mAb FG-3019 enhances chemotherapy response without increasing drug delivery in murine ductal pancreas cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 12325–12330. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.; Qin, Y.; Ji, S.; Shi, X.; Dai, W.; Fan, G.; Li, S.; Xu, W.; Liu, W.; Liu, M.; et al. MTAP deficiency-induced metabolic reprogramming creates a vulnerability to cotargeting de novo purine synthesis and glycolysis in pancreatic cancer. Cancer Res. 2021, 81, 4964–4980. [Google Scholar] [CrossRef]
- Xiao, Y.; Qin, T.; Sun, L.; Qian, W.; Li, J.; Duan, W.; Lei, J.; Wang, Z.; Ma, J.; Li, X.; et al. Resveratrol ameliorates the malignant progression of pancreatic cancer by inhibiting hypoxia-induced pancreatic stellate cell activation. Cell Transplant. 2020, 29, 963689720929987. [Google Scholar] [CrossRef]
- Estaras, M.; Gonzalez-Portillo, M.R.; Fernandez-Bermejo, M.; Mateos, J.M.; Vara, D.; Blanco-Fernandez, G.; Lopez-Guerra, D.; Roncero, V.; Salido, G.M.; Gonzalez, A. Melatonin induces apoptosis and modulates cyclin expression and MAPK phosphorylation in pancreatic stellate cells subjected to hypoxia. Int. J. Mol. Sci. 2021, 22, 5555. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.J.; Jung, B.J.; Lee, S.H.; Yoo, H.S.; Shin, E.A.; Ko, H.J.; Chang, S.; Kim, S.Y.; Jeon, S.M. A clinical drug library screen identifies clobetasol propionate as an NRF2 inhibitor with potential therapeutic efficacy in KEAP1 mutant lung cancer. Oncogene 2017, 36, 5285–5295. [Google Scholar] [CrossRef]
- Singh, A.; Venkannagari, S.; Oh, K.H.; Zhang, Y.Q.; Rohde, J.M.; Liu, L.; Nimmagadda, S.; Sudini, K.; Brimacombe, K.R.; Gajghate, S.; et al. Small molecule inhibitor of NRF2 selectively intervenes therapeutic resistance in KEAP1-deficient NSCLC tumors. ACS Chem. Biol. 2016, 11, 3214–3225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, B.; Augustine, J.J.; Kang, Y.; Roife, D.; Li, X.; Deng, J.; Tan, L.; Rusling, L.A.; Weinstein, J.N.; Lorenzi, P.L.; et al. Compound NSC84167 selectively targets NRF2-activated pancreatic cancer by inhibiting asparagine synthesis pathway. Cell Death Dis. 2021, 12, 693. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, K.; Tsujita, T.; Hayashi, M.; Ojima, A.; Keleku-Lukwete, N.; Katsuoka, F.; Otsuki, A.; Kikuchi, H.; Oshima, Y.; Suzuki, M.; et al. Halofuginone enhances the chemo-sensitivity of cancer cells by suppressing NRF2 accumulation. Free Radic. Biol. Med. 2017, 103, 236–247. [Google Scholar] [CrossRef]
- Matsumoto, R.; Hamada, S.; Tanaka, Y.; Taguchi, K.; Yamamoto, M.; Masamune, A. Nuclear factor erythroid 2-related factor 2 depletion sensitizes pancreatic cancer cells to gemcitabine via aldehyde dehydrogenase 3a1 repression. J. Pharmacol. Exp. Ther. 2021, 379, 33–40. [Google Scholar] [CrossRef]
- Suzuki, S.; Yamamoto, M.; Sanomachi, T.; Togashi, K.; Sugai, A.; Seino, S.; Yoshioka, T.; Okada, M.; Kitanaka, C. Dexamethasone sensitizes cancer stem cells to gemcitabine and 5-Fluorouracil by increasing reactive oxygen species production through NRF2 reduction. Life 2021, 11, 885. [Google Scholar] [CrossRef]
- Chen, X.; Jiang, Z.; Zhou, C.; Chen, K.; Li, X.; Wang, Z.; Wu, Z.; Ma, J.; Ma, Q.; Duan, W. Activation of Nrf2 by sulforaphane inhibits high glucose-induced progression of pancreatic cancer via AMPK dependent signaling. Cell. Physiol. Biochem. 2018, 50, 1201–1215. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Wu, S.; Ye, S.; Huang, H.; Zhou, Y.; Zhou, H.; Wu, S.; Mao, Y.; Shangguan, F.; Lan, L.; et al. Dimethyl fumarate induces metabolic crisie to suppress pancreatic carcinoma. Front. Pharmacol. 2021, 12, 617714. [Google Scholar] [CrossRef] [PubMed]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galan-Cobo, A.; Sitthideatphaiboon, P.; Qu, X.; Poteete, A.; Pisegna, M.A.; Tong, P.; Chen, P.H.; Boroughs, L.K.; Rodriguez, M.L.M.; Zhang, W.; et al. LKB1 and KEAP1/NRF2 pathways cooperatively promote metabolic reprogramming with enhanced glutamine dependence in KRAS-mutant lung adenocarcinoma. Cancer Res. 2019, 79, 3251–3267. [Google Scholar] [CrossRef]
- Hamada, S.; Matsumoto, R.; Tanaka, Y.; Taguchi, K.; Yamamoto, M.; Masamune, A. Nrf2 activation sensitizes K-Ras mutant pancreatic cancer cells to glutaminase inhibition. Int. J. Mol. Sci. 2021, 22, 1870. [Google Scholar] [CrossRef] [PubMed]
- Abdalla, M.Y.; Ahmad, I.M.; Rachagani, S.; Banerjee, K.; Thompson, C.M.; Maurer, H.C.; Olive, K.P.; Bailey, K.L.; Britigan, B.E.; Kumar, S. Enhancing responsiveness of pancreatic cancer cells to gemcitabine treatment under hypoxia by heme oxygenase-1 inhibition. Transl. Res. 2019, 207, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Kuper, A.; Baumann, J.; Gopelt, K.; Baumann, M.; Sanger, C.; Metzen, E.; Kranz, P.; Brockmeier, U. Overcoming hypoxia-induced resistance of pancreatic and lung tumor cells by disrupting the PERK-NRF2-HIF-axis. Cell Death Dis. 2021, 12, 82. [Google Scholar] [CrossRef]
- Setton, J.; Zinda, M.; Riaz, N.; Durocher, D.; Zimmermann, M.; Koehler, M.; Reis-Filho, J.S.; Powell, S.N. Synthetic lethality in cancer therapeutics: The next generation. Cancer Discov. 2021, 11, 1626–1635. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamada, S.; Matsumoto, R.; Masamune, A. HIF-1 and NRF2; Key Molecules for Malignant Phenotypes of Pancreatic Cancer. Cancers 2022, 14, 411. https://doi.org/10.3390/cancers14020411
Hamada S, Matsumoto R, Masamune A. HIF-1 and NRF2; Key Molecules for Malignant Phenotypes of Pancreatic Cancer. Cancers. 2022; 14(2):411. https://doi.org/10.3390/cancers14020411
Chicago/Turabian StyleHamada, Shin, Ryotaro Matsumoto, and Atsushi Masamune. 2022. "HIF-1 and NRF2; Key Molecules for Malignant Phenotypes of Pancreatic Cancer" Cancers 14, no. 2: 411. https://doi.org/10.3390/cancers14020411
APA StyleHamada, S., Matsumoto, R., & Masamune, A. (2022). HIF-1 and NRF2; Key Molecules for Malignant Phenotypes of Pancreatic Cancer. Cancers, 14(2), 411. https://doi.org/10.3390/cancers14020411