
Chromosomal Instability, Selection and Competition: Factors That Shape the Level of Karyotype Intra-Tumor Heterogeneity

{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Numerical and Structural CIN Generates New Karyotypes
3. Cell intrinsic Selection for Karyotypes
3.1. Aneuploidy Tolerance
3.2. Aneuploidy and Cell Fitness
3.3. Tissue Specificity of CNVs
3.4. Divergence of CNVs during Carcinogenesis
4. Selection for Karyotypes by the Microenvironment
4.1. Immunoediting
4.2. CIN Modulates the Tumor Immune Microenvironment
4.3. Hypoxia
5. Competition between Malignant Cells
5.1. Cell Competition and Relative Fitness
5.2. Genetic Drift and Spatial Constraints
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Reiter, J.G.; Baretti, M.; Gerold, J.M.; Makohon-Moore, A.P.; Daud, A.; Iacobuzio-Donahue, C.A.; Azad, N.S.; Kinzler, K.W.; Nowak, M.A.; Vogelstein, B. An analysis of genetic heterogeneity in untreated cancers. Nat. Rev. Cancer 2019, 19, 639–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mroz, E.A.; Rocco, J.W. MATH, a novel measure of intratumor genetic heterogeneity, is high in poor-outcome classes of head and neck squamous cell carcinoma. Oral. Oncol. 2013, 49, 211–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andor, N.; Graham, T.A.; Jansen, M.; Xia, L.C.; Aktipis, C.A.; Petritsch, C.; Ji, H.P.; Maley, C.C. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat. Med. 2016, 22, 105–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Dijk, E.; van den Bosch, T.; Lenos, K.J.; El Makrini, K.; Nijman, L.E.; van Essen, H.F.B.; Lansu, N.; Boekhout, M.; Hageman, J.H.; Fitzgerald, R.C.; et al. Chromosomal copy number heterogeneity predicts survival rates across cancers. Nat. Commun. 2021, 12, 3188. [Google Scholar] [CrossRef] [PubMed]
- Dentro, S.C.; Leshchiner, I.; Haase, K.; Tarabichi, M.; Wintersinger, J.; Deshwar, A.G.; Yu, K.; Rubanova, Y.; Macintyre, G.; Demeulemeester, J.; et al. Characterizing genetic intra-tumor heterogeneity across 2,658 human cancer genomes. Cell 2021, 184, 2239–2254. [Google Scholar] [CrossRef]
- Greaves, M. Evolutionary determinants of cancer. Cancer Discov. 2015, 5, 806–820. [Google Scholar] [CrossRef] [Green Version]
- Ramon, Y.C.S.; Sese, M.; Capdevila, C.; Aasen, T.; De Mattos-Arruda, L.; Diaz-Cano, S.J.; Hernandez-Losa, J.; Castellvi, J. Clinical implications of intratumor heterogeneity: Challenges and opportunities. J. Mol. Med. 2020, 98, 161–177. [Google Scholar] [CrossRef] [Green Version]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [Green Version]
- Landau, D.A.; Carter, S.L.; Stojanov, P.; McKenna, A.; Stevenson, K.; Lawrence, M.S.; Sougnez, C.; Stewart, C.; Sivachenko, A.; Wang, L.; et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 2013, 152, 714–726. [Google Scholar] [CrossRef]
- Zhang, J.; Fujimoto, J.; Zhang, J.; Wedge, D.C.; Song, X.; Zhang, J.; Seth, S.; Chow, C.W.; Cao, Y.; Gumbs, C.; et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 2014, 346, 256–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Bruin, E.C.; McGranahan, N.; Mitter, R.; Salm, M.; Wedge, D.C.; Yates, L.; Jamal-Hanjani, M.; Shafi, S.; Murugaesu, N.; Rowan, A.J.; et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 2014, 346, 251–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maley, C.C.; Galipeau, P.C.; Finley, J.C.; Wongsurawat, V.J.; Li, X.; Sanchez, C.A.; Paulson, T.G.; Blount, P.L.; Risques, R.A.; Rabinovitch, P.S.; et al. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat. Genet. 2006, 38, 468–473. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Gonen, M.; Kim, H.J.; Michor, F.; Polyak, K. Cellular and genetic diversity in the progression of in situ human breast carcinomas to an invasive phenotype. J. Clin. Investig. 2010, 120, 636–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holland, A.J.; Cleveland, D.W. Boveri revisited: Chromosomal instability, aneuploidy and tumorigenesis. Nat. Rev. Mol. Cell Bio. 2009, 10, 478–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef]
- Yung, W.K.; Shapiro, J.R.; Shapiro, W.R. Heterogeneous chemosensitivities of subpopulations of human glioma cells in culture. Cancer Res. 1982, 42, 992–998. [Google Scholar]
- Isaacs, J.T.; Wake, N.; Coffey, D.S.; Sandberg, A.A. Genetic instability coupled to clonal selection as a mechanism for tumor progression in the Dunning R-3327 rat prostatic adenocarcinoma system. Cancer Res. 1982, 42, 2353–2371. [Google Scholar]
- Taylor, A.M.; Shih, J.; Ha, G.; Gao, G.F.; Zhang, X.; Berger, A.C.; Schumacher, S.E.; Wang, C.; Hu, H.; Liu, J.; et al. Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell 2018, 33, 676–689. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, B.; Fong, C.; Luthra, A.; Smith, S.A.; DiNatale, R.G.; Nandakumar, S.; Walch, H.; Chatila, W.K.; Madupuri, R.; Kundra, R.; et al. Genomic characterization of metastatic patterns from prospective clinical sequencing of 25,000 patients. Cell 2022, 185, 563–575. [Google Scholar] [CrossRef]
- Hieronymus, H.; Murali, R.; Tin, A.; Yadav, K.; Abida, W.; Moller, H.; Berney, D.; Scher, H.; Carver, B.; Scardino, P.; et al. Tumor copy number alteration burden is a pan-cancer prognostic factor associated with recurrence and death. Elife 2018, 7, e37294. [Google Scholar] [CrossRef] [PubMed]
- Heppner, G.H. Tumor heterogeneity. Cancer Res 1984, 44, 2259–2265. [Google Scholar] [PubMed]
- Bakhoum, S.F.; Landau, D.A. Chromosomal Instability as a Driver of Tumor Heterogeneity and Evolution. Cold Spring Harb. Perspect. Med. 2017, 7, a029611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakhoum, S.F.; Cantley, L.C. The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment. Cell 2018, 174, 1347–1360. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Seehawer, M.; Polyak, K. Untangling the web of intratumour heterogeneity. Nat. Cell Biol. 2022, 24, 1192–1201. [Google Scholar] [CrossRef]
- Turajlic, S.; Xu, H.; Litchfield, K.; Rowan, A.; Horswell, S.; Chambers, T.; O’Brien, T.; Lopez, J.I.; Watkins, T.B.K.; Nicol, D.; et al. Deterministic Evolutionary Trajectories Influence Primary Tumor Growth: TRACERx Renal. Cell 2018, 173, 595–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watkins, T.B.K.; Lim, E.L.; Petkovic, M.; Elizalde, S.; Birkbak, N.J.; Wilson, G.A.; Moore, D.A.; Gronroos, E.; Rowan, A.; Dewhurst, S.M.; et al. Pervasive chromosomal instability and karyotype order in tumour evolution. Nature 2020, 587, 126–132. [Google Scholar] [CrossRef]
- Pectasides, E.; Stachler, M.D.; Derks, S.; Liu, Y.; Maron, S.; Islam, M.; Alpert, L.; Kwak, H.; Kindler, H.; Polite, B.; et al. Genomic Heterogeneity as a Barrier to Precision Medicine in Gastroesophageal Adenocarcinoma. Cancer Discov. 2018, 8, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Bolhaqueiro, A.C.F.; Ponsioen, B.; Bakker, B.; Klaasen, S.J.; Kucukkose, E.; van Jaarsveld, R.H.; Vivie, J.; Verlaan-Klink, I.; Hami, N.; Spierings, D.C.J.; et al. Ongoing chromosomal instability and karyotype evolution in human colorectal cancer organoids. Nat. Genet. 2019, 51, 824–834. [Google Scholar] [CrossRef]
- Minussi, D.C.; Nicholson, M.D.; Ye, H.; Davis, A.; Wang, K.; Baker, T.; Tarabichi, M.; Sei, E.; Du, H.; Rabbani, M.; et al. Breast tumours maintain a reservoir of subclonal diversity during expansion. Nature 2021, 592, 302–308. [Google Scholar] [CrossRef]
- Bakker, B.; Taudt, A.; Belderbos, M.E.; Porubsky, D.; Spierings, D.C.; de Jong, T.V.; Halsema, N.; Kazemier, H.G.; Hoekstra-Wakker, K.; Bradley, A.; et al. Single-cell sequencing reveals karyotype heterogeneity in murine and human malignancies. Genome. Biol. 2016, 17, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Verma, A.; Naveed, U.; Bakhoum, S.F.; Khosravi, P.; Elemento, O. Deep learning predicts chromosomal instability from histopathology images. iScience 2021, 24, 102394. [Google Scholar] [CrossRef] [PubMed]
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhsng, C.Z.; Wala, J.; Mermel, C.H.; et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, S.L.; Cibulskis, K.; Helman, E.; McKenna, A.; Shen, H.; Zack, T.; Laird, P.W.; Onofrio, R.C.; Winckler, W.; Weir, B.A.; et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 2012, 30, 413–421. [Google Scholar] [CrossRef]
- Van Loo, P.; Nordgard, S.H.; Lingjaerde, O.C.; Russnes, H.G.; Rye, I.H.; Sun, W.; Weigman, V.J.; Marynen, P.; Zetterberg, A.; Naume, B.; et al. Allele-specific copy number analysis of tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 16910–16915. [Google Scholar] [CrossRef] [Green Version]
- Sansregret, L.; Vanhaesebroeck, B.; Swanton, C. Determinants and clinical implications of chromosomal instability in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 139–150. [Google Scholar] [CrossRef] [Green Version]
- Kalkan, B.M.; Ozcan, S.C.; Quintyne, N.J.; Reed, S.L.; Acilan, C. Keep Calm and Carry on with Extra Centrosomes. Cancers 2022, 14, 442. [Google Scholar] [CrossRef]
- Thompson, S.L.; Bakhoum, S.F.; Compton, D.A. Mechanisms of chromosomal instability. Curr. Biol. 2010, 20, R285–R295. [Google Scholar] [CrossRef] [Green Version]
- Iemura, K.; Yoshizaki, Y.; Kuniyasu, K.; Tanaka, K. Attenuated Chromosome Oscillation as a Cause of Chromosomal Instability in Cancer Cells. Cancers 2021, 13, 4531. [Google Scholar] [CrossRef]
- Gemble, S.; Basto, R. CHRONOCRISIS: When Cell Cycle Asynchrony Generates DNA Damage in Polyploid Cells. Bioessays 2020, 42, e2000105. [Google Scholar] [CrossRef]
- Miles, D.M.; Desdouets, C.; Geli, V. Histone stress: An unexplored source of chromosomal instability in cancer? Curr. Genet. 2019, 65, 1081–1088. [Google Scholar] [CrossRef]
- Dewhurst, S.M.; McGranahan, N.; Burrell, R.A.; Rowan, A.J.; Gronroos, E.; Endesfelder, D.; Joshi, T.; Mouradov, D.; Gibbs, P.; Ward, R.L.; et al. Tolerance of Whole-Genome Doubling Propagates Chromosomal Instability and Accelerates Cancer Genome Evolution. Cancer Discov. 2014, 4, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.; Bloomfield, M.; Levi, H.; Keuper, K.; Bernhard, S.V.; Baudoin, N.C.; Leor, G.; Eliezer, Y.; Giam, M.; Wong, C.K.; et al. Whole-Genome Duplication Shapes the Aneuploidy Landscape of Human Cancers. Cancer Res. 2022, 82, 1736–1752. [Google Scholar] [CrossRef]
- Li, Y.; Roberts, N.D.; Wala, J.A.; Shapira, O.; Schumacher, S.E.; Kumar, K.; Khurana, E.; Waszak, S.; Korbel, J.O.; Haber, J.E.; et al. Patterns of somatic structural variation in human cancer genomes. Nature 2020, 578, 112–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siri, S.O.; Martino, J.; Gottifredi, V. Structural Chromosome Instability: Types, Origins, Consequences, and Therapeutic Opportunities. Cancers 2021, 13, 3056. [Google Scholar] [CrossRef] [PubMed]
- Orr, B.; Godek, K.M.; Compton, D. Aneuploidy. Curr. Biol. 2015, 25, R538–R542. [Google Scholar] [CrossRef] [Green Version]
- Pentzold, C.; Kokal, M.; Pentzold, S.; Weise, A. Sites of chromosomal instability in the context of nuclear architecture and function. Cell Mol. Life Sci. 2021, 78, 2095–2103. [Google Scholar] [CrossRef]
- Kjeldsen, E. Congenital Aneuploidy in Klinefelter Syndrome with B-Cell Acute Lymphoblastic Leukemia Might Be Associated with Chromosomal Instability and Reduced Telomere Length. Cancers 2022, 14, 2316. [Google Scholar] [CrossRef]
- Davoli, T.; Uno, H.; Wooten, E.C.; Elledge, S.J. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017, 355, eaaf8399. [Google Scholar] [CrossRef] [Green Version]
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Campbell, P.J.; Stratton, M.R. Deciphering Signatures of Mutational Processes Operative in Human Cancer. Cell Rep. 2013, 3, 246–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steele, C.D.; Abbasi, A.; Islam, S.M.A.; Bowes, A.L.; Khandekar, A.; Haase, K.; Hames-Fathi, S.; Ajayi, D.; Verfaillie, A.; Dhami, P.; et al. Signatures of copy number alterations in human cancer. Nature 2022, 606, 984–991. [Google Scholar] [CrossRef]
- Drews, R.M.; Hernando, B.; Tarabichi, M.; Haase, K.; Lesluyes, T.; Smith, P.S.; Morrill Gavarro, L.; Couturier, D.L.; Liu, L.; Schneider, M.; et al. A pan-cancer compendium of chromosomal instability. Nature 2022, 606, 976–983. [Google Scholar] [CrossRef]
- Wang, S.; Li, H.; Song, M.; Tao, Z.; Wu, T.; He, Z.; Zhao, X.; Wu, K.; Liu, X.S. Copy number signature analysis tool and its application in prostate cancer reveals distinct mutational processes and clinical outcomes. PLoS Genet. 2021, 17, e1009557. [Google Scholar] [CrossRef]
- Macintyre, G.; Goranova, T.E.; De Silva, D.; Ennis, D.; Piskorz, A.M.; Eldridge, M.; Sie, D.; Lewsley, L.A.; Hanif, A.; Wilson, C.; et al. Copy number signatures and mutational processes in ovarian carcinoma. Nat. Genet. 2018, 50, 1262–1270. [Google Scholar] [CrossRef]
- Sheltzer, J.M.; Ko, J.H.; Replogle, J.M.; Habibe Burgos, N.C.; Chung, E.S.; Meehl, C.M.; Sayles, N.M.; Passerini, V.; Storchova, Z.; Amon, A. Single-chromosome Gains Commonly Function as Tumor Suppressors. Cancer Cell 2017, 31, 240–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, B.R.; Prabhu, V.R.; Hunter, K.E.; Glazier, C.M.; Whittaker, C.A.; Housman, D.E.; Amon, A. Aneuploidy Affects Proliferation and Spontaneous Immortalization in Mammalian Cells. Science 2008, 322, 703–709. [Google Scholar] [CrossRef] [Green Version]
- Hassold, T.; Hunt, P. To err (meiotically) is human: The genesis of human aneuploidy. Nat. Rev. Genet. 2001, 2, 280–291. [Google Scholar] [CrossRef]
- Taylor, A.I. Autosomal trisomy syndromes: A detailed study of 27 cases of Edwards’ syndrome and 27 cases of Patau’s syndrome. J. Med. Genet. 1968, 5, 227–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auwerx, C.; Lepamets, M.; Sadler, M.C.; Patxot, M.; Stojanov, M.; Baud, D.; Magi, R.; Estonian Biobank Research, T.; Porcu, E.; Reymond, A.; et al. The individual and global impact of copy-number variants on complex human traits. Am. J. Hum. Genet. 2022, 109, 647–668. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, D.; Fisher, E.M. Mouse autosomal trisomy: Two’s company, three’s a crowd. Trends Genet. 1999, 15, 241–247. [Google Scholar] [CrossRef]
- McClintock, B. A Cytological and Genetical Study of Triploid Maize. Genetics 1929, 14, 180–222. [Google Scholar] [CrossRef] [PubMed]
- Sheltzer, J.M.; Amon, A. The aneuploidy paradox: Costs and benefits of an incorrect karyotype. Trends Genet. 2011, 27, 446–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, E.M.; Dephoure, N.; Panneerselvam, A.; Tucker, C.M.; Whittaker, C.A.; Gygi, S.P.; Dunham, M.J.; Amon, A. Identification of aneuploidy-tolerating mutations. Cell 2010, 143, 71–83. [Google Scholar] [CrossRef] [Green Version]
- Vasudevan, A.; Schukken, K.M.; Sausville, E.L.; Girish, V.; Adebambo, O.A.; Sheltzer, J.M. Aneuploidy as a promoter and suppressor of malignant growth. Nat. Rev. Cancer 2021, 21, 89–103. [Google Scholar] [CrossRef]
- Aylon, Y.; Oren, M. p53: Guardian of ploidy. Mol. Oncol. 2011, 5, 315–323. [Google Scholar] [CrossRef]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef] [Green Version]
- Narkar, A.; Johnson, B.A.; Bharne, P.; Zhu, J.; Padmanaban, V.; Biswas, D.; Fraser, A.; Iglesias, P.A.; Ewald, A.J.; Li, R. On the role of p53 in the cellular response to aneuploidy. Cell Rep. 2021, 34, 108892. [Google Scholar] [CrossRef]
- Lopez-Garcia, C.; Sansregret, L.; Domingo, E.; McGranahan, N.; Hobor, S.; Birkbak, N.J.; Horswell, S.; Gronroos, E.; Favero, F.; Rowan, A.J.; et al. BCL9L Dysfunction Impairs Caspase-2 Expression Permitting Aneuploidy Tolerance in Colorectal Cancer. Cancer Cell 2017, 31, 79–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simoes-Sousa, S.; Littler, S.; Thompson, S.L.; Minshall, P.; Whalley, H.; Bakker, B.; Belkot, K.; Moralli, D.; Bronder, D.; Tighe, A.; et al. The p38alpha Stress Kinase Suppresses Aneuploidy Tolerance by Inhibiting Hif-1alpha. Cell Rep. 2018, 25, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Godek, K.M.; Venere, M.; Wu, Q.L.; Mills, K.D.; Hickey, W.F.; Rich, J.N.; Compton, D.A. Chromosomal Instability Affects the Tumorigenicity of Glioblastoma Tumor-Initiating Cells. Cancer Discov. 2016, 6, 532–545. [Google Scholar] [CrossRef] [Green Version]
- van Wely, K.H.M.; Martinez, C. Linking stem cells to chromosomal instability. Oncoimmunology 2012, 1, 195–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moyer, A.J.; Gardiner, K.; Reeves, R.H. All Creatures Great and Small: New Approaches for Understanding Down Syndrome Genetics. Trends Genet. 2021, 37, 444–459. [Google Scholar] [CrossRef]
- Duijf, P.H.; Schultz, N.; Benezra, R. Cancer cells preferentially lose small chromosomes. Int. J. Cancer 2013, 132, 2316–2326. [Google Scholar] [CrossRef]
- Dong, G.; Mao, Q.; Yu, D.; Zhang, Y.; Qiu, M.; Dong, G.; Chen, Q.; Xia, W.; Wang, J.; Xu, L.; et al. Integrative analysis of copy number and transcriptional expression profiles in esophageal cancer to identify a novel driver gene for therapy. Sci. Rep. 2017, 7, 42060. [Google Scholar] [CrossRef] [Green Version]
- Pavelka, N.; Rancati, G.; Zhu, J.; Bradford, W.D.; Saraf, A.; Florens, L.; Sanderson, B.W.; Hattem, G.L.; Li, R. Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature 2010, 468, 321–325. [Google Scholar] [CrossRef] [Green Version]
- Dephoure, N.; Hwang, S.; O’Sullivan, C.; Dodgson, S.E.; Gygi, S.P.; Amon, A.; Torres, E.M. Quantitative proteomic analysis reveals posttranslational responses to aneuploidy in yeast. Elife 2014, 3, e03023. [Google Scholar] [CrossRef]
- Group, P.T.C.; Calabrese, C.; Davidson, N.R.; Demircioglu, D.; Fonseca, N.A.; He, Y.; Kahles, A.; Lehmann, K.V.; Liu, F.; Shiraishi, Y.; et al. Genomic basis for RNA alterations in cancer. Nature 2020, 578, 129–136. [Google Scholar] [CrossRef] [Green Version]
- Fehrmann, R.S.; Karjalainen, J.M.; Krajewska, M.; Westra, H.J.; Maloney, D.; Simeonov, A.; Pers, T.H.; Hirschhorn, J.N.; Jansen, R.C.; Schultes, E.A.; et al. Gene expression analysis identifies global gene dosage sensitivity in cancer. Nat. Genet. 2015, 47, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Serin Harmanci, A.; Harmanci, A.O.; Zhou, X. CaSpER identifies and visualizes CNV events by integrative analysis of single-cell or bulk RNA-sequencing data. Nat. Commun. 2020, 11, 89. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Bai, S.; Henderson, Y.C.; Lin, Y.; Schalck, A.; Yan, Y.; Kumar, T.; Hu, M.; Sei, E.; Davis, A.; et al. Delineating copy number and clonal substructure in human tumors from single-cell transcriptomes. Nat. Biotechnol. 2021, 39, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Prestel, M.; Feller, C.; Becker, P.B. Dosage compensation and the global re-balancing of aneuploid genomes. Genome. Biol. 2010, 11, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schukken, K.M.; Sheltzer, J.M. Extensive protein dosage compensation in aneuploid human cancers. Genome. Res. 2022, 32, 1254–1270. [Google Scholar] [CrossRef] [PubMed]
- Hose, J.; Yong, C.M.; Sardi, M.; Wang, Z.; Newton, M.A.; Gasch, A.P. Dosage compensation can buffer copy-number variation in wild yeast. Elife 2015, 4, e05462. [Google Scholar] [CrossRef]
- Cheng, J.Q.; Demeulemeester, J.; Wedge, D.C.; Vollan, H.K.M.; Pitt, J.J.; Russnes, H.G.; Pandey, B.P.; Nilsen, G.; Nord, S.; Bignell, G.R.; et al. Pan-cancer analysis of homozygous deletions in primary tumours uncovers rare tumour suppressors. Nat. Commun. 2017, 8, 1221. [Google Scholar] [CrossRef] [Green Version]
- Priestley, P.; Baber, J.; Lolkema, M.P.; Steeghs, N.; de Bruijn, E.; Shale, C.; Duyvesteyn, K.; Haidari, S.; van Hoeck, A.; Onstenk, W.; et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 2019, 575, 210–216. [Google Scholar] [CrossRef] [Green Version]
- Solimini, N.L.; Xu, Q.; Mermel, C.H.; Liang, A.C.; Schlabach, M.R.; Luo, J.; Burrows, A.E.; Anselmo, A.N.; Bredemeyer, A.L.; Li, M.Z.; et al. Recurrent hemizygous deletions in cancers may optimize proliferative potential. Science 2012, 337, 104–109. [Google Scholar] [CrossRef] [Green Version]
- Davoli, T.; Xu, A.W.; Mengwasser, K.E.; Sack, L.M.; Yoon, J.C.; Park, P.J.; Elledge, S.J. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 2013, 155, 948–962. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Tsai, H.J.; Gordon, M.R.; Li, R. Cellular Stress Associated with Aneuploidy. Dev. Cell 2018, 44, 420–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buccitelli, C.; Selbach, M. mRNAs, proteins and the emerging principles of gene expression control. Nat. Rev. Genet. 2020, 21, 630–644. [Google Scholar] [CrossRef]
- Ben-David, U.; Amon, A. Context is everything: Aneuploidy in cancer. Nat. Rev. Genet. 2020, 21, 44–62. [Google Scholar] [CrossRef] [PubMed]
- Ried, T.; Hu, Y.; Difilippantonio, M.J.; Ghadimi, B.M.; Grade, M.; Camps, J. The consequences of chromosomal aneuploidy on the transcriptome of cancer cells. Biochim. Biophys. Acta 2012, 1819, 784–793. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.; Van Hoeck, A.; Cuppen, E. Machine learning-based tissue of origin classification for cancer of unknown primary diagnostics using genome-wide mutation features. Nat. Commun. 2022, 13, 4013. [Google Scholar] [CrossRef]
- Patkar, S.; Heselmeyer-Haddad, K.; Auslander, N.; Hirsch, D.; Camps, J.; Bronder, D.; Brown, M.; Chen, W.D.; Lokanga, R.; Wangsa, D.; et al. Hard wiring of normal tissue-specific chromosome-wide gene expression levels is an additional factor driving cancer type-specific aneuploidies. Genome Med. 2021, 13, 93. [Google Scholar] [CrossRef]
- Wang, D.; Niu, X.H.; Wang, Z.J.; Song, C.L.; Huang, Z.; Chen, K.N.; Duan, J.C.; Bai, H.; Xu, J.C.; Zhao, J.; et al. Multiregion Sequencing Reveals the Genetic Heterogeneity and Evolutionary History of Osteosarcoma and Matched Pulmonary Metastases. Cancer Res. 2019, 79, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Mamlouk, S.; Childs, L.H.; Aust, D.; Heim, D.; Melching, F.; Oliveira, C.; Wolf, T.; Durek, P.; Schumacher, D.; Blaker, H.; et al. DNA copy number changes define spatial patterns of heterogeneity in colorectal cancer. Nat. Commun. 2017, 8, 14093. [Google Scholar] [CrossRef] [Green Version]
- Sack, L.M.; Davoli, T.; Li, M.Z.; Li, Y.; Xu, Q.; Naxerova, K.; Wooten, E.C.; Bernardi, R.J.; Martin, T.D.; Chen, T.; et al. Profound Tissue Specificity in Proliferation Control Underlies Cancer Drivers and Aneuploidy Patterns. Cell 2018, 173, 499–514. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Nguyen, N.P.; Turner, K.; Wu, S.; Gujar, A.D.; Luebeck, J.; Liu, J.; Deshpande, V.; Rajkumar, U.; Namburi, S.; et al. Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers. Nat. Genet. 2020, 52, 891–897. [Google Scholar] [CrossRef]
- Hoadley, K.A.; Yau, C.; Wolf, D.M.; Cherniack, A.D.; Tamborero, D.; Ng, S.; Leiserson, M.D.M.; Niu, B.; McLellan, M.D.; Uzunangelov, V.; et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 2014, 158, 929–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hugen, N.; Simmer, F.; Mekenkamp, L.J.; Koopman, M.; van den Broek, E.; de Wilt, J.H.; Punt, C.J.; Ylstra, B.; Meijer, G.A.; Nagtegaal, I.D. Reduced rate of copy number aberrations in mucinous colorectal carcinoma. Oncotarget 2015, 6, 25715–25725. [Google Scholar] [CrossRef] [PubMed]
- Valtorta, E.; Misale, S.; Sartore-Bianchi, A.; Nagtegaal, I.D.; Paraf, F.; Lauricella, C.; Dimartino, V.; Hobor, S.; Jacobs, B.; Ercolani, C.; et al. KRAS gene amplification in colorectal cancer and impact on response to EGFR-targeted therapy. Int. J. Cancer 2013, 133, 1259–1265. [Google Scholar] [CrossRef]
- Baker, S.J.; Fearon, E.R.; Nigro, J.M.; Hamilton, S.R.; Preisinger, A.C.; Jessup, J.M.; vanTuinen, P.; Ledbetter, D.H.; Barker, D.F.; Nakamura, Y.; et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science 1989, 244, 217–221. [Google Scholar] [CrossRef]
- Ciriello, G.; Cerami, E.; Sander, C.; Schultz, N. Mutual exclusivity analysis identifies oncogenic network modules. Genome. Res. 2012, 22, 398–406. [Google Scholar] [CrossRef] [Green Version]
- Gross, A.M.; Orosco, R.K.; Shen, J.P.; Egloff, A.M.; Carter, H.; Hofree, M.; Choueiri, M.; Coffey, C.S.; Lippman, S.M.; Hayes, D.N.; et al. Multi-tiered genomic analysis of head and neck cancer ties TP53 mutation to 3p loss. Nat. Genet. 2014, 46, 939–943. [Google Scholar] [CrossRef] [Green Version]
- Klijn, C.; Bot, J.; Adams, D.J.; Reinders, M.; Wessels, L.; Jonkers, J. Identification of networks of co-occurring, tumor-related DNA copy number changes using a genome-wide scoring approach. PLoS Comput. Biol. 2010, 6, e1000631. [Google Scholar] [CrossRef] [Green Version]
- Bredel, M.; Scholtens, D.M.; Harsh, G.R.; Bredel, C.; Chandler, J.P.; Renfrow, J.J.; Yadav, A.K.; Vogel, H.; Scheck, A.C.; Tibshirani, R.; et al. A network model of a cooperative genetic landscape in brain tumors. JAMA 2009, 302, 261–275. [Google Scholar] [CrossRef]
- Nakayama, K.; Nakayama, N.; Jinawath, N.; Salani, R.; Kurman, R.J.; Shih Ie, M.; Wang, T.L. Amplicon profiles in ovarian serous carcinomas. Int. J. Cancer 2007, 120, 2613–2617. [Google Scholar] [CrossRef]
- Kester, L.; de Barbanson, B.; Lyubimova, A.; Chen, L.-T.; van der Schrier, V.; Alemany, A.; Mooijman, D.; Peterson-Maduro, J.; Drost, J.; de Ridder, J.; et al. Integration of multiple lineage measurements from the same cell reconstructs parallel tumor evolution. Cell Genom. 2022, 2, 100096. [Google Scholar] [CrossRef]
- Schouten, P.C.; Grigoriadis, A.; Kuilman, T.; Mirza, H.; Watkins, J.A.; Cooke, S.A.; van Dyk, E.; Severson, T.M.; Rueda, O.M.; Hoogstraat, M.; et al. Robust BRCA1-like classification of copy number profiles of samples repeated across different datasets and platforms. Mol. Oncol. 2015, 9, 1274–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baslan, T.; Morris, J.P.; Zhao, Z.; Reyes, J.; Ho, Y.J.; Tsanov, K.M.; Bermeo, J.; Tian, S.; Zhang, S.; Askan, G.; et al. Ordered and deterministic cancer genome evolution after p53 loss. Nature 2022, 608, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Easwaran, H.; Tsai, H.C.; Baylin, S.B. Cancer epigenetics: Tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol. Cell 2014, 54, 716–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waddington, C.E. The Strategy of the Genes; Allen & Unwin: London, UK, 1957. [Google Scholar]
- Laughney, A.M.; Elizalde, S.; Genovese, G.; Bakhoum, S.F. Dynamics of Tumor Heterogeneity Derived from Clonal Karyotypic Evolution. Cell Rep. 2015, 12, 809–820. [Google Scholar] [CrossRef] [Green Version]
- Gao, R.L.; Davis, A.; McDonald, T.O.; Sei, E.; Shi, X.Q.; Wang, Y.; Tsai, P.C.; Casasent, A.; Waters, J.; Zhang, H.; et al. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Nat. Genet. 2016, 48, 1119–1130. [Google Scholar] [CrossRef] [Green Version]
- Gatenby, R.A.; Brown, J. Mutations, evolution and the central role of a self-defined fitness function in the initiation and progression of cancer. Biochim. Biophys. Acta Rev. Cancer 2017, 1867, 162–166. [Google Scholar] [CrossRef] [Green Version]
- Maley, C.C.; Aktipis, A.; Graham, T.A.; Sottoriva, A.; Boddy, A.M.; Janiszewska, M.; Silva, A.S.; Gerlinger, M.; Yuan, Y.; Pienta, K.J.; et al. Classifying the evolutionary and ecological features of neoplasms. Nat. Rev. Cancer 2017, 17, 605–619. [Google Scholar] [CrossRef]
- Drost, J.; van Jaarsveld, R.H.; Ponsioen, B.; Zimberlin, C.; van Boxtel, R.; Buijs, A.; Sachs, N.; Overmeer, R.M.; Offerhaus, G.J.; Begthel, H.; et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature 2015, 521, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Eirew, P.; Steif, A.; Khattra, J.; Ha, G.; Yap, D.; Farahani, H.; Gelmon, K.; Chia, S.; Mar, C.; Wan, A.; et al. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature 2015, 518, 422–426. [Google Scholar] [CrossRef] [Green Version]
- Woo, X.Y.; Giordano, J.; Srivastava, A.; Zhao, Z.M.; Lloyd, M.W.; de Bruijn, R.; Suh, Y.S.; Patidar, R.; Chen, L.; Scherer, S.; et al. Conservation of copy number profiles during engraftment and passaging of patient-derived cancer xenografts. Nat. Genet. 2021, 53, 86–99. [Google Scholar] [CrossRef]
- Sun, H.; Cao, S.; Mashl, R.J.; Mo, C.K.; Zaccaria, S.; Wendl, M.C.; Davies, S.R.; Bailey, M.H.; Primeau, T.M.; Hoog, J.; et al. Comprehensive characterization of 536 patient-derived xenograft models prioritizes candidatesfor targeted treatment. Nat. Commun. 2021, 12, 5086. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Ha, G.; Tseng, Y.Y.; Greenwald, N.F.; Oh, C.; Shih, J.; McFarland, J.M.; Wong, B.; Boehm, J.S.; Beroukhim, R.; et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat. Genet. 2017, 49, 1567–1575. [Google Scholar] [CrossRef] [PubMed]
- Hoge, A.C.H.; Getz, M.; Zimmer, A.; Ko, M.J.; Raz, L.; Beroukhim, R.; Golub, T.R.; Ha, G.; Ben-David, U. DNA-based copy number analysis confirms genomic evolution of PDX models. NPJ Precis. Oncol. 2022, 6, 1–7. [Google Scholar] [CrossRef]
- Ben-David, U.; Siranosian, B.; Ha, G.; Tang, H.; Oren, Y.; Hinohara, K.; Strathdee, C.A.; Dempster, J.; Lyons, N.J.; Burns, R.; et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature 2018, 560, 325–330. [Google Scholar] [CrossRef]
- Ippolito, M.R.; Martis, V.; Martin, S.; Tijhuis, A.E.; Hong, C.; Wardenaar, R.; Dumont, M.; Zerbib, J.; Spierings, D.C.J.; Fachinetti, D.; et al. Gene copy-number changes and chromosomal instability induced by aneuploidy confer resistance to chemotherapy. Dev. Cell 2021, 56, 2440–2454.e6. [Google Scholar] [CrossRef] [PubMed]
- Lukow, D.A.; Sausville, E.L.; Suri, P.; Chunduri, N.K.; Wieland, A.; Leu, J.; Smith, J.C.; Girish, V.; Kumar, A.A.; Kendall, J.; et al. Chromosomal instability accelerates the evolution of resistance to anti-cancer therapies. Dev. Cell 2021, 56, 2427–2439. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelova, M.; Mlecnik, B.; Vasaturo, A.; Bindea, G.; Fredriksen, T.; Lafontaine, L.; Buttard, B.; Morgand, E.; Bruni, D.; Jouret-Mourin, A.; et al. Evolution of Metastases in Space and Time under Immune Selection. Cell 2018, 175, 751. [Google Scholar] [CrossRef] [Green Version]
- Milo, I.; Bedora-Faure, M.; Garcia, Z.; Thibaut, R.; Perie, L.; Shakhar, G.; Deriano, L.; Bousso, P. The immune system profoundly restricts intratumor genetic heterogeneity. Sci. Immunol. 2018, 3, eaat1435. [Google Scholar] [CrossRef]
- Kloor, M.; von Knebel Doeberitz, M. The Immune Biology of Microsatellite-Unstable Cancer. Trends Cancer 2016, 2, 121–133. [Google Scholar] [CrossRef] [Green Version]
- Rosenthal, R.; Cadieux, E.L.; Salgado, R.; Bakir, M.A.; Moore, D.A.; Hiley, C.T.; Lund, T.; Tanic, M.; Reading, J.L.; Joshi, K.; et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 2019, 567, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Anagnostou, V.; Smith, K.N.; Forde, P.M.; Niknafs, N.; Bhattacharya, R.; White, J.; Zhang, T.; Adleff, V.; Phallen, J.; Wali, N.; et al. Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Discov. 2017, 7, 264–276. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Rosenthal, R.; Hiley, C.T.; Rowan, A.J.; Watkins, T.B.K.; Wilson, G.A.; Birkbak, N.J.; Veeriah, S.; Van Loo, P.; Herrero, J.; et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 2017, 171, 1259–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pages, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammerl, D.; Martens, J.W.M.; Timmermans, M.; Smid, M.; Trapman-Jansen, A.M.; Foekens, R.; Isaeva, O.I.; Voorwerk, L.; Balcioglu, H.E.; Wijers, R.; et al. Spatial immunophenotypes predict response to anti-PD1 treatment and capture distinct paths of T cell evasion in triple negative breast cancer. Nat. Commun. 2021, 12, 5668. [Google Scholar] [CrossRef]
- Nguyen, P.H.D.; Ma, S.; Phua, C.Z.J.; Kaya, N.A.; Lai, H.L.H.; Lim, C.J.; Lim, J.Q.; Wasser, M.; Lai, L.; Tam, W.L.; et al. Intratumoural immune heterogeneity as a hallmark of tumour evolution and progression in hepatocellular carcinoma. Nat. Commun. 2021, 12, 227. [Google Scholar] [CrossRef]
- Derks, S.; de Klerk, L.K.; Xu, X.; Fleitas, T.; Liu, K.X.; Liu, Y.; Dietlein, F.; Margolis, C.; Chiaravalli, A.M.; Da Silva, A.C.; et al. Characterizing diversity in the tumor-immune microenvironment of distinct subclasses of gastroesophageal adenocarcinomas. Ann. Oncol. 2020, 31, 1011–1020. [Google Scholar] [CrossRef]
- Wellenstein, M.D.; de Visser, K.E. Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity 2018, 48, 399–416. [Google Scholar] [CrossRef] [Green Version]
- Layer, J.P.; Kronmuller, M.T.; Quast, T.; van den Boorn-Konijnenberg, D.; Effern, M.; Hinze, D.; Althoff, K.; Schramm, A.; Westermann, F.; Peifer, M.; et al. Amplification of N-Myc is associated with a T-cell-poor microenvironment in metastatic neuroblastoma restraining interferon pathway activity and chemokine expression. Oncoimmunology 2017, 6, e1320626. [Google Scholar] [CrossRef] [Green Version]
- Kortlever, R.M.; Sodir, N.M.; Wilson, C.H.; Burkhart, D.L.; Pellegrinet, L.; Swigart, L.B.; Littlewood, T.D.; Evan, G.I. Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell 2017, 171, 1301–1315. [Google Scholar] [CrossRef]
- Casey, S.C.; Tong, L.; Li, Y.L.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gutgemann, I.; Eilers, M.; et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santaguida, S.; Richardson, A.; Iyer, D.R.; M’Saad, O.; Zasadil, L.; Knouse, K.A.; Wong, Y.L.; Rhind, N.; Desai, A.; Amon, A. Chromosome Mis-segregation Generates Cell-Cycle-Arrested Cells with Complex Karyotypes that Are Eliminated by the Immune System. Dev. Cell 2017, 41, 638–651.e5. [Google Scholar] [CrossRef] [PubMed]
- Vashi, N.; Bakhoum, S.F. The Evolution of STING Signaling and Its Involvement in Cancer. Trends Biochem. Sci. 2021, 46, 446–460. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.Z.; Liang, H.; Rao, E.Y.; Zheng, W.X.; Huang, X.N.; Deng, L.F.; Zhang, Y.; Yu, X.S.; Xu, M.; Mauceri, H.; et al. Non-canonical NF-kappa B Antagonizes STING Sensor-Mediated DNA Sensing in Radiotherapy. Immunity 2018, 49, 490–503.e4. [Google Scholar] [CrossRef] [Green Version]
- Benci, J.L.; Xu, B.H.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C.; et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016, 167, 1540–1554.e12. [Google Scholar] [CrossRef] [Green Version]
- Bristow, R.G.; Hill, R.P. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 2008, 8, 180–192. [Google Scholar] [CrossRef]
- Tang, M.; Bolderson, E.; O’Byrne, K.J.; Richard, D.J. Tumor Hypoxia Drives Genomic Instability. Front. Cell Dev. Biol. 2021, 9, 626229. [Google Scholar] [CrossRef]
- Haider, S.; McIntyre, A.; van Stiphout, R.G.; Winchester, L.M.; Wigfield, S.; Harris, A.L.; Buffa, F.M. Genomic alterations underlie a pan-cancer metabolic shift associated with tumour hypoxia. Genome Biol 2016, 17, 140. [Google Scholar] [CrossRef] [Green Version]
- Black, J.C.; Atabakhsh, E.; Kim, J.; Biette, K.M.; Van Rechem, C.; Ladd, B.; Burrowes, P.D.; Donado, C.; Mattoo, H.; Kleinstiver, B.P.; et al. Hypoxia drives transient site-specific copy gain and drug-resistant gene expression. Genes Dev. 2015, 29, 1018–1031. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Fu, X.; Lopez, J.I.; Rowan, A.; Au, L.; Fendler, A.; Hazell, S.; Xu, H.; Horswell, S.; Shepherd, S.T.C.; et al. Selection of metastasis competent subclones in the tumour interior. Nat. Ecol. Evol. 2021, 5, 1033–1045. [Google Scholar] [CrossRef]
- Graham, N.A.; Minasyan, A.; Lomova, A.; Cass, A.; Balanis, N.G.; Friedman, M.; Chan, S.N.; Zhao, S.; Delgado, A.; Go, J.; et al. Recurrent patterns of DNA copy number alterations in tumors reflect metabolic selection pressures. Mol. Syst. Biol. 2017, 13, 914. [Google Scholar] [CrossRef]
- Bollen, Y.; Stelloo, E.; van Leenen, P.; van den Bos, M.; Ponsioen, B.; Lu, B.; van Roosmalen, M.J.; Bolhaqueiro, A.C.F.; Kimberley, C.; Mossner, M.; et al. Reconstructing single-cell karyotype alterations in colorectal cancer identifies punctuated and gradual diversification patterns. Nat. Genet. 2021, 53, 1187–1195. [Google Scholar] [CrossRef]
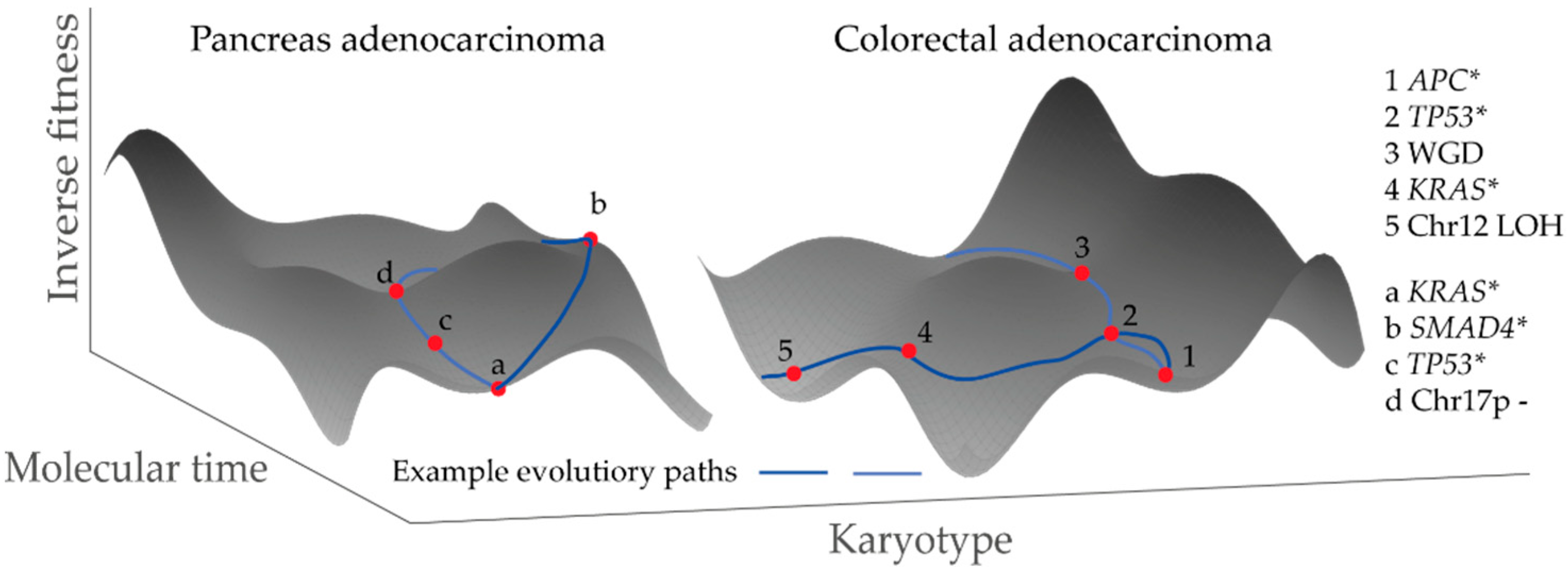
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Vermeulen, L.; Morrissey, E.; van der Heijden, M.; Nicholson, A.M.; Sottoriva, A.; Buczacki, S.; Kemp, R.; Tavare, S.; Winton, D.J. Defining stem cell dynamics in models of intestinal tumor initiation. Science 2013, 342, 995–998. [Google Scholar] [CrossRef] [PubMed]
- Moreno, E. Is cell competition relevant to cancer? Nat. Rev. Cancer 2008, 8, 141–147. [Google Scholar] [CrossRef] [PubMed]
- de la Cova, C.; Abril, M.; Bellosta, P.; Gallant, P.; Johnston, L.A. Drosophila myc regulates organ size by inducing cell competition. Cell 2004, 117, 107–116. [Google Scholar] [CrossRef] [Green Version]
- Di Giacomo, S.; Sollazzo, M.; de Biase, D.; Ragazzi, M.; Bellosta, P.; Pession, A.; Grifoni, D. Human Cancer Cells Signal Their Competitive Fitness Through MYC Activity. Sci. Rep. 2017, 7, 12568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalkat, M.; De Melo, J.; Hickman, K.A.; Lourenco, C.; Redel, C.; Resetca, D.; Tamachi, A.; Tu, W.B.; Penn, L.Z. MYC Deregulation in Primary Human Cancers. Genes 2017, 8, 151. [Google Scholar] [CrossRef] [Green Version]
- Yum, M.K.; Han, S.; Fink, J.; Wu, S.S.; Dabrowska, C.; Trendafilova, T.; Mustata, R.; Chatzeli, L.; Azzarelli, R.; Pshenichnaya, I.; et al. Tracing oncogene-driven remodelling of the intestinal stem cell niche. Nature 2021, 594, 442–447. [Google Scholar] [CrossRef]
- van Neerven, S.M.; de Groot, N.E.; Nijman, L.E.; Scicluna, B.P.; van Driel, M.S.; Lecca, M.C.; Warmerdam, D.O.; Kakkar, V.; Moreno, L.F.; Vieira Braga, F.A.; et al. Apc-mutant cells act as supercompetitors in intestinal tumour initiation. Nature 2021, 594, 436–441. [Google Scholar] [CrossRef]
- Flanagan, D.J.; Pentinmikko, N.; Luopajarvi, K.; Willis, N.J.; Gilroy, K.; Raven, A.P.; McGarry, L.; Englund, J.I.; Webb, A.T.; Scharaw, S.; et al. NOTUM from Apc-mutant cells biases clonal competition to initiate cancer. Nature 2021, 594, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Di Gregorio, A.; Bowling, S.; Rodriguez, T.A. Cell Competition and Its Role in the Regulation of Cell Fitness from Development to Cancer. Dev. Cell 2016, 38, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Tabassum, D.P.; Altrock, P.M.; Almendro, V.; Michor, F.; Polyak, K. Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature 2014, 514, 54–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janiszewska, M.; Tabassum, D.P.; Castano, Z.; Cristea, S.; Yamamoto, K.N.; Kingston, N.L.; Murphy, K.C.; Shu, S.; Harper, N.W.; Del Alcazar, C.G.; et al. Subclonal cooperation drives metastasis by modulating local and systemic immune microenvironments. Nat. Cell Biol. 2019, 21, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Gerlee, P.; Altrock, P.M.; Malik, A.; Krona, C.; Nelander, S. Autocrine signaling can explain the emergence of Allee effects in cancer cell populations. PLoS Comput. Biol. 2022, 18, e1009844. [Google Scholar] [CrossRef]
- Morrow, R.J.; Allam, A.H.; Yeo, B.; Deb, S.; Murone, C.; Lim, E.; Johnstone, C.N.; Ernst, M. Paracrine IL-6 Signaling Confers Proliferation between Heterogeneous Inflammatory Breast Cancer Sub-Clones. Cancers 2022, 14, 2292. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.E.; Howard, G.; Mo, W.; Strasser, M.K.; Lima, E.; Huang, S.; Brock, A. Cancer cell population growth kinetics at low densities deviate from the exponential growth model and suggest an Allee effect. PLoS Biol. 2019, 17, e3000399. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Jimenez, F.; Muinos, F.; Sentis, I.; Deu-Pons, J.; Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H.; et al. A compendium of mutational cancer driver genes. Nat. Rev. Cancer 2020, 20, 555–572. [Google Scholar] [CrossRef]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 174, 1034–1035. [Google Scholar] [CrossRef] [Green Version]
- Martincorena, I.; Raine, K.M.; Gerstung, M.; Dawson, K.J.; Haase, K.; Van Loo, P.; Davies, H.; Stratton, M.R.; Campbell, P.J. Universal Patterns of Selection in Cancer and Somatic Tissues. Cell 2017, 171, 1029–1041.e21. [Google Scholar] [CrossRef]
- Williams, M.J.; Werner, B.; Barnes, C.P.; Graham, T.A.; Sottoriva, A. Identification of neutral tumor evolution across cancer types. Nat. Genet. 2016, 48, 238–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, S.P.; Hu, Z.; Yang, Z.Y.; Yang, F.; Li, Y.W.; Lin, P.; Chen, K.; Dong, L.L.; Cao, L.H.; Tao, Y.; et al. Extremely high genetic diversity in a single tumor points to prevalence of non-Darwinian cell evolution. Proc. Natl. Acad. Sci. USA 2015, 112, E6496–E6505. [Google Scholar] [CrossRef]
- Durrett, R.; Foo, J.; Leder, K.; Mayberry, J.; Michor, F. Intratumor Heterogeneity in Evolutionary Models of Tumor Progression. Genetics 2011, 188, 461–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naugler, C.T. Population genetics of cancer cell clones: Possible implications of cancer stem cells. Biol. Med. Model 2010, 7, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, M.; Ackerman, M.S.; Gout, J.F.; Long, H.; Sung, W.; Thomas, W.K.; Foster, P.L. Genetic drift, selection and the evolution of the mutation rate. Nat. Rev. Genet. 2016, 17, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Silander, O.K.; Tenaillon, O.; Chao, L. Understanding the evolutionary fate of finite populations: The dynamics of mutational effects. PLoS Biol. 2007, 5, e94. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, K.A.; Barber, L.J.; Davies, M.N.; Ashenden, M.; Sottoriva, A.; Gerlinger, M. Cancer Evolution and the Limits of Predictability in Precision Cancer Medicine. Trends Cancer 2016, 2, 49–63. [Google Scholar] [CrossRef] [Green Version]
- Sottoriva, A.; Kang, H.; Ma, Z.C.; Graham, T.A.; Salomon, M.P.; Zhao, J.S.; Marjoram, P.; Siegmund, K.; Press, M.F.; Shibata, D.; et al. A Big Bang model of human colorectal tumor growth. Nat. Genet. 2015, 47, 209–216. [Google Scholar] [CrossRef] [Green Version]
- van der Heijden, M.; Miedema, D.M.; Waclaw, B.; Veenstra, V.L.; Lecca, M.C.; Nijman, L.E.; van Dijk, E.; van Neerven, S.M.; Lodestijn, S.C.; Lenos, K.J.; et al. Spatiotemporal regulation of clonogenicity in colorectal cancer xenografts. Proc. Natl. Acad. Sci. USA 2019, 116, 6140–6145. [Google Scholar] [CrossRef] [Green Version]
- Fusco, D.; Gralka, M.; Kayser, J.; Anderson, A.; Hallatschek, O. Excess of mutational jackpot events in expanding populations revealed by spatial Luria-Delbruck experiments. Nat. Commun. 2016, 7, 12760. [Google Scholar] [CrossRef] [Green Version]
- West, J.; Schenck, R.O.; Gatenbee, C.; Robertson-Tessi, M.; Anderson, A.R.A. Normal tissue architecture determines the evolutionary course of cancer. Nat. Commun. 2021, 12, 2060. [Google Scholar] [CrossRef] [PubMed]
- Noble, R.; Burri, D.; Le Sueur, C.; Lemant, J.; Viossat, Y.; Kather, J.N.; Beerenwinkel, N. Spatial structure governs the mode of tumour evolution. Nat. Ecol. Evol. 2022, 6, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Zhao, Y.; Lopez, J.I.; Rowan, A.; Au, L.; Fendler, A.; Hazell, S.; Xu, H.; Horswell, S.; Shepherd, S.T.C.; et al. Spatial patterns of tumour growth impact clonal diversification in a computational model and the TRACERx Renal study. Nat. Ecol. Evol. 2022, 6, 88–102. [Google Scholar] [CrossRef] [PubMed]
- Waclaw, B.; Bozic, I.; Pittman, M.E.; Hruban, R.H.; Vogelstein, B.; Nowak, M.A. A spatial model predicts that dispersal and cell turnover limit intratumour heterogeneity. Nature 2015, 525, 261–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chkhaidze, K.; Heide, T.; Werner, B.; Williams, M.J.; Huang, W.; Caravagna, G.; Graham, T.A.; Sottoriva, A. Spatially constrained tumour growth affects the patterns of clonal selection and neutral drift in cancer genomic data. PLoS Comput. Biol. 2019, 15, e1007243. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van den Bosch, T.; Derks, S.; Miedema, D.M. Chromosomal Instability, Selection and Competition: Factors That Shape the Level of Karyotype Intra-Tumor Heterogeneity. Cancers 2022, 14, 4986. https://doi.org/10.3390/cancers14204986
van den Bosch T, Derks S, Miedema DM. Chromosomal Instability, Selection and Competition: Factors That Shape the Level of Karyotype Intra-Tumor Heterogeneity. Cancers. 2022; 14(20):4986. https://doi.org/10.3390/cancers14204986
Chicago/Turabian Stylevan den Bosch, Tom, Sarah Derks, and Daniël M. Miedema. 2022. "Chromosomal Instability, Selection and Competition: Factors That Shape the Level of Karyotype Intra-Tumor Heterogeneity" Cancers 14, no. 20: 4986. https://doi.org/10.3390/cancers14204986