
Dissecting the Immunological Profiles in NSD3-Amplified LUSC through Integrative Multi-Scale Analyses

 ,
,  , , ,
, , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Retrieval and Preprocessing
2.2. Identification of Differentially Expressed Genes (DEGs) and Functional Enrichment Analysis
2.3. Estimation of the Immunological Profiles of the TME in LUSC
2.4. Ethics Statements
2.5. Immunohistochemistry of LUSC Tissue Microarray
2.6. Semiquantitative Scoring
2.7. Classification of Immune Subtypes
2.8. Prediction of Clinical Drug Response
2.9. Statistical Analysis
3. Results
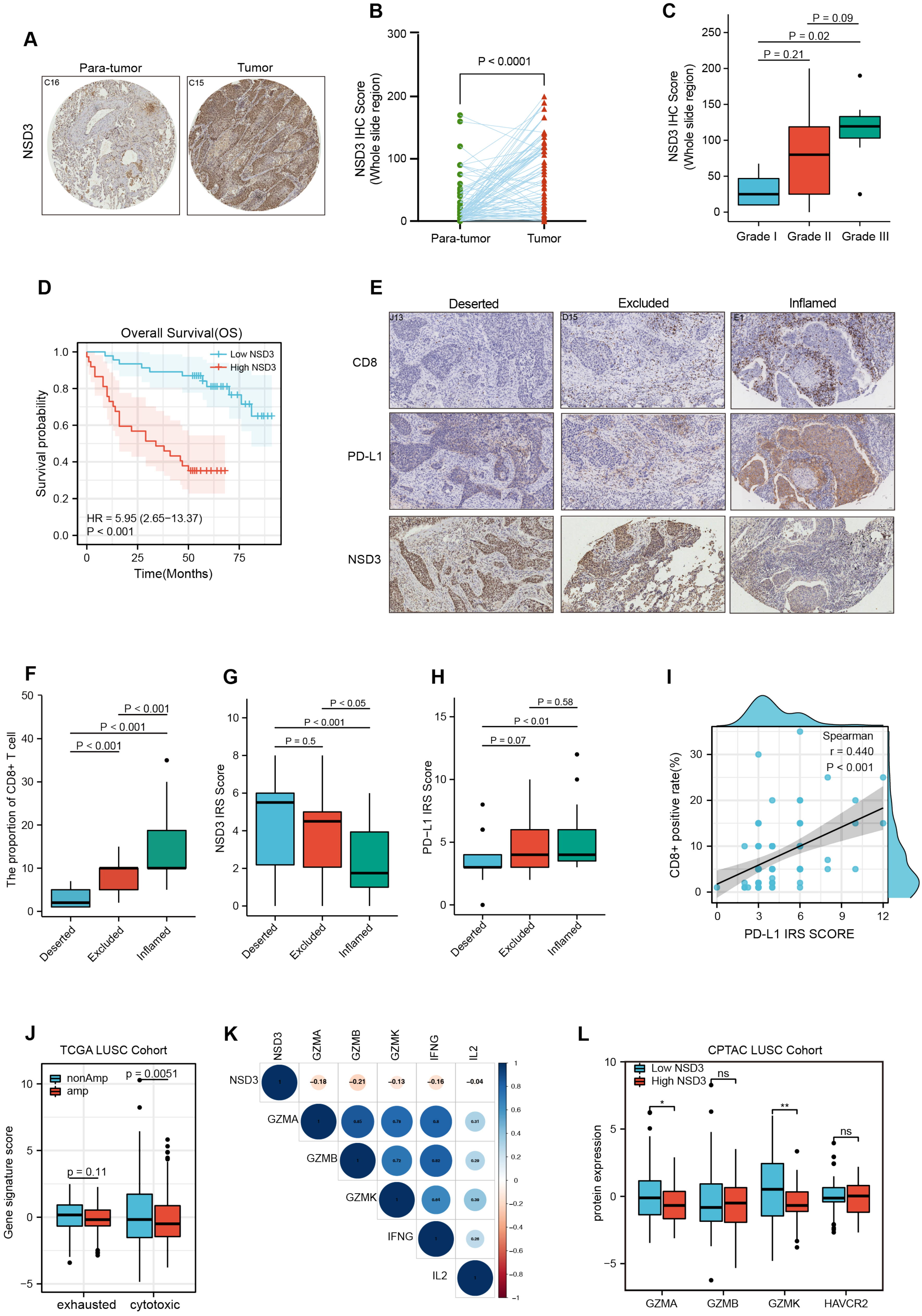
3.1. NSD3 Is a Critical Genetic Driver of LUSC Tumorigenesis
3.2. NSD3 Shapes a Non-Inflamed TIME in LUSC
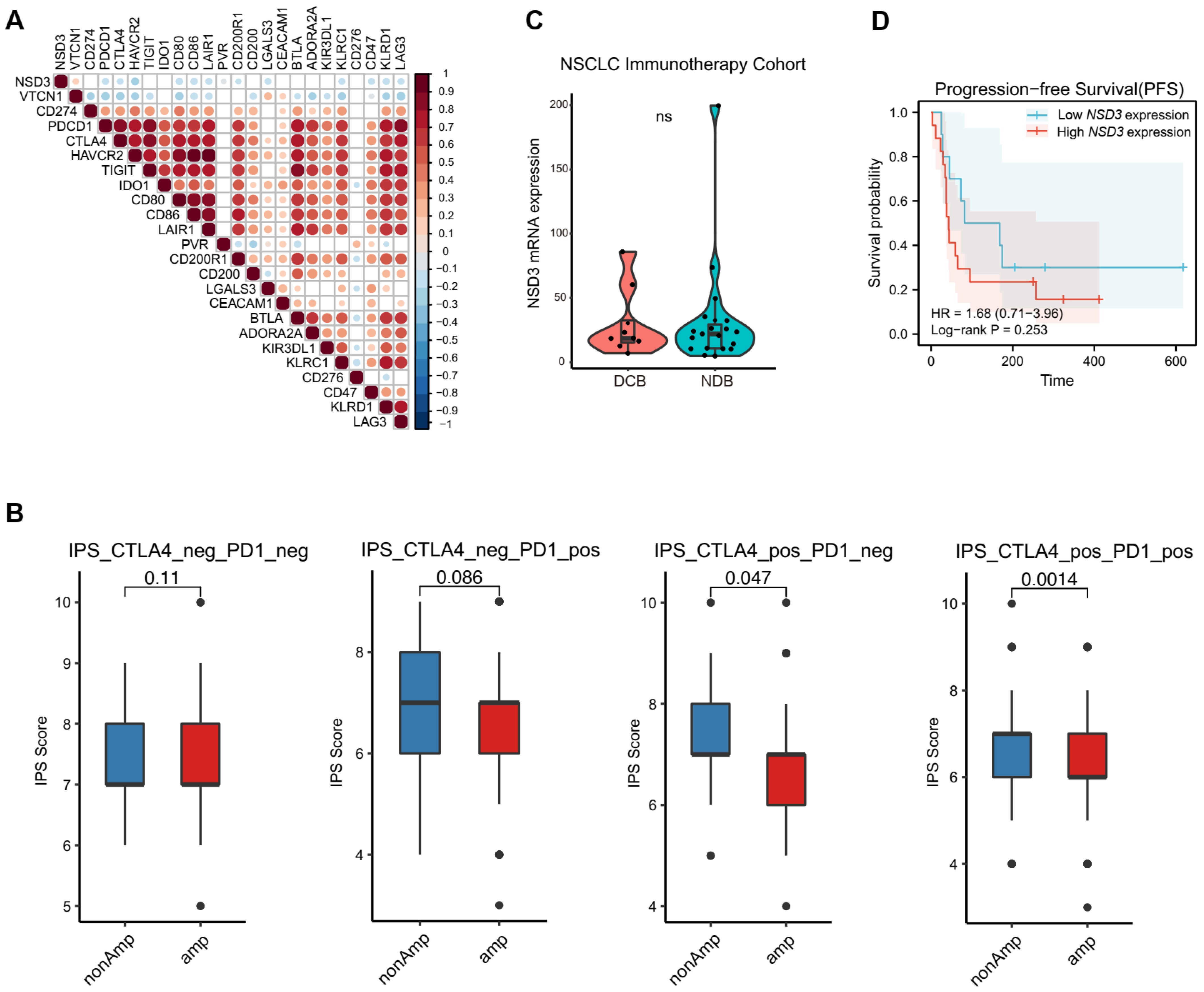
3.3. The Association between NSD3 and Immune Infiltrates in LUSC
3.4. Immunotherapy Outcome Prediction by NSD3
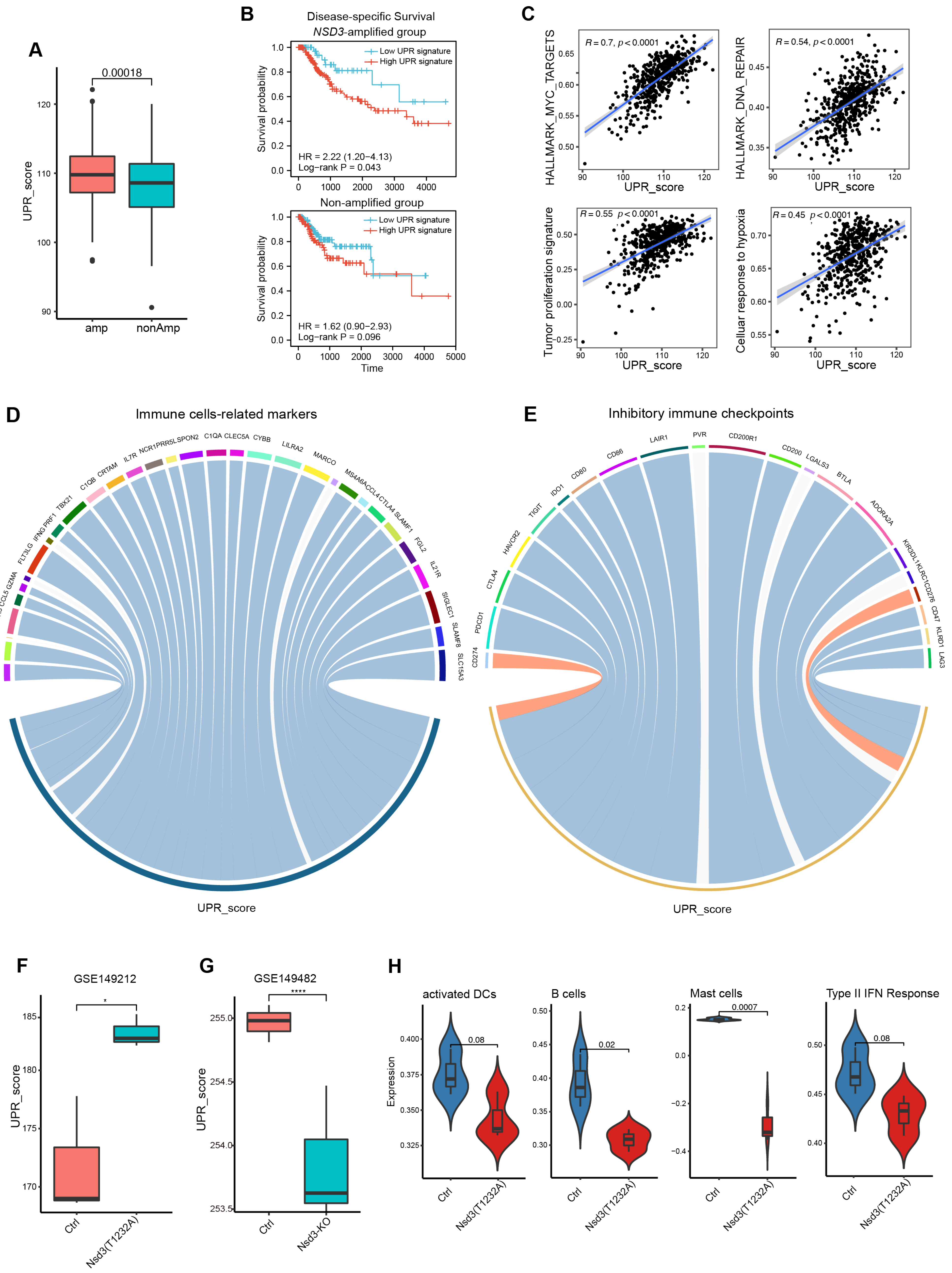
3.5. Internal Links between the Unfolded Protein Response and Non-Immunogenic Feature of NSD3-Amplified LUSC
3.6. Exploration of Potential Therapeutic Targets for NSD3-Amplified LUSC
4. Discussion
5. Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Pan, Y.; Han, H.; Labbe, K.E.; Zhang, H.; Wong, K.-K. Recent advances in preclinical models for lung squamous cell carcinoma. Oncogene 2021, 40, 2817–2829. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Hammerman, P.S.; Kim, J.; Yoon, J.-A.; Lee, Y.; Sun, J.-M.; Wilkerson, M.D.; Pedamallu, C.S.; Cibulskis, K.; Yoo, Y.K.; et al. Integrative and Comparative Genomic Analysis of Lung Squamous Cell Carcinomas in East Asian Patients. J. Clin. Oncol. 2014, 32, 121–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paik, P.K.; Pillai, R.N.; Lathan, C.S.; Velasco, S.A.; Papadimitrakopoulou, V. New Treatment Options in Advanced Squamous Cell Lung Cancer. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, e198–e206. [Google Scholar] [CrossRef]
- Weiss, J.; Sos, M.L.; Seidel, D.; Peifer, M.; Zander, T.; Heuckmann, J.M.; Ullrich, R.T.; Menon, R.; Maier, S.; Soltermann, A.; et al. Frequent and Focal FGFR1 Amplification Associates with Therapeutically Tractable FGFR1 Dependency in Squamous Cell Lung Cancer. Sci. Transl. Med. 2010, 2, 62ra93. [Google Scholar] [CrossRef] [Green Version]
- Rooney, C.; Geh, C.; Williams, V.; Heuckmann, J.M.; Menon, R.; Schneider, P.; Al-Kadhimi, K.; Dymond, M.; Smith, N.R.; Baker, D.; et al. Characterization of FGFR1 Locus in sqNSCLC Reveals a Broad and Heterogeneous Amplicon. PLoS ONE 2016, 11, e0149628. [Google Scholar] [CrossRef]
- Babina, I.S.; Turner, N.C. Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 2017, 17, 318–332. [Google Scholar] [CrossRef]
- Paik, P.K.; Shen, R.; Berger, M.F.; Ferry, D.; Soria, J.-C.; Mathewson, A.; Rooney, C.; Smith, N.R.; Cullberg, M.; Kilgour, E.; et al. A Phase Ib Open-Label Multicenter Study of AZD4547 in Patients with Advanced Squamous Cell Lung Cancers. Clin. Cancer Res. 2017, 23, 5366–5373. [Google Scholar] [CrossRef] [Green Version]
- Yuan, G.; Flores, N.M.; Hausmann, S.; Lofgren, S.M.; Kharchenko, V.; Angulo-Ibanez, M.; Sengupta, D.; Lu, X.; Czaban, I.; Azhibek, D.; et al. Elevated NSD3 histone methylation activity drives squamous cell lung cancer. Nature 2021, 590, 504–508. [Google Scholar] [CrossRef]
- Turner-Ivey, B.; Smith, E.L.; Rutkovsky, A.C.; Spruill, L.S.; Mills, J.N.; Ethier, S.P. Development of mammary hyperplasia, dysplasia, and invasive ductal carcinoma in transgenic mice expressing the 8p11 amplicon oncogene NSD3. Breast Cancer Res. Treat. 2017, 164, 349–358. [Google Scholar] [CrossRef] [Green Version]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Fares, C.M.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 147–164. [Google Scholar] [CrossRef]
- Shembrey, C.; Huntington, N.D.; Hollande, F. Impact of Tumor and Immunological Heterogeneity on the Anti-Cancer Immune Response. Cancers 2019, 11, 1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Lin, Y.; Wang, Y.; Wang, S.; Yang, Y.; Mu, X.; Chen, Y.; Gao, Z. Characterization of the Tumor Immune Microenvironment in Lung Squamous Cell Carcinoma Using Imaging Mass Cytometry. Front. Oncol. 2021, 11, 620989. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, Q.; Xu, X.; Xie, B.; Zhao, Y.; Li, N.; Cao, X. The methyltransferase NSD3 promotes antiviral innate immunity via direct lysine methylation of IRF3. J. Exp. Med. 2017, 214, 3597–3610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satpathy, S.; Krug, K.; Beltran, P.M.J.; Savage, S.R.; Petralia, F.; Kumar-Sinha, C.; Dou, Y.; Reva, B.; Kane, M.H.; Avanessian, S.C.; et al. A proteogenomic portrait of lung squamous cell carcinoma. Cell 2021, 184, 4348–4371.e40. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Kim, H.S.; Kim, J.Y.; Sun, J.-M.; Ahn, J.S.; Ahn, M.-J.; Park, K.; Esteller, M.; Lee, S.-H.; Choi, J.K. DNA methylation loss promotes immune evasion of tumours with high mutation and copy number load. Nat. Commun. 2019, 10, 4278. [Google Scholar] [CrossRef] [Green Version]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576.e16. [Google Scholar] [CrossRef]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Yu, A.; Othmane, B.; Qiu, D.; Li, H.; Li, C.; Liu, P.; Ren, W.; Chen, M.; Gong, G.; et al. Siglec15 shapes a non-inflamed tumor microenvironment and predicts the molecular subtype in bladder cancer. Theranostics 2021, 11, 3089–3108. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef]
- Racle, J.; De Jonge, K.; Baumgaertner, P.; Speiser, D.E.; Gfeller, D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. eLife 2017, 6, e26476. [Google Scholar] [CrossRef] [PubMed]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautes-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 218. [Google Scholar] [CrossRef]
- Finotello, F.; Mayer, C.; Plattner, C.; Laschober, G.; Rieder, D.; Hackl, H.; Krogsdam, A.; Loncova, Z.; Posch, W.; Wilflingseder, D.; et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med. 2019, 11, 34. [Google Scholar] [CrossRef] [Green Version]
- Ru, B.; Wong, C.N.; Tong, Y.; Zhong, J.Y.; Zhong, S.S.W.; Wu, W.C.; Chu, K.C.; Wong, C.Y.; Lau, C.Y.; Chen, I.; et al. TISIDB: An integrated repository portal for tumor-immune system interactions. Bioinformatics 2019, 35, 4200–4202. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal Dynamics of Intratumoral Immune Cells Reveal the Immune Landscape in Human Cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Deng, C.; Pang, B.; Zhang, X.; Liu, W.; Liao, G.; Yuan, H.; Cheng, P.; Li, F.; Long, Z.; et al. TIP: A Web Server for Resolving Tumor Immunophenotype Profiling. Cancer Res. 2018, 78, 6575–6580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auslander, N.; Zhang, G.; Lee, J.S.; Frederick, D.T.; Miao, B.; Moll, T.; Tian, T.; Wei, Z.; Madan, S.; Sullivan, R.J.; et al. Robust prediction of response to immune checkpoint blockade therapy in metastatic melanoma. Nat. Med. 2018, 24, 1545–1549. [Google Scholar] [CrossRef] [PubMed]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Geeleher, P.; Cox, N.; Huang, R.S. pRRophetic: An R Package for Prediction of Clinical Chemotherapeutic Response from Tumor Gene Expression Levels. PLoS ONE 2014, 9, e107468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grootjans, J.; Kaser, A.; Kaufman, R.J.; Blumberg, R.S. The unfolded protein response in immunity and inflammation. Nat. Rev. Immunol. 2016, 16, 469–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanetti, M.; Xian, S.; Dosset, M.; Carter, H. The Unfolded Protein Response at the Tumor-Immune Interface. Front. Immunol. 2022, 13, 823157. [Google Scholar] [CrossRef]
- Mok, T.S.K.; Wu, Y.-L.; Kudaba, I.; Kowalski, D.M.; Cho, B.C.; Turna, H.Z.; Castro, G., Jr.; Srimuninnimit, V.; Laktionov, K.K.; Bondarenko, I.; et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): A randomised, open-label, controlled, phase 3 trial. Lancet 2019, 393, 1819–1830. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Ciuleanu, T.-E.; Cobo, M.; Schenker, M.; Zurawski, B.; Menezes, J.; Richardet, E.; Bennouna, J.; Felip, E.; Juan-Vidal, O.; et al. First-line nivolumab plus ipilimumab combined with two cycles of chemotherapy in patients with non-small-cell lung cancer (CheckMate 9LA): An international, randomised, open-label, phase 3 trial. Lancet Oncol. 2021, 22, 198–211. [Google Scholar] [CrossRef]
- Duffy, M.J.; Crown, J. Biomarkers for Predicting Response to Immunotherapy with Immune Checkpoint Inhibitors in Cancer Patients. Clin. Chem. 2019, 65, 1228–1238. [Google Scholar] [CrossRef]
- Borcoman, E.; Nandikolla, A.; Long, G.; Goel, S.; Le Tourneau, C. Patterns of Response and Progression to Immunotherapy. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 169–178. [Google Scholar] [CrossRef]
- Vitale, I.; Shema, E.; Loi, S.; Galluzzi, L. Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat. Med. 2021, 27, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Liang, S.-Q.; Yang, H.; Lüthi, U.; Riether, C.; Berezowska, S.; Marti, T.M.; Hall, S.R.R.; Bruggmann, R.; Kocher, G.J.; et al. Increased sensitivity to apoptosis upon endoplasmic reticulum stress-induced activation of the unfolded protein response in chemotherapy-resistant malignant pleural mesothelioma. Br. J. Cancer 2018, 119, 65–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerezo, M.; Lehraiki, A.; Millet, A.; Rouaud, F.; Plaisant, M.; Jaune, E.; Botton, T.; Ronco, C.; Abbe, P.; Amdouni, H.; et al. Compounds Triggering ER Stress Exert Anti-Melanoma Effects and Overcome BRAF Inhibitor Resistance. Cancer Cell 2016, 29, 805–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Yang, H.; Berezowska, S.; Gao, Y.; Liang, S.-Q.; Marti, T.M.; Hall, S.R.R.; Dorn, P.; Kocher, G.J.; Schmid, R.A.; et al. Endoplasmic Reticulum Stress Signaling as a Therapeutic Target in Malignant Pleural Mesothelioma. Cancers 2019, 11, 1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetz, C.; Chevet, E.; Oakes, S.A. Proteostasis control by the unfolded protein response. Nat. Cell Biol. 2015, 17, 829–838. [Google Scholar] [CrossRef] [Green Version]
- Jeong, G.-Y.; Park, M.K.; Choi, H.-J.; An, H.W.; Park, Y.-U.; Choi, H.-J.; Park, J.; Kim, H.-Y.; Son, T.; Lee, H.; et al. NSD3-Induced Methylation of H3K36 Activates NOTCH Signaling to Drive Breast Tumor Initiation and Metastatic Progression. Cancer Res. 2021, 81, 77–90. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [Green Version]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef] [Green Version]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell–Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Canellas, A.; Hernando-Momblona, X.; et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cubillos-Ruiz, J.R.; Silberman, P.C.; Rutkowski, M.R.; Chopra, S.; Perales-Puchalt, A.; Song, M.; Zhang, S.; Bettigole, S.E.; Gupta, D.; Holcomb, K.; et al. ER Stress Sensor XBP1 Controls Anti-tumor Immunity by Disrupting Dendritic Cell Homeostasis. Cell 2015, 161, 1527–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Condamine, T.; Kumar, V.; Ramachandran, I.R.; Youn, J.-I.; Celis, E.; Finnberg, N.; El-Deiry, W.; Winograd, R.; Vonderheide, R.H.; English, N.R.; et al. ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R–mediated apoptosis. J. Clin. Investig. 2014, 124, 2626–2639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thevenot, P.T.; Sierra, R.A.; Raber, P.L.; Al-Khami, A.A.; Trillo-Tinoco, J.; Zarreii, P.; Ochoa, A.C.; Cui, Y.; Del Valle, L.; Rodriguez, P.C. The Stress-Response Sensor Chop Regulates the Function and Accumulation of Myeloid-Derived Suppressor Cells in Tumors. Immunity 2014, 41, 389–401. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Li, A.; Song, N.-J.; Riesenberg, B.P.; Li, Z. The Emerging Roles of Endoplasmic Reticulum Stress in Balancing Immunity and Tolerance in Health and Diseases: Mechanisms and Opportunities. Front. Immunol. 2019, 10, 3154. [Google Scholar] [CrossRef] [Green Version]
- Pol, J.; Vacchelli, E.; Aranda, F.; Castoldi, F.; Eggermont, A.; Cremer, I.; Sautes-Fridman, C.; Fucikova, J.; Galon, J.; Spisek, R.; et al. Trial Watch: Immunogenic cell death inducers for anticancer chemotherapy. OncoImmunology 2015, 4, e1008866. [Google Scholar] [CrossRef]
- Mohamed, E.; Cao, Y.; Rodriguez, P.C. Endoplasmic reticulum stress regulates tumor growth and anti-tumor immunity: A promising opportunity for cancer immunotherapy. Cancer Immunol. Immunother. 2017, 66, 1069–1078. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Cubillos-Ruiz, J.R. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat. Rev. Cancer 2021, 21, 71–88. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, D.; Liu, S.; Wu, X.; Marti, T.M.; Dorn, P.; Schmid, R.A.; Peng, R.-W.; Shu, Y. Dissecting the Immunological Profiles in NSD3-Amplified LUSC through Integrative Multi-Scale Analyses. Cancers 2022, 14, 4997. https://doi.org/10.3390/cancers14204997
Xu D, Liu S, Wu X, Marti TM, Dorn P, Schmid RA, Peng R-W, Shu Y. Dissecting the Immunological Profiles in NSD3-Amplified LUSC through Integrative Multi-Scale Analyses. Cancers. 2022; 14(20):4997. https://doi.org/10.3390/cancers14204997
Chicago/Turabian StyleXu, Duo, Shengchen Liu, Xi Wu, Thomas M. Marti, Patrick Dorn, Ralph A. Schmid, Ren-Wang Peng, and Yongqian Shu. 2022. "Dissecting the Immunological Profiles in NSD3-Amplified LUSC through Integrative Multi-Scale Analyses" Cancers 14, no. 20: 4997. https://doi.org/10.3390/cancers14204997