
Atypical Macropinocytosis Contributes to Malignant Progression: A Review of Recent Evidence in Endometrioid Endometrial Cancer Cells


Abstract
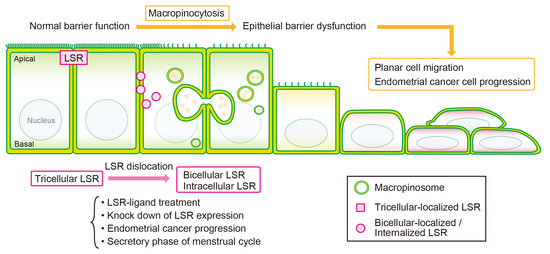
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Uptake Mechanisms of Extracellular Components
3. A Universal Role of Macropinocytosis
4. Disruption of Epithelial Homeostasis Triggers Cancer Malignancy
4.1. Regulation of Epithelial Homeostasis by Cell Polarity
4.2. Regulation of Epithelial Homeostasis by Cell Density
5. LSR-Related Molecular Mechanisms Leading to Disruption of Endometrial Homeostasis
5.1. Disruption of Endometrial Homeostasis by TGF-β Signaling
5.2. Disruption of Endometrial Homeostasis by Hippo-Yap Pathway
6. Pathophysiological Changes in Endometriosis and Endometrial Carcinoma Are Accompanied by Changes in LSR Localization
7. Internalization Pathways of Tight Junction Proteins
8. Regulation of Cell Motility by Macropinocytosis Induced by LSR Ligand
9. SLC9A1-Mediated Macropinocytosis and Its Associated Regulation of Cancer Malignancy
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- King, J.S.; Kay, R.R. The Origins and Evolution of Macropinocytosis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2019, 374, 20180158. [Google Scholar] [CrossRef] [PubMed]
- Hoeller, O.; Bolourani, P.; Clark, J.; Stephens, L.R.; Hawkins, P.T.; Weiner, O.D.; Weeks, G.; Kay, R.R. Two Distinct Functions for PI3-Kinases in Macropinocytosis. J. Cell Sci. 2013, 126, 4296–4307. [Google Scholar] [CrossRef]
- Recouvreux, M.V.; Commisso, C. Macropinocytosis: A Metabolic Adaptation to Nutrient Stress in Cancer. Front. Endocrinol. 2017, 8, 261. [Google Scholar] [CrossRef] [PubMed]
- Kohno, T.; Konno, T.; Kikuchi, S.; Kondoh, M.; Kojima, T. Translocation of LSR from Tricellular Corners Causes Macropinocytosis at Cell-Cell Interface as a Trigger for Breaking out of Contact Inhibition. FASEB J. 2021, 35, e21742. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Jiang, W.; Kim, B.Y.S.; Zhang, C.C.; Fu, Y.-X.; Weissman, I.L. Phagocytosis Checkpoints as New Targets for Cancer Immunotherapy. Nat. Rev. Cancer 2019, 19, 568–586. [Google Scholar] [CrossRef]
- Uribe-Querol, E.; Rosales, C. Phagocytosis: Our Current Understanding of a Universal Biological Process. Front. Immunol. 2020, 11, 1066. [Google Scholar] [CrossRef] [PubMed]
- Schlam, D.; Bagshaw, R.D.; Freeman, S.A.; Collins, R.F.; Pawson, T.; Fairn, G.D.; Grinstein, S. Phosphoinositide 3-Kinase Enables Phagocytosis of Large Particles by Terminating Actin Assembly through Rac/Cdc42 GTPase-Activating Proteins. Nat. Commun. 2015, 6, 8623. [Google Scholar] [CrossRef]
- Bohdanowicz, M.; Grinstein, S. Role of Phospholipids in Endocytosis, Phagocytosis, and Macropinocytosis. Physiol. Rev. 2013, 93, 69–106. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.G. Mycobacterium Tuberculosis: Here Today, and Here Tomorrow. Nat. Rev. Mol. Cell Biol. 2001, 2, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Matlung, H.L.; Szilagyi, K.; Barclay, N.A.; van den Berg, T.K. The CD47-SIRPα Signaling Axis as an Innate Immune Checkpoint in Cancer. Immunol. Rev. 2017, 276, 145–164. [Google Scholar] [CrossRef] [PubMed]
- Logtenberg, M.E.W.; Scheeren, F.A.; Schumacher, T.N. The CD47-SIRPα Immune Checkpoint. Immunity 2020, 52, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Ni, T.; Wang, J.; Liu, Y.; Fan, Q.; Wang, Y.; Huang, T.; Chu, Y.; Sun, X.; Wang, Y. CD47 Blockade Inhibits Tumor Progression through Promoting Phagocytosis of Tumor Cells by M2 Polarized Macrophages in Endometrial Cancer. J. Immunol. Res. 2018, 2018, 6156757. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; Kavaliauskiene, S.; Skotland, T. Clathrin-Independent Endocytosis: An Increasing Degree of Complexity. Histochem. Cell Biol. 2018, 150, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Young, A. Structural Insights into the Clathrin Coat. Semin. Cell Dev. Biol. 2007, 18, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Rothberg, K.G.; Heuser, J.E.; Donzell, W.C.; Ying, Y.-S.; Glenney, J.R.; Anderson, R.G.W. Caveolin, a Protein Component of Caveolae Membrane Coats. Cell 1992, 68, 673–682. [Google Scholar] [CrossRef]
- Rennick, J.J.; Johnston, A.P.R.; Parton, R.G. Key Principles and Methods for Studying the Endocytosis of Biological and Nanoparticle Therapeutics. Nat. Nanotechnol. 2021, 16, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Kerr, M.C.; Teasdale, R.D. Defining Macropinocytosis. Traffic 2009, 10, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.P.; Mintern, J.D.; Gleeson, P.A. Macropinocytosis in Different Cell Types: Similarities and Differences. Membranes 2020, 10, 177. [Google Scholar] [CrossRef] [PubMed]
- Buckley, C.M.; King, J.S. Drinking Problems: Mechanisms of Macropinosome Formation and Maturation. FEBS J. 2017, 284, 3778–3790. [Google Scholar] [CrossRef] [PubMed]
- Saito, N.; Sawai, S. Three-Dimensional Morphodynamic Simulations of Macropinocytic Cups. iScience 2021, 24, 103087. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.; Helenius, A. Virus Entry by Macropinocytosis. Nat. Cell Biol. 2009, 11, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.P.; Gleeson, P.A. Macropinocytosis: An Endocytic Pathway for Internalising Large Gulps. Immunol. Cell Biol. 2011, 89, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Paluck, A.; Osan, J.; Hollingsworth, L.; Talukdar, S.N.; Saegh, A.A.; Mehedi, M. Role of ARP2/3 Complex-Driven Actin Polymerization in RSV Infection. Pathogens 2021, 11, 26. [Google Scholar] [CrossRef] [PubMed]
- Norbury, C.C. Drinking a Lot Is Good for Dendritic Cells. Immunology 2006, 117, 443–451. [Google Scholar] [CrossRef] [PubMed]
- García-Pérez, B.E.; de la Cruz-López, J.J.; Castañeda-Sánchez, J.I.; Muñóz-Duarte, A.R.; Hernández-Pérez, A.D.; Villegas-Castrejón, H.; García-Latorre, E.; Caamal-Ley, A.; Luna-Herrera, J. Macropinocytosis Is Responsible for the Uptake of Pathogenic and Non-Pathogenic Mycobacteria by B Lymphocytes (Raji Cells). BMC Microbiol. 2012, 12, 246. [Google Scholar] [CrossRef]
- Charpentier, J.C.; Chen, D.; Lapinski, P.E.; Turner, J.; Grigorova, I.; Swanson, J.A.; King, P.D. Macropinocytosis Drives T Cell Growth by Sustaining the Activation of MTORC1. Nat. Commun. 2020, 11, 180. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Noss, E.H.; Hsu, V.W.; Brenner, M.B. Integrins Traffic Rapidly via Circular Dorsal Ruffles and Macropinocytosis during Stimulated Cell Migration. J. Cell Biol. 2011, 193, 61–70. [Google Scholar] [CrossRef]
- Mettlen, M.; Platek, A.; van der Smissen, P.; Carpentier, S.; Amyere, M.; Lanzetti, L.; de Diesbach, P.; Tyteca, D.; Courtoy, P.J. Src Triggers Circular Ruffling and Macropinocytosis at the Apical Surface of Polarized MDCK Cells. Traffic 2006, 7, 589–603. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Pacitto, R.; Sesi, C.; Kotula, L.; Swanson, J.A. Dorsal Ruffles Enhance Activation of Akt by Growth Factors. J. Cell Sci. 2018, 131, jcs220517. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Pacitto, R.; Inoki, K.; Swanson, J. Macropinocytosis, MTORC1 and Cellular Growth Control. Cell. Mol. Life Sci. 2018, 75, 1227–1239. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Zhu, T.; Guan, K.-L.L. TSC2 Mediates Cellular Energy Response to Control Cell Growth and Survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Condon, K.J.; Sabatini, D.M. Nutrient Regulation of MTORC1 at a Glance. J. Cell Sci. 2019, 132, jcs222570. [Google Scholar] [CrossRef] [PubMed]
- Basagiannis, D.; Zografou, S.; Murphy, C.; Fotsis, T.; Morbidelli, L.; Ziche, M.; Bleck, C.; Mercer, J.; Christoforidis, S. VEGF Induces Signalling and Angiogenesis by Directing VEGFR2 Internalisation through Macropinocytosis. J. Cell Sci. 2016, 129, 4091–4104. [Google Scholar] [CrossRef]
- Elfenbein, A.; Lanahan, A.; Zhou, T.X.; Yamasaki, A.; Tkachenko, E.; Matsuda, M.; Simons, M. Syndecan 4 Regulates FGFR1 Signaling in Endothelial Cells by Directing Macropinocytosis. Sci. Signal 2012, 5, ra36. [Google Scholar] [CrossRef]
- Valdivia, A.; Goicoechea, S.M.; Awadia, S.; Zinn, A.; Garcia-Mata, R. Regulation of Circular Dorsal Ruffles, Macropinocytosis, and Cell Migration by RhoG and Its Exchange Factor, Trio. Mol. Biol. Cell 2017, 28, 1768–1781. [Google Scholar] [CrossRef] [PubMed]
- Zihni, C.; Balda, M.S.; Matter, K. Signalling at Tight Junctions during Epithelial Differentiation and Microbial Pathogenesis. J. Cell Sci. 2014, 127, 3401–3413. [Google Scholar] [CrossRef] [PubMed]
- Takai, Y.; Nakanishi, H. Nectin and Afadin: Novel Organizers of Intercellular Junctions. J. Cell Sci. 2003, 116, 17–27. [Google Scholar] [CrossRef]
- Zihni, C.; Mills, C.; Matter, K.; Balda, M.S. Tight Junctions: From Simple Barriers to Multifunctional Molecular Gates. Nat. Rev. Mol. Cell Biol. 2016, 17, 564–580. [Google Scholar] [CrossRef]
- Citi, S. The Mechanobiology of Tight Junctions. Biophys. Rev. 2019, 11, 783–793. [Google Scholar] [CrossRef]
- Nishimura, T.; Takeichi, M. Shroom3-Mediated Recruitment of Rho Kinases to the Apical Cell Junctions Regulates Epithelial and Neuroepithelial Planar Remodeling. Development 2008, 135, 1493–1502. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.W.; Gerstenzang, E.; Ossipova, O.; Sokol, S.Y. Lulu Regulates Shroom-Induced Apical Constriction during Neural Tube Closure. PLoS ONE 2013, 8, e81854. [Google Scholar] [CrossRef] [PubMed]
- Hurd, T.W.; Gao, L.; Roh, M.H.; Macara, I.G.; Margolis, B. Direct Interaction of Two Polarity Complexes Implicated in Epthelial Tight Junction Assembly. Nat. Cell Biol. 2003, 5, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Higashi, T.; Tokuda, S.; Kitajiri, S.; Masuda, S.; Nakamura, H.; Oda, Y.; Furuse, M. Analysis of the “angulin” Proteins LSR, ILDR1 and ILDR2--Tricellulin Recruitment, Epithelial Barrier Function and Implication in Deafness Pathogenesis. J. Cell Sci. 2013, 126, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Tsukita, S.; Yamazaki, Y.; Katsuno, T.; Tamura, A.; Tsukita, S. Tight Junction-Based Epithelial Microenvironment and Cell Proliferation. Oncogene 2008, 27, 6930–6938. [Google Scholar] [CrossRef] [PubMed]
- Yen, F.T.; Roitel, O.; Bonnard, L.; Notet, V.; Pratte, D.; Stenger, C.; Magueur, E.; Bihain, B.E. Lipolysis Stimulated Lipoprotein Receptor: A Novel Molecular Link between Hyperlipidemia, Weight Gain, and Atherosclerosis in Mice. J. Biol. Chem. 2008, 283, 25650–25659. [Google Scholar] [CrossRef] [PubMed]
- Stenger, C.; Hanse, M.; Pratte, D.; Mbala, M.L.; Akbar, S.; Koziel, V.; Escanyé, M.C.; Kriem, B.; Malaplate-Armand, C.; Olivier, J.L.; et al. Up-Regulation of Hepatic Lipolysis Stimulated Lipoprotein Receptor by Leptin: A Potential Lever for Controlling Lipid Clearance during the Postprandial Phase. FASEB J. 2010, 24, 4218–4228. [Google Scholar] [CrossRef] [PubMed]
- Kohno, T.; Kikuchi, S.; Ninomiya, T.; Kojima, T. The Bicellular Tensile Force Sorts the Localization of LSRs in Bicellular and Tricellular Junctions. Ann. N. Y. Acad. Sci. 2017, 1397, 185–194. [Google Scholar] [CrossRef]
- Kohno, T.; Konno, T.; Kojima, T. Role of Tricellular Tight Junction Protein Lipolysis-Stimulated Lipoprotein Receptor (LSR) in Cancer Cells. Int. J. Mol. Sci. 2019, 20, 3555. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.A.; Jiang, W.G. Loss of Tight Junction Barrier Function and Its Role in Cancer Metastasis. Biochim. Biophys. Acta Biomembr. 2009, 1788, 872–891. [Google Scholar] [CrossRef] [PubMed]
- Guillot, C.; Lecuit, T. Mechanics of Epithelial Tissue Homeostasis and Morphogenesis. Science (1979) 2013, 340, 1185–1189. [Google Scholar] [CrossRef]
- Loza, A.J.; Koride, S.; Schimizzi, G.V.; Li, B.; Sun, S.X.; Longmore, G.D. Cell Density and Actomyosin Contractility Control the Organization of Migrating Collectives within an Epithelium. Mol. Biol. Cell 2016, 27, 3459–3470. [Google Scholar] [CrossRef] [PubMed]
- Gumbiner, B.M.; Kim, N.G. The Hippo-YAP Signaling Pathway and Contact Inhibition of Growth. J. Cell Sci. 2014, 127, 709–717. [Google Scholar] [CrossRef]
- Mendonsa, A.M.; Na, T.-Y.; Gumbiner, B.M. E-Cadherin in Contact Inhibition and Cancer. Oncogene 2018, 37, 4769–4780. [Google Scholar] [CrossRef] [PubMed]
- McClatchey, A.I.; Yap, A.S. Contact Inhibition (of Proliferation) Redux. Curr. Opin. Cell Biol. 2012, 24, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.A.; Nelson, W.J.; Chavez, N. Cell–Cell Junctions Organize Structural and Signaling Networks. Cold Spring Harb. Perspect. Biol. 2018, 10, a029181. [Google Scholar] [CrossRef]
- Eisenhoffer, G.T.; Rosenblatt, J. Bringing Balance by Force: Live Cell Extrusion Controls Epithelial Cell Numbers. Trends Cell Biol. 2013, 23, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Stramer, B.; Mayor, R. Mechanisms and in Vivo Functions of Contact Inhibition of Locomotion. Nat. Rev. Mol. Cell Biol. 2017, 18, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Buckley, C.E.; St Johnston, D. Apical–Basal Polarity and the Control of Epithelial Form and Function. Nat. Rev. Mol. Cell Biol. 2022, 23, 559–577. [Google Scholar] [CrossRef] [PubMed]
- Haigis, K.M. KRAS Alleles: The Devil Is in the Detail. Trends Cancer 2017, 3, 686–697. [Google Scholar] [CrossRef]
- Li, S.; Balmain, A.; Counter, C.M. A Model for RAS Mutation Patterns in Cancers: Finding the Sweet Spot. Nat. Rev. Cancer 2018, 18, 767–777. [Google Scholar] [CrossRef]
- Byron, S.A.; Gartside, M.; Powell, M.A.; Wellens, C.L.; Gao, F.; Mutch, D.G.; Goodfellow, P.J.; Pollock, P.M. FGFR2 Point Mutations in 466 Endometrioid Endometrial Tumors: Relationship with MSI, KRAS, PIK3CA, CTNNB1 Mutations and Clinicopathological Features. PLoS ONE 2012, 7, e30801. [Google Scholar] [CrossRef]
- Inoue, S.; Hirota, Y.; Ueno, T.; Fukui, Y.; Yoshida, E.; Hayashi, T.; Kojima, S.; Takeyama, R.; Hashimoto, T.; Kiyono, T.; et al. Uterine Adenomyosis Is an Oligoclonal Disorder Associated with KRAS Mutations. Nat. Commun. 2019, 10, 5785. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Nishida, M.; Miyazaki, Y.; Sugita, M.; Arai, Y.; Oki, A.; Kono, K.; Tsunoda, H.; Kasahara, K.; Kubo, T. Establishment of a Cisplatin-Resistant New Human Endometrial Adenocarcinoma Cell Line, Sawano Cells. Hum. Cell 1995, 8, 67–72. [Google Scholar] [PubMed]
- Ray, I.; Meira, L.B.; Michael, A.; Ellis, P.E. Adipocytokines and Disease Progression in Endometrial Cancer: A Systematic Review. Cancer Metastasis Rev. 2022, 41, 211–242. [Google Scholar] [CrossRef]
- Konno, T.; Takano, K.; Kaneko, Y.; Kakuki, T.; Nomura, K.; Yajima, R.; Kakiuchi, A.; Kohno, T.; Himi, T.; Kojima, T. Guanylate Binding Protein-1-Mediated Epithelial Barrier in Human Salivary Gland Duct Epithelium. Exp. Cell Res. 2018, 371, 31–41. [Google Scholar] [CrossRef]
- Kyuno, T.; Kyuno, D.; Kohno, T.; Konno, T.; Kikuchi, S.; Arimoto, C.; Yamaguchi, H.; Imamura, M.; Kimura, Y.; Kondoh, M.; et al. Tricellular Tight Junction Protein LSR/Angulin-1 Contributes to the Epithelial Barrier and Malignancy in Human Pancreatic Cancer Cell Line. Histochem. Cell Biol. 2020, 153, 5–16. [Google Scholar] [CrossRef]
- Kodera, Y.; Kohno, T.; Konno, T.; Arai, W.; Tsujiwaki, M.; Shindo, Y.; Chiba, H.; Miyakawa, M.; Tanaka, H.; Sakuma, Y.; et al. HMGB1 Enhances Epithelial Permeability via P63/TGF-β Signaling in Lung and Terminal Bronchial Epithelial Cells. Tissue Barriers 2020, 8, 1805997. [Google Scholar] [CrossRef]
- Kodera, Y.; Chiba, H.; Konno, T.; Kohno, T.; Takahashi, H.; Kojima, T. HMGB1-Downregulated Angulin-1/LSR Induces Epithelial Barrier Disruption via Claudin-2 and Cellular Metabolism via AMPK in Airway Epithelial Calu-3 Cells. Biochem. Biophys. Res. Commun. 2020, 527, 553–560. [Google Scholar] [CrossRef]
- Miyakawa, M.; Konno, T.; Kohno, T.; Kikuchi, S.; Tanaka, H.; Kojima, T. Increase in Epithelial Permeability and Cell Metabolism by High Mobility Group Box 1, Inflammatory Cytokines and TPEN in Caco-2 Cells as a Novel Model of Inflammatory Bowel Disease. Int. J. Mol. Sci. 2020, 21, 8434. [Google Scholar] [CrossRef]
- Shindo, Y.; Arai, W.; Konno, T.; Kohno, T.; Kodera, Y.; Chiba, H.; Miyajima, M.; Sakuma, Y.; Watanabe, A.; Kojima, T. Effects of Histone Deacetylase Inhibitors Tricostatin A and Quisinostat on Tight Junction Proteins of Human Lung Adenocarcinoma A549 Cells and Normal Lung Epithelial Cells. Histochem. Cell Biol. 2021, 155, 637–653. [Google Scholar] [CrossRef]
- Ohwada, K.; Konno, T.; Kohno, T.; Nakano, M.; Ohkuni, T.; Miyata, R.; Kakuki, T.; Kondoh, M.; Takano, K.; Kojima, T. Effects of HMGB1 on Tricellular Tight Junctions via TGF-β Signaling in Human Nasal Epithelial Cells. Int. J. Mol. Sci. 2021, 22, 8390. [Google Scholar] [CrossRef] [PubMed]
- Kojima, T.; Shindo, Y.; Konno, T.; Kodera, Y.; Arai, W.; Miyakawa, M.; Ohwada, K.; Tanaka, H.; Tsujiwaki, M.; Sakuma, Y.; et al. Dysfunction of Epithelial Permeability Barrier Induced by HMGB1 in 2.5D Cultures of Human Epithelial Cells. Tissue Barriers 2022, 10, 1972760. [Google Scholar] [CrossRef]
- Nezhat, F.; Datta, M.S.; Hanson, V.; Pejovic, T.; Nezhat, C.; Nezhat, C. The Relationship of Endometriosis and Ovarian Malignancy: A Review. Fertil. Steril. 2008, 90, 1559–1570. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.-J.; Yang, H.-L.; Shao, J.; Mei, J.; Chang, K.-K.; Zhu, R.; Li, M.-Q. Anti-Inflammatory Cytokines in Endometriosis. Cell. Mol. Life Sci. 2019, 76, 2111–2132. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Hong, W. The Emerging Role of the Hippo Pathway in Cell Contact Inhibition, Organ Size Control, and Cancer Development in Mammals. Cancer Cell 2008, 13, 188–192. [Google Scholar] [CrossRef]
- Halder, G.; Dupont, S.; Piccolo, S. Transduction of Mechanical and Cytoskeletal Cues by YAP and TAZ. Nat. Rev. Mol. Cell Biol. 2012, 13, 591–600. [Google Scholar] [CrossRef]
- Shimada, H.; Abe, S.; Kohno, T.; Satohisa, S.; Konno, T.; Takahashi, S.; Hatakeyama, T.; Arimoto, C.; Kakuki, T.; Kaneko, Y.; et al. Loss of Tricellular Tight Junction Protein LSR Promotes Cell Invasion and Migration via Upregulation of TEAD1/AREG in Human Endometrial Cancer. Sci. Rep. 2017, 7, 37049. [Google Scholar] [CrossRef]
- Konno, T.; Kohno, T.; Okada, T.; Shimada, H.; Satohisa, S.; Kikuchi, S.; Saito, T.; Kojima, T. ASPP2 Suppression Promotes Malignancy via LSR and YAP in Human Endometrial Cancer. Histochem. Cell Biol. 2020, 154, 197–213. [Google Scholar] [CrossRef]
- Bao, Y.; Nakagawa, K.; Yang, Z.; Ikeda, M.; Withanage, K.; Ishigami-Yuasa, M.; Okuno, Y.; Hata, S.; Nishina, H.; Hata, Y. A Cell-Based Assay to Screen Stimulators of the Hippo Pathway Reveals the Inhibitory Effect of Dobutamine on the YAP-Dependent Gene Transcription. J. Biochem. 2011, 150, 199–208. [Google Scholar] [CrossRef]
- Zheng, H.X.; Wu, L.N.; Xiao, H.; Du, Q.; Liang, J.F. Inhibitory Effects of Dobutamine on Human Gastric Adenocarcinoma. World J. Gastroenterol. 2014, 20, 17092–17099. [Google Scholar] [CrossRef]
- Paul, S.; Xie, S.; Yao, X.; Dey, A. Transcriptional Regulation of the Hippo Pathway: Current Understanding and Insights from Single-Cell Technologies. Cells 2022, 11, 2225. [Google Scholar] [CrossRef] [PubMed]
- Someya, M.; Kojima, T.; Ogawa, M.; Ninomiya, T.; Nomura, K.; Takasawa, A.; Murata, M.; Tanaka, S.; Saito, T.; Sawada, N. Regulation of Tight Junctions by Sex Hormones in Normal Human Endometrial Epithelial Cells and Uterus Cancer Cell Line Sawano. Cell Tissue Res. 2013, 354, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Shimada, H.; Satohisa, S.; Kohno, T.; Takahashi, S.; Hatakeyama, T.; Konno, T.; Tsujiwaki, M.; Saito, T.; Kojima, T. The Roles of Tricellular Tight Junction Protein Lipolysis-Stimulated Lipoprotein Receptor in Malignancy of Human Endometrial Cancer Cells. Oncotarget 2016, 7, 27735–27752. [Google Scholar] [CrossRef] [PubMed]
- Grund, S.; Grümmer, R. Direct Cell-Cell Interactions in the Endometrium and in Endometrial Pathophysiology. Int. J. Mol. Sci. 2018, 19, 2227. [Google Scholar] [CrossRef] [PubMed]
- Leech, A.O.; Cruz, R.G.B.; Hill, A.D.K.; Hopkins, A.M. Paradigms Lost—An Emerging Role for over-Expression of Tight Junction Adhesion Proteins in Cancer Pathogenesis. Ann. Transl. Med. 2015, 3, 184. [Google Scholar] [CrossRef] [PubMed]
- Shimada, H.; Satohisa, S.; Kohno, T.; Konno, T.; Takano, K.; Takahashi, S.; Hatakeyama, T.; Arimoto, C.; Saito, T.; Kojima, T. Downregulation of Lipolysis-Stimulated Lipoprotein Receptor Promotes Cell Invasion via Claudin-1-Mediated Matrix Metalloproteinases in Human Endometrial Cancer. Oncol. Lett. 2017, 14, 6776–6782. [Google Scholar] [CrossRef][Green Version]
- Wang, H.B.; Wang, P.Y.; Wang, X.; Wan, Y.L.; Liu, Y.C. Butyrate Enhances Intestinal Epithelial Barrier Function via Up-Regulation of Tight Junction Protein Claudin-1 Transcription. Dig. Dis. Sci. 2012, 57, 3126–3135. [Google Scholar] [CrossRef]
- Dufresne, J.; Cyr, D.G. Activation of an SP Binding Site Is Crucial for the Expression of Claudin 1 in Rat Epididymal Principal Cells. Biol. Reprod. 2007, 76, 825–832. [Google Scholar] [CrossRef][Green Version]
- Honda, H.; Pazin, M.J.; Ji, H.; Wernyj, R.P.; Morin, P.J. Crucial Roles of Sp1 and Epigenetic Modifications in the Regulation of the Cldn4 Promoter in Ovarian Cancer Cells. J. Biol. Chem. 2006, 281, 21433–21444. [Google Scholar] [CrossRef]
- Luk, J.M.; Tong, M.K.; Mok, B.W.; Tam, P.C.; Yeung, W.S.B.; Lee, K.F. Sp1 Site Is Crucial for the Mouse Claudin-19 Gene Expression in the Kidney Cells. FEBS Lett. 2004, 578, 251–256. [Google Scholar] [CrossRef][Green Version]
- Oku, N.; Sasabe, E.; Ueta, E.; Yamamoto, T.; Osaki, T. Tight Junction Protein Claudin-1 Enhances the Invasive Activity of Oral Squamous Cell Carcinoma Cells by Promoting Cleavage of Laminin-5 Γ2 Chain via Matrix Metalloproteinase (MMP)-2 and Membrane-Type MMP-1. Cancer Res. 2006, 66, 5251–5257. [Google Scholar] [CrossRef] [PubMed]
- van Itallie, C.M.; Fanning, A.S.; Anderson, J.M.; Boireau, S.; Samuel, M.S.; Pannequin, J.; Ryan, J.L.; Choquet, A.; Chapuis, H.; Rebillard, X.; et al. Claudin-1 Regulates Cellular Transformation and Metastatic Behavior in Colon Cancer. J. Clin. Investig. 2005, 115, 1765–1776. [Google Scholar] [CrossRef]
- Castro-Castro, A.; Marchesin, V.; Monteiro, P.; Lodillinsky, C.; Rossé, C.; Chavrier, P. Cellular and Molecular Mechanisms of MT1-MMP-Dependent Cancer Cell Invasion. Annu. Rev. Cell Dev. Biol. 2016, 32, 555–576. [Google Scholar] [CrossRef]
- Brown, G.T.; Murray, G.I. Current Mechanistic Insights into the Roles of Matrix Metalloproteinases in Tumour Invasion and Metastasis. J. Pathol. 2015, 237, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Gaide Chevronnay, H.P.; Selvais, C.; Emonard, H.; Galant, C.; Marbaix, E.; Henriet, P. Regulation of Matrix Metalloproteinases Activity Studied in Human Endometrium as a Paradigm of Cyclic Tissue Breakdown and Regeneration. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2012, 1824, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Pandit, A.; das Mahapatra, P.; Saha, P.; Srivastava, A.K.; Swarnakar, S. Interleukin-1β Activated C-FOS Transcription Factor Binds Preferentially to a Specific Allele of the Matrix Metalloproteinase-13 Promoter and Increases Susceptibility to Endometriosis. J. Cell Physiol. 2022, 237, 3095–3108. [Google Scholar] [CrossRef]
- Mwaura, A.N.; Riaz, M.A.; Maoga, J.B.; Mecha, E.; Omwandho, C.O.A.; Scheiner-Bobis, G.; Meinhold-Heerlein, I.; Konrad, L. Role of Betaglycan in TGF-β Signaling and Wound Healing in Human Endometriotic Epithelial Cells and in Endometriosis. Biology 2022, 11, 513. [Google Scholar] [CrossRef]
- Paz, H.; Pathak, N.; Yang, J. Invading One Step at a Time: The Role of Invadopodia in Tumor Metastasis. Oncogene 2014, 33, 4193–4202. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.W.; SEO, S.T.; IM, M.A.; WON, H.-R.; LIU, L.; OH, C.; JIN, Y.L.; PIAO, Y.; KIM, H.J.; KIM, J.T.; et al. Claudin-1 Mediates Progression by Regulating EMT through AMPK/TGF-β Signaling in Head and Neck Squamous Cell Carcinoma. Transl. Res. 2022, 247, 58–78. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Pascal, L.E.; Li, F.; Chen, W.; Dhir, R.; Balasubramani, G.K.; DeFranco, D.B.; Yoshimura, N.; He, D.; Wang, Z. Tight Junction Protein Claudin-1 Is Downregulated by TGF-β1 via MEK Signaling in Benign Prostatic Epithelial Cells. Prostate 2020, 80, 1203–1215. [Google Scholar] [CrossRef] [PubMed]
- Stamatovic, S.M.; Johnson, A.M.; Sladojevic, N.; Keep, R.F.; Andjelkovic, A.V. Endocytosis of Tight Junction Proteins and the Regulation of Degradation and Recycling. Ann. N. Y. Acad. Sci. 2017, 1397, 54–65. [Google Scholar] [CrossRef]
- Bruewer, M.; Utech, M.; Ivanov, A.I.; Hopkins, A.M.; Parkos, C.A.; Nusrat, A. Interferon-γ Induces Internalization of Epithelial Tight Junction Proteins via a Macropinocytosis-like Process. FASEB J. 2005, 19, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.M.; Walsh, S.V.; Verkade, P.; Boquet, P.; Nusrat, A. Constitutive Activation of Rho Proteins by CNF-1 Influences Tight Junction Structure and Epithelial Barrier Function. J. Cell Sci. 2003, 116, 725–742. [Google Scholar] [CrossRef]
- Shen, L.; Turner, J.R. Actin Depolymerization Disrupts Tight Junctions via Caveolae-Mediated Endocytosis. Mol. Biol. Cell 2005, 16, 3919–3936. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, M.; Kubo, A.; Furuse, M.; Tsukita, S. A Peculiar Internalization of Claudins, Tight Junction-Specific Adhesion Molecules, during the Intercellular Movement of Epithelial Cells. J. Cell Sci. 2004, 117, 1247–1257. [Google Scholar] [CrossRef]
- Yamaki, T.; Ohtake, K.; Ichikawa, K.; Uchida, M.; Uchida, H.; Ohshima, S.; Juni, K.; Kobayashi, J.; Morimoto, Y.; Natsume, H. Poly-l-Arginine-Induced Internalization of Tight Junction Proteins Increases the Paracellular Permeability of the Caco-2 Cell Monolayer to Hydrophilic Macromolecules. Biol. Pharm. Bull. 2013, 36, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Felinski, E.A.; Antonetti, D.A. Occludin Phosphorylation and Ubiquitination Regulate Tight Junction Trafficking and Vascular Endothelial Growth Factor-Induced Permeability. J. Biol. Chem. 2009, 284, 21036–21046. [Google Scholar] [CrossRef] [PubMed]
- Krug, S.M.; Hayaishi, T.; Iguchi, D.; Watari, A.; Takahashi, A.; Fromm, M.; Nagahama, M.; Takeda, H.; Okada, Y.; Sawasaki, T.; et al. Angubindin-1, a Novel Paracellular Absorption Enhancer Acting at the Tricellular Tight Junction. J. Control. Release 2017, 260, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Pernet-Gallay, K.; Jouneau, P.H.; Bertrand, A.; Delaroche, J.; Farion, R.; Rémy, C.; Barbier, E.L. Vascular Permeability in the RG2 Glioma Model Can Be Mediated by Macropinocytosis and Be Independent of the Opening of the Tight Junction. J. Cereb. Blood Flow Metab. 2017, 37, 1264–1275. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, M.; Takehara, M.; Kobayashi, K. Interaction of Clostridium Perfringens Iota Toxin and Lipolysis-Stimulated Lipoprotein Receptor (LSR). Toxins 2018, 10, 405. [Google Scholar] [CrossRef] [PubMed]
- Sohet, F.; Lin, C.; Munji, R.N.; Lee, S.Y.; Ruderisch, N.; Soung, A.; Arnold, T.D.; Derugin, N.; Vexler, Z.S.; Yen, F.T.; et al. LSR/Angulin-1 Is a Tricellular Tight Junction Protein Involved in Blood-Brain Barrier Formation. J. Cell Biol. 2015, 208, 703–711. [Google Scholar] [CrossRef]
- Zeniya, S.; Kuwahara, H.; Daizo, K.; Watari, A.; Kondoh, M.; Yoshida-Tanaka, K.; Kaburagi, H.; Asada, K.; Nagata, T.; Nagahama, M.; et al. Angubindin-1 Opens the Blood–Brain Barrier in Vivo for Delivery of Antisense Oligonucleotide to the Central Nervous System. J. Control. Release 2018, 283, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Konno, T.; Kohno, T.; Kikuchi, S.; Shimada, H.; Satohisa, S.; Saito, T.; Kondoh, M.; Kojima, T. Epithelial Barrier Dysfunction and Cell Migration Induction via JNK/Cofilin/Actin by Angubindin-1. Tissue Barriers 2020, 8, 1695475. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Jacobson, K.; Schaller, M.D. MAP Kinases and Cell Migration. J. Cell Sci. 2004, 117, 4619–4628. [Google Scholar] [CrossRef]
- Magi, S.; Tashiro, E.; Imoto, M. A Chemical Genomic Study Identifying Diversity in Cell Migration Signaling in Cancer Cells. Sci. Rep. 2012, 2, 823. [Google Scholar] [CrossRef]
- Ichimizu, S.; Watanabe, H.; Maeda, H.; Hamasaki, K.; Ikegami, K.; Chuang, V.T.G.; Kinoshita, R.; Nishida, K.; Shimizu, T.; Ishima, Y.; et al. Cell-Penetrating Mechanism of Intracellular Targeting Albumin: Contribution of Macropinocytosis Induction and Endosomal Escape. J. Control. Release 2019, 304, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Bretou, M.; Sáez, P.J.; Sanséau, D.; Maurin, M.; Lankar, D.; Chabaud, M.; Spampanato, C.; Malbec, O.; Barbier, L.; Muallem, S.; et al. Lysosome Signaling Controls the Migration of Dendritic Cells. Sci. Immunol. 2017, 2, eaak9573. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Manzanares, M.; Ma, X.; Adelstein, R.S.; Horwitz, A.R. Non-Muscle Myosin II Takes Centre Stage in Cell Adhesion and Migration. Nat. Rev. Mol. Cell Biol. 2009, 10, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Masereel, B. An Overview of Inhibitors of Na+/H+ Exchanger. Eur. J. Med. Chem. 2003, 38, 547–554. [Google Scholar] [CrossRef]
- Fliegel, L. Structural and Functional Changes in the Na+ /H+ Exchanger Isoform 1, Induced by Erk1/2 Phosphorylation. Int. J. Mol. Sci. 2019, 20, 2378. [Google Scholar] [CrossRef] [PubMed]
- Donowitz, M.; Ming Tse, C.; Fuster, D. SLC9/NHE Gene Family, a Plasma Membrane and Organellar Family of Na+/H+ Exchangers. Mol. Aspects Med. 2013, 34, 236–251. [Google Scholar] [CrossRef] [PubMed]
- Koivusalo, M.; Welch, C.; Hayashi, H.; Scott, C.C.; Kim, M.; Alexander, T.; Touret, N.; Hahn, K.M.; Grinstein, S. Amiloride Inhibits Macropinocytosis by Lowering Submembranous PH and Preventing Rac1 and Cdc42 Signaling. J. Cell Biol. 2010, 188, 547–563. [Google Scholar] [CrossRef]
- Rotty, J.D.; Wu, C.; Bear, J.E. New Insights into the Regulation and Cellular Functions of the ARP2/3 Complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Mehedi, M.; McCarty, T.; Martin, S.E.; le Nouën, C.; Buehler, E.; Chen, Y.-C.; Smelkinson, M.; Ganesan, S.; Fischer, E.R.; Brock, L.G.; et al. Actin-Related Protein 2 (ARP2) and Virus-Induced Filopodia Facilitate Human Respiratory Syncytial Virus Spread. PLoS Pathog. 2016, 12, e1006062. [Google Scholar] [CrossRef] [PubMed]
- Denker, S.P.; Huang, D.C.; Orlowski, J.; Furthmayr, H.; Barber, D.L. Direct Binding of the Na-H Exchanger NHE1 to ERM Proteins Regulates the Cortical Cytoskeleton and Cell Shape Independently of H+ Translocation. Mol. Cell 2000, 6, 1425–1436. [Google Scholar] [CrossRef]
- Tominaga, T.; Ishizaki, T.; Narumiya, S.; Barber, D.L. P160ROCK Mediates RhoA Activation of Na-H Exchange. EMBO J. 1998, 17, 4712–4722. [Google Scholar] [CrossRef]
- Hendus-Altenburger, R.; Pedraz-Cuesta, E.; Olesen, C.W.; Papaleo, E.; Schnell, J.A.; Hopper, J.T.S.; Robinson, C.V.; Pedersen, S.F.; Kragelund, B.B. The Human Na+/H+ Exchanger 1 Is a Membrane Scaffold Protein for Extracellular Signal-Regulated Kinase 2. BMC Biol. 2016, 14, 31. [Google Scholar] [CrossRef]
- Amith, S.R.; Wilkinson, J.M.; Baksh, S.; Fliegel, L. The Na+/H+ Exchanger (NHE1) as a Novel Co-Adjuvant Target in Paclitaxel Therapy of Triple-Negative Breast Cancer Cells. Oncotarget 2015, 6, 1262–1275. [Google Scholar] [CrossRef]
- Kaminota, T.; Yano, H.; Shiota, K.; Nomura, N.; Yaguchi, H.; Kirino, Y.; Ohara, K.; Tetsumura, I.; Sanada, T.; Ugumori, T.; et al. Elevated Na+/H+ Exchanger-1 Expression Enhances the Metastatic Collective Migration of Head and Neck Squamous Cell Carcinoma Cells. Biochem. Biophys. Res. Commun. 2017, 486, 101–107. [Google Scholar] [CrossRef]
- Guan, X.; Luo, L.; Begum, G.; Kohanbash, G.; Song, Q.; Rao, A.; Amankulor, N.; Sun, B.; Sun, D.; Jia, W. Elevated Na/H Exchanger 1 (SLC9A1) Emerges as a Marker for Tumorigenesis and Prognosis in Gliomas. J. Exp. Clin. Cancer Res. 2018, 37, 255. [Google Scholar] [CrossRef]
- Yamada, T.; Yoshida, T.; Kawamoto, A.; Mitsuoka, K.; Iwasaki, K.; Tsuge, H. Cryo-EM Structures Reveal Translocational Unfolding in the Clostridial Binary Iota Toxin Complex. Nat. Struct. Mol. Biol. 2020, 27, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Amith, S.R.; Fong, S.; Baksh, S.; Fliegel, L. Na+/H+ Exchange in the Tumour Microenvironment: Does NHE1 Drive Breast Cancer Carcinogenesis? Int. J. Dev. Biol. 2015, 59, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Levine, D.A. Integrated Genomic Characterization of Endometrial Carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Torres-Flores, J.M.; Arias, C.F. Tight Junctions Go Viral! Viruses 2015, 7, 5145–5154. [Google Scholar] [CrossRef]
- Commisso, C.; Davidson, S.M.; Soydaner-Azeloglu, R.G.; Parker, S.J.; Kamphorst, J.J.; Hackett, S.; Grabocka, E.; Nofal, M.; Drebin, J.A.; Thompson, C.B.; et al. Macropinocytosis of Protein Is an Amino Acid Supply Route in Ras-Transformed Cells. Nature 2013, 497, 633–637. [Google Scholar] [CrossRef]
- Davidson, S.M.; Jonas, O.; Keibler, M.A.; Hou, H.W.; Luengo, A.; Mayers, J.R.; Wyckoff, J.; del Rosario, A.M.; Whitman, M.; Chin, C.R.; et al. Direct Evidence for Cancer-Cell-Autonomous Extracellular Protein Catabolism in Pancreatic Tumors. Nat. Med. 2017, 23, 235–241. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kohno, T.; Kojima, T. Atypical Macropinocytosis Contributes to Malignant Progression: A Review of Recent Evidence in Endometrioid Endometrial Cancer Cells. Cancers 2022, 14, 5056. https://doi.org/10.3390/cancers14205056
Kohno T, Kojima T. Atypical Macropinocytosis Contributes to Malignant Progression: A Review of Recent Evidence in Endometrioid Endometrial Cancer Cells. Cancers. 2022; 14(20):5056. https://doi.org/10.3390/cancers14205056
Chicago/Turabian StyleKohno, Takayuki, and Takashi Kojima. 2022. "Atypical Macropinocytosis Contributes to Malignant Progression: A Review of Recent Evidence in Endometrioid Endometrial Cancer Cells" Cancers 14, no. 20: 5056. https://doi.org/10.3390/cancers14205056
APA StyleKohno, T., & Kojima, T. (2022). Atypical Macropinocytosis Contributes to Malignant Progression: A Review of Recent Evidence in Endometrioid Endometrial Cancer Cells. Cancers, 14(20), 5056. https://doi.org/10.3390/cancers14205056