



Autophagy in Hematological Malignancies

 , , ,
, , ,  , and
, and 
Abstract
:Simple Summary
Abstract
1. Introduction
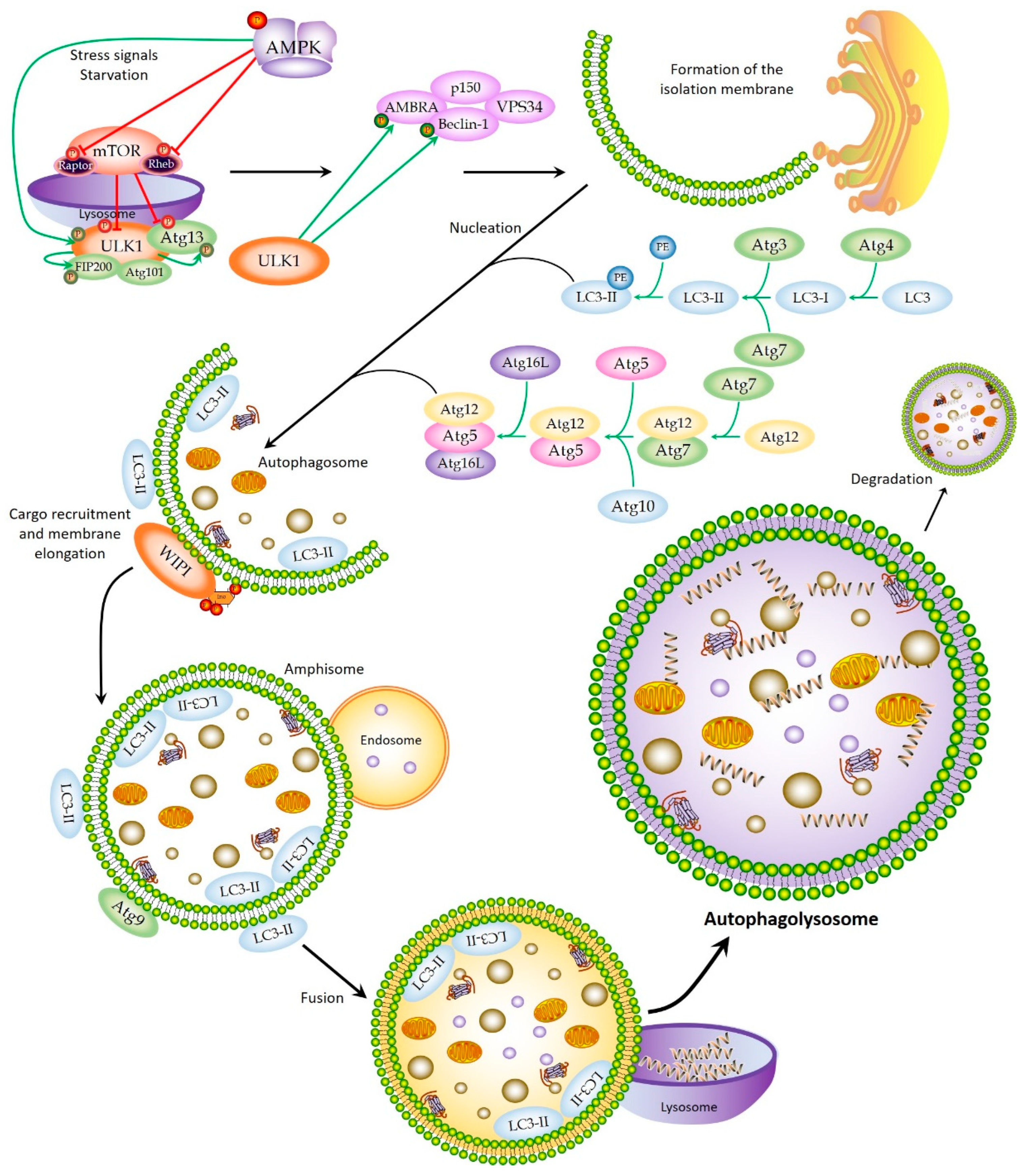
2. Autophagic Machinery and Process
2.1. Initiation
2.2. Nucleation and Elongation
2.3. Autophagolysosome Formation
2.4. Autophagy Regulatory Drugs
3. The Crosstalk between Autophagy and Proteasome
4. The Role of Autophagy in Hematopoiesis
5. Autophagy in Hematological Malignancies
5.1. Autophagy and Multiple Myeloma
5.2. Autophagy and Leukemia
5.2.1. The Role of Autophagy in Chronic Lymphocytic Leukemia
5.2.2. Role of Autophagy in Acute Lymphocytic Leukemia
5.2.3. Role of Autophagy in Chronic Myeloid Leukemia
5.2.4. Role of Autophagy in Acute Myeloid Leukemia
5.3. Autophagy and Lymphomas
5.4. Autophagy and Hodgkin’s Lymphoma
5.5. Autophagy and Non-Hodgkin’s Lymphoma
5.5.1. Autophagy and B-Cell Lymphoma
5.5.2. Burkitt’s Lymphoma
5.5.3. Mantle Cell Lymphoma
5.5.4. Primary Effusion Lymphoma
5.5.5. Diffuse Large B-Cell Lymphoma
5.5.6. Follicular Lymphoma
5.5.7. Autophagy and T-Cell Lymphoma
Cutaneous Cell Lymphoma
Peripheral T-Cell Lymphoma

{kind=link}
| Disease | Therapy | Effect on Autophagy | Organism/Cell Types | Reference |
|---|---|---|---|---|
| MM | Betulinic acid | Inhibition | MM cell lines | [88] |
| Melphalan | Inhibition | MM cell lines and MM cells resistant to melphalan | [89] | |
| Metformin | Activation | MM cell lines | [91] | |
| NVP-BEZ235 | Activation | MM cell lines | [92] | |
| Asiatoside | Activation | MM cells resistant to BTZ | [93] | |
| BTZ and HCQ | Activation | MM cell lines | [95] | |
| Carfilzomib + CQ/HCQ | Activation | MM cell lines | [96] | |
| ACY-121561 | Inhibition | MM cell lines and ANBL-6 BTZ sensitive and resistant cells | [97] | |
| CLL | MGCD0103 | Inhibition | CLL cell lines | [103] |
| Obotoclax | Activation | Human pre-B acute lymphocytic leukemic cell lines and fludarabine-resistant cells | [105] | |
| Venetoclax | Activation | CLL cell lines | [106] | |
| Flavopiridol | Activation | CLL cell lines | [106] | |
| Cyclophosphamide + flavopiridol + rituximab | Activation | CLL cell lines | [108] | |
| Dasatinib | Activation | CLL cell lines | [112] | |
| 3-MA and CQ (Vorinostat) | Inhibition | Primary CLL cells | [115] | |
| ALL | Resistance to Glucocorticoids | Inhibition | B-ALL cell lines | [120] |
| Dexamethasone and MEK inhibitor | Activation | B-ALL cell lines | [122] | |
| BTZ (Resistance) | Inhibition | B-ALL cell lines | [124] | |
| L-Asparaginase | Inhibition | B-ALL cell lines | [125] | |
| 20(S)-Ginsenoside Rh2 | Inhibition | B-ALL cell lines | [126] | |
| Thymosaponin A-III | Activation | T-ALL cell lines | [127] | |
| NVP-BKM120 | Activation | T-ALL cell lines | [128] | |
| BTZ + Obotoclax | Inhibition | T-ALL cell lines | [129] | |
| Quinacrine | Inhibition | T-ALL cell lines | [130] | |
| Dihydroceramides C22:0 and C24:0 | Inhibition | T-ALL cell lines | [131] | |
| 3-MA | Inhibition | T-ALL cell lines | [131] | |
| CML | 3-MA and CQ | Inhibition | BCR-ABL+ cell lines | [135] |
| Imatinib | Activation | BCR-ABL+ cell lines | [137] | |
| Ponatinib | Activation | BCR-ABL+ cell lines | [137] | |
| Nilotinib | Activation | BCR-ABL+ cell lines | [132] | |
| Dasatinib | Activation | BCR-ABL+ cell lines | [132] | |
| Ponatinib + HCQ | Inhibition | Primary CML cells | [138] | |
| Lys05 | Inhibition | Primary CML cells | [139] | |
| PIK-III | Inhibition | Primary CML cells | [140] | |
| Diosgenin | Activation | CML cell lines | [140] | |
| 20(S)-Ginsenoside rh2 | Activation | K562 and U937 cells | [141] | |
| AML | BTZ | Activation | FLT3-ITD | [146] |
| shRNAs | Inhibition | AML cells (Murine model) | [147] | |
| Quizartinib | Inhibition | AML cells | [147] | |
| Trans-retinoic acid (ATRA) therapy | Activation | AML cell lines | [149] | |
| Alkaloid matrine | Activation | AML cell lines | [150] | |
| Bafilomycin-A | Inhibition | AML cells | [151] | |
| Rapamycin | Activation | AML cells | [151] | |
| Typhaneoside (TYP) | Activation | AML cells | [152] | |
| Cytarabine | Activation | AML cells | [155] | |
| JL1037 | Activation | AML cells | [156] | |
| JL1037 + CQ | Activation | AML cells | [156] | |
| HL | CQ | Inhibition | Murine model/Myc-induced model of lymphoma | [161] |
| ATG5 short hairpin RNA (shRNA) | Inhibition | Myc-induced model of lymphoma | [162] | |
| DPN | Activation | HL cells (in vitro) | [163] | |
| LBH589 (panobinostat) | Activation | HL cell lines | [164] | |
| Melatonin | Activation | HL cell lines | [165] | |
| NHL, Burkitt’s lymphoma | Artesunate | Activation | Raji cells | [170] |
| Ouabain | Activation | Raji cells | [172] | |
| chLym-1 | Activation | Raji cells | [173] | |
| Rituximab–monomethyl auristatin E | Activation | NHL cells | [174] | |
| CQ | Inhibition | HL cell lines | [174] | |
| Phototherapy | Inhibition | Raji Cells | [175] | |
| 3-MA | Inhibition | Raji Cells | [175] | |
| Valproic acid + temsirolimus | Activation | Murine xenograft model | [176] | |
| Vismodegib | Activation/Inhibition | Raji cells | [178] | |
| NHL, mantle cell lymphoma | Everolimus | Inhibition | Clinical models of MCL | [182] |
| FTY720 + milatuzumab | Inhibition | MCL cell lines | [183] | |
| BTZ | Inhibition | MCL cell lines and patient samples | [184] | |
| Temsirolimus | Activation | MCL cell lines | [185] | |
| Vorinostat | Activation | MCL cell lines | [185] | |
| Temsirolimus + vorinostat | Activation | MCL cell lines | [185] | |
| NHL, primary effusion lymphoma | CQ | Inhibition | PEL cell lines | [188] |
| AG490 | Activation | PEL cells | [189] | |
| Quercetin | Activation | PEL cells | [190] | |
| BTZ | Activation | PEL cells | [191] | |
| NHL, diffuse large B-cell lymphoma | SIRT3 KO | Activation | DLBCL cells | [194] |
| SIRT3 | Inhibition | DLBCL cells | [194] | |
| ATG5 knockdown | Inhibition | DLBCL cells | [194] | |
| MALAT-1 (lncRNA) | Inhibition | DLBCL cells (Mice) | [195] | |
| CUL4B | Activation | JNK cells | [197] | |
| NHL, follicular lymphoma | R-CHOP | Activation | FL cells | [199] |
| R-bendamustine | Activation | FL cells | [199] | |
| Bafilomycin A | Inhibition | FL cells | [202] | |
| NHL, T-cell lymphoma | miR-449 | Inhibition | T-cell lymphoma | [205] |
| Sinensetin | Inhibition | Jurkat cells | [207] | |
| BCYRN1 | Activation | Extranodal NK/T-cell lymphoma cells | [208] | |
| Crizotinib and CQ | Activation | NHL cells | [210] | |
| Crizotinib | Activation | [210] | ||
| Metformin | Activation | [211] | ||
| Metformin + doxorubicin or temsirolimus | Activation | NHL cells | [211] | |
| Fenugreek extract | Activation | Jurkat cells | [212] |
6. Germline Variation in Autophagy-Related Genes
6.1. Common Germline Variation Affecting the Risk of Developing Hematological Malignancies
6.2. Common Germline Variation Affecting Disease Progression, Drug Response, and Patient Survival in Hematological Malignancies
| Disease | Autophagy-Related Genes | References |
|---|---|---|
| MM | ATG2B, ATG4, ATG5, ATG7, ATG14, Akt, Beclin-1, Bnip3, CDCA7L, CBX7, DNMT3A, FOXO3a, KLF2, LC3, Linc00515, miR-140-5p, mTORC1, mTORC2, NBK/Bik, p62, PRDM1, STAT3, ULK1, ULK4, Vsp34, WAC | [86,89,90,91,92,93,96,98,232,233,235,238] |
| CLL | AMPK, ATG5, ATG7, Bag-1, Bax, Beclin-1, Bim, CD38, LAMP2, LC3B, mcl-1, P53, p62, SLAMF1, ZAP70 | [102,104,105,109,110,111,112] |
| ALL | ATG12, Beclin-1, BCL2A1, Bax, LC3-II, MYC, ULK2 | [120,122,127] |
| CML | ATG3, ATG7 | [135,138] |
| AML | ATF4, ATG3, ATG4D, ATG5, ATG7, Beclin-1, CXCR4, LC3, p62, ULK1 | [147,148,149,150,155] |
| HL | ATG1, ATG5, Beclin-1, DRAM2, LC3, PRDM1, RORC | [161,162,163,165] |
| NHL, B-cell lymphoma | Beclin-1 | [168,169] |
| NHL, Burkitt’s lymphoma | BAFF, LC3 | [171,175] |
| NHL, mantle cell lymphoma | ATG5, Caspase-3, CD74, LC3 | [181,183,185] |
| NHL, primary effusion lymphoma | ATG5, Beclin-1, STAT3 | [189,190,191] |
| NHL, diffuse large B-cell lymphoma | ATG5, CUL4B, LC3, MALAT-1, p62, PCYT1A, SIRT3 | [194,195,196,197] |
| NHL, follicular lymphoma | ATP6V1B2, BCL-2, LC3A | [200,201,202] |
| NHL, T-cell lymphoma | MiR-449, BCL-2, BCYRN1 | [205,208,210] |
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Taylor, J.; Xiao, W.; Abdel-Wahab, O. Diagnosis and classification of hematologic malignancies on the basis of genetics. Blood 2017, 130, 410–423. [Google Scholar] [CrossRef] [Green Version]
- Djavaheri-Mergny, M.; Giuriato, S.; Tschan, M.P.; Humbert, M. Therapeutic Modulation of Autophagy in Leukaemia and Lymphoma. Cells 2019, 8, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shlush, L.I. Age-related clonal hematopoiesis. Blood 2018, 131, 496–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomasetti, C. Drug Resistance. Adv. Exp. Med. Biol. 2014, 844, 303–316. [Google Scholar] [CrossRef] [PubMed]
- Costas, M.A.; Rubio, M.F. Autophagy. A strategy for cell survival. Medicina 2017, 77, 314–320. [Google Scholar] [PubMed]
- Deter, R.L.; Baudhuin, P.; De Duve, C. Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. J. Cell Biol. 1967, 35, C11–C16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glick, D.; Barth, S.; MacLeod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auberger, P.; Puissant, A. Autophagy, a key mechanism of oncogenesis and resistance in leukemia. Blood 2017, 129, 547–552. [Google Scholar] [CrossRef]
- Chun, Y.; Kim, J. Autophagy: An Essential Degradation Program for Cellular Homeostasis and Life. Cells 2018, 7, 278. [Google Scholar] [CrossRef] [PubMed]
- Bialik, S.; Dasari, S.K.; Kimchi, A. Autophagy-dependent cell death—Where, how and why a cell eats itself to death. J. Cell Sci. 2018, 131, jcs215152. [Google Scholar] [CrossRef] [Green Version]
- Riffelmacher, T.; Simon, A.K. Mechanistic roles of autophagy in hematopoietic differentiation. FEBS J. 2017, 284, 1008–1020. [Google Scholar] [CrossRef]
- Li, Y.; Hahn, T.; Garrison, K.; Cui, Z.-H.; Thorburn, A.; Thorburn, J.; Hu, H.-M.; Akporiaye, E.T. The Vitamin E Analogue α-TEA Stimulates Tumor Autophagy and Enhances Antigen Cross-Presentation. Cancer Res. 2012, 72, 3535–3545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Z.; Liang, S.; Hu, J.; Jin, W.; Zhan, Q.; Zhao, K. Autophagy as a target for hematological malignancy therapy. Blood Rev. 2016, 30, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Shahrabi, S.; Paridar, M.; Zeinvand-Lorestani, M.; Jalili, A.; Zibara, K.; Abdollahi, M.; Khosravi, A. Autophagy regulation and its role in normal and malignant hematopoiesis. J. Cell. Physiol. 2019, 234, 21746–21757. [Google Scholar] [CrossRef] [PubMed]
- Nazio, F.; Bordi, M.; Cianfanelli, V.; Locatelli, F.; Cecconi, F. Autophagy and cancer stem cells: Molecular mechanisms and therapeutic applications. Cell Death Differ. 2019, 26, 690–702. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the Integrated Stress Response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Niewerth, D.; Jansen, G.; Assaraf, Y.G.; Zweegman, S.; Kaspers, G.J.; Cloos, J. Molecular basis of resistance to proteasome inhibitors in hematological malignancies. Drug Resist. Updat. 2015, 18, 18–35. [Google Scholar] [CrossRef]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravanan, P.; Srikumar, I.F.; Talwar, P. Autophagy: The spotlight for cellular stress responses. Life Sci. 2017, 188, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Noda, T. Regulation of Autophagy through TORC1 and mTORC1. Biomolecules 2017, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Devereaux, K.; Dall’Armi, C.; Alcazar-Roman, A.; Ogasawara, Y.; Zhou, X.; Wang, F.; Yamamoto, A.; De Camilli, P.; Di Paolo, G. Regulation of Mammalian Autophagy by Class II and III PI 3-Kinases through PI3P Synthesis. PLoS ONE 2013, 8, e76405. [Google Scholar] [CrossRef] [Green Version]
- Puissant, A.; Robert, G.; Auberger, P. Targeting autophagy to fight hematopoietic malignancies. Cell Cycle 2010, 9, 3470–3478. [Google Scholar] [CrossRef] [Green Version]
- Müller, A.J.; Proikas-Cezanne, T. Function of human WIPI proteins in autophagosomal rejuvenation of endomembranes? FEBS Lett. 2015, 589, 1546–1551. [Google Scholar] [CrossRef] [Green Version]
- Nencioni, A.; Cea, M.; Montecucco, F.; Longo, V.D.; Patrone, F.; Carella, A.M.; Holyoake, T.L.; Helgason, G.V. Autophagy in blood cancers: Biological role and therapeutic implications. Haematologica 2013, 98, 1335–1343. [Google Scholar] [CrossRef]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin–proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 2013, 24, 92–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McEwan, D.G.; Popovic, D.; Gubas, A.; Terawaki, S.; Suzuki, H.; Stadel, D.; Coxon, F.P.; de Stegmann, D.M.; Bhogaraju, S.; Maddi, K.; et al. PLEKHM1 Regulates Autophagosome-Lysosome Fusion through HOPS Complex and LC3/GABARAP Proteins. Mol. Cell 2015, 57, 39–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, P.; Mizushima, N. Autophagy and human diseases. Cell Res. 2014, 24, 69–79. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Feng, H.; Wang, N.; Zhang, N.; Liao, H.-H. Alternative autophagy: Mechanisms and roles in different diseases. Cell Commun. Signal. 2022, 20, 43. [Google Scholar] [CrossRef]
- Jin, S.; Wei, J.; You, L.; Liu, H.; Qian, W. Autophagy regulation and its dual role in blood cancers: A novel target for therapeutic development (Review). Oncol. Rep. 2018, 39, 2473–2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thoreen, C.C.; Kang, S.A.; Chang, J.W.; Liu, Q.; Zhang, J.; Gao, Y.; Reichling, L.J.; Sim, T.; Sabatini, D.M.; Gray, N.S. An ATP-competitive Mammalian Target of Rapamycin Inhibitor Reveals Rapamycin-resistant Functions of mTORC1. J. Biol. Chem. 2009, 284, 8023–8032, Correction in J. Biol. Chem. 2020, 295, 2886. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Tan, M.; Cai, W.; Wang, B.; He, P.; Zhang, X. Arsenic trioxide induces autophagic cell death in osteosarcoma cells via the ROS-TFEB signaling pathway. Biochem. Biophys. Res. Commun. 2018, 496, 167–175. [Google Scholar] [CrossRef]
- Kumar, S.; Guru, S.K.; Pathania, A.S.; Manda, S.; Kumar, A.; Bharate, S.B.; Vishwakarma, R.A.; Malik, F.; Bhushan, S. Fascaplysin Induces Caspase Mediated Crosstalk between Apoptosis and Autophagy through the Inhibition of PI3K/AKT/mTOR Signaling Cascade in Human Leukemia HL-60 Cells. J. Cell. Biochem. 2015, 116, 985–997. [Google Scholar] [CrossRef]
- Pasquier, B. Autophagy inhibitors. Cell. Mol. Life Sci. 2016, 73, 985–1001. [Google Scholar] [CrossRef]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [Green Version]
- Mohsen, S.; Sobash, P.T.; Algwaiz, G.F.; Nasef, N.; Al-Zeidaneen, S.A.; Karim, N.A. Autophagy Agents in Clinical Trials for Cancer Therapy: A Brief Review. Curr. Oncol. 2022, 29, 1695–1708. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.H.; Kwon, A.Y.T. Crosstalk and Interplay between the Ubiquitin-Proteasome System and Autophagy. Mol. Cells 2017, 40, 441–449. [Google Scholar] [CrossRef] [Green Version]
- Nam, T.; Han, J.H.; Devkota, S.; Lee, H.-W. Emerging Paradigm of Crosstalk between Autophagy and the Ubiquitin-Proteasome System. Mol. Cells 2017, 40, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Le, W.-D. Autophagy and Ubiquitin-Proteasome System. Adv. Exp. Med. Biol. 2019, 1206, 527–550. [Google Scholar] [CrossRef] [PubMed]
- Robak, T.; Huang, H.; Jin, J.; Zhu, J.; Liu, T.; Samoilova, O.; Pylypenko, H.; Verhoef, G.; Siritanaratkul, N.; Osmanov, E.; et al. Bortezomib-Based Therapy for Newly Diagnosed Mantle-Cell Lymphoma. N. Engl. J. Med. 2015, 372, 944–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varshavsky, A. The Ubiquitin System, Autophagy, and Regulated Protein Degradation. Annu. Rev. Biochem. 2017, 86, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Varshavsky, A. The Ubiquitin System, an Immense Realm. Annu. Rev. Biochem. 2012, 81, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Varshavsky, A. The ubiquitin system. Trends Biochem. Sci. 1997, 22, 383–387. [Google Scholar] [CrossRef]
- Varshavsky, A. Discovery of the Biology of the Ubiquitin System. JAMA 2014, 311, 1969–1970. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Koike, M.; Sou, Y.-S.; Ueno, T.; Hara, T.; Mizushima, N.; Iwata, J.; Ezaki, J.; Murata, S.; et al. Homeostatic Levels of p62 Control Cytoplasmic Inclusion Body Formation in Autophagy-Deficient Mice. Cell 2007, 131, 1149–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramarski, L.; Arbely, E. Translational read-through promotes aggregation and shapes stop codon identity. Nucleic Acids Res. 2020, 48, 3747–3760. [Google Scholar] [CrossRef] [PubMed]
- Minoia, M.; Boncoraglio, A.; Vinet, J.; Morelli, F.F.; Brunsting, J.F.; Poletti, A.; Krom, S.; Reits, E.; Kampinga, H.H.; Carra, S. BAG3 Induces the Sequestration of Proteasomal Clients into Cytoplasmic Puncta: Implications for a Proteasome-to-Autophagy Switch. Autophagy 2014, 10, 1603–1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- B’Chir, W.; Maurin, A.C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic. Acids Res. 2013, 41, 7683–7699. [Google Scholar] [CrossRef] [PubMed]
- Di Lernia, G.; Leone, P.; Solimando, A.G.; Buonavoglia, A.; Saltarella, I.; Ria, R.; Ditonno, P.; Silvestris, N.; Crudele, L.; Vacca, A.; et al. Bortezomib Treatment Modulates Autophagy in Multiple Myeloma. J. Clin. Med. 2020, 9, 552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dytfeld, D.; Matuszak, M.; Lewandowski, K.; Komarnicki, M. Bortezomib in combination with thalidomide and dexamethasone—A successful treatment regimen in refractory extramedullary multiple myeloma. Ann. Hematol. 2008, 87, 253–254. [Google Scholar] [CrossRef] [PubMed]
- Field-Smith, A.; Morgan, G.; Davies, F. Bortezomib (Velcadetrade mark) in the Treatment of Multiple Myeloma. Ther. Clin. Risk Manag. 2006, 2, 271–279. [Google Scholar] [CrossRef] [Green Version]
- Robak, T. Bortezomib in the treatment of mantle cell lymphoma. Future Oncol. 2015, 11, 2807–2818. [Google Scholar] [CrossRef] [PubMed]
- Tanday, S. Bortezomib treatment for patients with mantle-cell lymphoma. Lancet Oncol. 2015, 16, e162. [Google Scholar] [CrossRef]
- Harada, M.; Hanada, S.; Toivola, D.M.; Ghori, N.; Omary, M.B. Autophagy activation by rapamycin eliminates mouse Mallory-Denk bodies and blocks their proteasome inhibitor-mediated formation. Hepatology 2008, 47, 2026–2035. [Google Scholar] [CrossRef]
- Ogawa, M. Hematopoiesis. J. Allergy Clin. Immunol. 1994, 94, 645–650. [Google Scholar] [CrossRef]
- Orkin, S.H.; Zon, L.I. Hematopoiesis: An Evolving Paradigm for Stem Cell Biology. Cell 2008, 132, 631–644. [Google Scholar] [CrossRef] [Green Version]
- Sotthibundhu, A.; Promjuntuek, W.; Liu, M.; Shen, S.; Noisa, P. Roles of autophagy in controlling stem cell identity: A perspective of self-renewal and differentiation. Cell Tissue Res. 2018, 374, 205–216. [Google Scholar] [CrossRef]
- Phadwal, K.; Watson, A.S.; Simon, A.K. Tightrope act: Autophagy in stem cell renewal, differentiation, proliferation, and aging. Cell. Mol. Life Sci. 2012, 70, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Vijayakumar, K.; Cho, G. Autophagy: An evolutionarily conserved process in the maintenance of stem cells and aging. Cell Biochem. Funct. 2019, 37, 452–458. [Google Scholar] [CrossRef]
- Cheung, T.H.T.; Rando, T.A. Molecular regulation of stem cell quiescence. Nat. Rev. Mol. Cell Biol. 2013, 14, 329–340. [Google Scholar] [CrossRef] [Green Version]
- Warr, M.R.; Binnewies, M.; Flach, J.; Reynaud, D.; Garg, T.; Malhotra, R.; Debnath, J.; Passegué, E. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 2013, 494, 323–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Vos, K.E.; Gomez-Puerto, C.; Coffer, P.J. Regulation of autophagy by Forkhead box (FOX) O transcription factors. Adv. Biol. Regul. 2012, 52, 122–136. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Budanov, A.V.; Park, E.J.; Birse, R.; Kim, T.E.; Perkins, G.A.; Ocorr, K.; Ellisman, M.H.; Bodmer, R.; Bier, E.; et al. Sestrin as a Feedback Inhibitor of TOR That Prevents Age-Related Pathologies. Science 2010, 327, 1223–1228. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-C.; Jeon, S.-M.; Bhaskar, P.T.; Nogueira, V.; Sundararajan, D.; Tonic, I.; Park, Y.; Hay, N. FoxOs Inhibit mTORC1 and Activate Akt by Inducing the Expression of Sestrin3 and Rictor. Dev. Cell 2010, 18, 592–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortensen, M.; Soilleux, E.J.; Djordjevic, G.; Tripp, R.; Lutteropp, M.; Sadighi-Akha, E.; Stranks, A.; Glanville, J.; Knight, S.; Jacobsen, S.E.; et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J. Exp. Med. 2011, 208, 455–467. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Zhang, S.; Yuan, N.; Wang, J.; Li, X.; Xu, F.; Lin, W.; Song, L.; Fang, Y.; Wang, Z.; et al. Hierarchal autophagic divergence of hematopoietic system. J. Biol. Chem. 2015, 290, 29240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Lee, J.Y.; Wei, H.; Tanabe, O.; Engel, J.D.; Morrison, S.J.; Guan, J.-L. FIP200 is required for the cell-autonomous maintenance of fetal hematopoietic stem cells. Blood 2010, 116, 4806–4814. [Google Scholar] [CrossRef] [Green Version]
- Watson, A.S.; Riffelmacher, T.; Stranks, A.; Williams, O.; de Boer, J.; Cain, K.; Macfarlane, M.; McGouran, J.; Kessler, B.; Khandwala, S.; et al. Autophagy limits proliferation and glycolytic metabolism in acute myeloid leukemia. Cell Death Discov. 2015, 1, 15008. [Google Scholar] [CrossRef] [Green Version]
- Joshi, A.; Kundu, M. Mitophagy in hematopoietic stem cells: The case for exploration. Autophagy 2013, 9, 1737–1749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannini, N.; Girotra, M.; Naveiras, O.; Nikitin, G.; Campos, V.; Giger, S.; Roch, A.; Auwerx, J.; Lutolf, M.P. Specification of haematopoietic stem cell fate via modulation of mitochondrial activity. Nat. Commun. 2016, 7, 13125. [Google Scholar] [CrossRef] [Green Version]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-Induced Autophagy Is Mediated through Hypoxia-Inducible Factor Induction of BNIP3 and BNIP3L via Their BH3 Domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Liu, Y.; Liu, R.; Ikenoue, T.; Guan, K.-L.; Liu, Y.; Zheng, P. TSC–mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J. Exp. Med. 2008, 205, 2397–2408. [Google Scholar] [CrossRef] [Green Version]
- Folmes, C.D.; Terzic, A. Energy metabolism in the acquisition and maintenance of stemness. Semin. Cell Dev. Biol. 2016, 52, 68–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazandjian, D. Multiple myeloma epidemiology and survival: A unique malignancy. Semin. Oncol. 2016, 43, 676–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabattini, E.; Bacci, F.; Sagramoso, C.; Pileri, S.A. WHO classification of tumours of haematopoietic and lymphoid tissues in 2008: An overview. Pathologica 2010, 102, 83–87. [Google Scholar] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; International Agency for Research on Cancer: Lyon, France, 2017; Volume 2. [Google Scholar]
- Aksenova, A.Y.; Zhuk, A.S.; Lada, A.G.; Zotova, I.V.; Stepchenkova, E.I.; Kostroma, I.I.; Gritsaev, S.V.; Pavlov, Y.I. Genome Instability in Multiple Myeloma: Facts and Factors. Cancers 2021, 13, 5949. [Google Scholar] [CrossRef]
- Yun, Z.; Zhichao, J.; Hao, Y.; Ou, J.; Ran, Y.; Wen, D.; Qun, S. Targeting autophagy in multiple myeloma. Leuk. Res. 2017, 59, 97–104. [Google Scholar] [CrossRef]
- Moriya, S.; Komatsu, S.; Yamasaki, K.; Kawai, Y.; Kokuba, H.; Hirota, A.; Che, X.-F.; Inazu, M.; Gotoh, A.; Hiramoto, M.; et al. Targeting the integrated networks of aggresome formation, proteasome, and autophagy potentiates ER stress-mediated cell death in multiple myeloma cells. Int. J. Oncol. 2014, 46, 474–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cea, M.; Cagnetta, A.; Fulciniti, M.; Tai, Y.-T.; Hideshima, T.; Chauhan, D.; Roccaro, A.; Sacco, A.; Calimeri, T.; Cottini, F.; et al. Targeting NAD+ salvage pathway induces autophagy in multiple myeloma cells via mTORC1 and extracellular signal-regulated kinase (ERK1/2) inhibition. Blood 2012, 120, 3519–3529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milan, E.; Fabbri, M.; Cenci, S. Autophagy in Plasma Cell Ontogeny and Malignancy. J. Clin. Immunol. 2016, 36 (Suppl. 1), 18–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milan, E.; Perini, T.; Resnati, M.; Orfanelli, U.; Oliva, L.; Raimondi, A.; Cascio, P.; Bachi, A.; Marcatti, M.; Ciceri, F.; et al. A plastic SQSTM1/p62-dependent autophagic reserve maintains proteostasis and determines proteasome inhibitor susceptibility in multiple myeloma cells. Autophagy 2015, 11, 1161–1178. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.-J.; Chen, Y.; He, J.; Yi, S.; Wen, L.; Zhao, J.; Zhang, B.-P.; Cui, G.-H. Betulinic acid inhibits autophagic flux and induces apoptosis in human multiple myeloma cells in vitro. Acta Pharmacol. Sin. 2012, 33, 1542–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, D.; Yang, C.; Zhang, Z.; Cong, Y.; Xiao, M. Knockdown of Linc00515 Inhibits Multiple Myeloma Autophagy and Chemoresistance by Upregulating miR-140-5p and Downregulating ATG14. Cell. Physiol. Biochem. 2018, 48, 2517–2527. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, L.; Zhang, Y.; Leng, Y.; Pei, X.-Y.; Lin, H.; Jones, R.; Orlowski, R.Z.; Dai, Y.; Grant, S. Targeting SQSTM1/p62 Induces Cargo Loading Failure and Converts Autophagy to Apoptosis via NBK/Bik. Mol. Cell. Biol. 2014, 34, 3435–3449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Xu, W.; Yan, Z.; Zhao, W.; Mi, J.; Li, J.; Yan, H. Metformin induces autophagy and G0/G1 phase cell cycle arrest in myeloma by targeting the AMPK/mTORC1 and mTORC2 pathways. J. Exp. Clin. Cancer Res. 2018, 37, 63. [Google Scholar] [CrossRef]
- Ma, Y.; Jin, Z.; Yu, K.; Liu, Q. NVP-BEZ235-induced autophagy as a potential therapeutic approach for multiple myeloma. Am. J. Transl. Res. 2019, 11, 87–105. [Google Scholar]
- Yingchun, L.; Huihan, W.; Rong, Z.; Guojun, Z.; Ying, Y.; Zhuogang, L. Antitumor Activity of Asiaticoside Against Multiple Myeloma Drug-Resistant Cancer Cells Is Mediated by Autophagy Induction, Activation of Effector Caspases, and Inhibition of Cell Migration, Invasion, and STAT-3 Signaling Pathway. Med. Sci. Monit. 2019, 25, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Pinto, V.; Bergantim, R.; Caires, H.R.; Seca, H.; Guimarães, J.E.; Vasconcelos, M.H. Multiple Myeloma: Available Therapies and Causes of Drug Resistance. Cancers 2020, 12, 407. [Google Scholar] [CrossRef] [PubMed]
- Vogl, D.T.; Stadtmauer, E.A.; Tan, K.-S.; Heitjan, D.F.; Davis, L.E.; Pontiggia, L.; Rangwala, R.; Piao, S.; Chang, Y.C.; Scott, E.C.; et al. Combined autophagy and proteasome inhibition: A phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 2014, 10, 1380–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarauta, V.; Jaime, P.; Gonzalo, O.; de Miguel, D.; Ramirez-Labrada, A.; Martínez-Lostao, L.; Anel, A.; Pardo, J.; Marzo, I.; Naval, J. Inhibition of autophagy with chloroquine potentiates carfilzomib-induced apoptosis in myeloma cells in vitro and in vivo. Cancer Lett. 2016, 382, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mishima, Y.; Santo, L.; Eda, H.; Cirstea, D.; Nemani, N.; Yee, A.J.; O’Donnell, E.; Selig, M.K.; Quayle, S.N.; Arastu-Kapur, S.; et al. Ricolinostat (ACY-1215) induced inhibition of aggresome formation accelerates carfilzomib-induced multiple myeloma cell death. Br. J. Haematol. 2015, 169, 423–434. [Google Scholar] [CrossRef]
- Jung, G.; Roh, J.; Lee, H.; Gil, M.; Yoon, D.H.; Suh, C.; Jang, S.; Park, C.-J.; Huh, J.; Park, C.-S. Autophagic Markers BECLIN 1 and LC3 Are Associated with Prognosis of Multiple Myeloma. Acta Haematol. 2015, 134, 17–24. [Google Scholar] [CrossRef]
- Sant, M.; Allemani, C.; Tereanu, C.; De Angelis, R.; Capocaccia, R.; Visser, O.; Marcos-Gragera, R.; Maynadié, M.; Simonetti, A.; Lutz, J.-M.; et al. Incidence of hematologic malignancies in Europe by morphologic subtype: Results of the HAEMACARE project. Blood 2010, 116, 3724–3734. [Google Scholar] [CrossRef] [Green Version]
- Hallek, M.; Shanafelt, T.D.; Eichhorst, B. Chronic lymphocytic leukaemia. Lancet 2018, 391, 1524–1537. [Google Scholar] [CrossRef]
- Sharma, K.; Le, N.; Alotaibi, M.; Gewirtz, D.A. Cytotoxic Autophagy in Cancer Therapy. Int. J. Mol. Sci. 2014, 15, 10034–10051. [Google Scholar] [CrossRef]
- Romero-Macías, J.-R.; Pascual-Serra, R.; Roche, O.; Ruiz-Marcos, F.; Serrano-Martínez, A.; González-Aguado, P.; Belandia, B.; Ruiz-Hidalgo, M.-J.; Sánchez-Prieto, R. Blockage of autophagic flux is associated with lymphocytosis and higher percentage of tumoral cells in chronic lymphocytic leukemia of B cells. Clin. Transl. Oncol. 2019, 21, 1280–1285. [Google Scholar] [CrossRef]
- El-Khoury, V.; Pierson, S.; Szwarcbart, E.; Brons, N.H.; Roland, O.; Cherrier-De Wilde, S.; Plawny, L.; Van Dyck, E.; Berchem, G. Disruption of autophagy by the histone deacetylase inhibitor MGCD0103 and its therapeutic implication in B-cell chronic lymphocytic leukemia. Leukemia 2014, 28, 1636–1646. [Google Scholar] [CrossRef] [Green Version]
- Faria, J.R.; Yamamoto, M.; Faria, R.M.; Kerbauy, J.; Oliveira, J.S. Fludarabine induces apoptosis in chronic lymphocytic leukemia—The role of P53, Bcl-2, Bax, Mcl-1, and Bag-1 proteins. Braz. J. Med. Biol. Res. 2006, 39, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Singh, K.; Mazumder, S.; Hill, B.T.; Kalaycio, M.; Almasan, A. BECN1 and BIM interactions with MCL-1 determine fludarabine resistance in leukemic B cells. Cell Death Dis. 2013, 4, e628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avsec, D.; Djordjevič, A.T.J.; Kandušer, M.; Podgornik, H.; Škerget, M.; Mlinarič-Raščan, I. Targeting Autophagy Triggers Apoptosis and Complements the Action of Venetoclax in Chronic Lymphocytic Leukemia Cells. Cancers 2021, 13, 4557. [Google Scholar] [CrossRef] [PubMed]
- Maddocks, K.; Wei, L.; Rozewski, D.; Jiang, Y.; Zhao, Y.; Adusumilli, M.; Pierceall, W.E.; Doykin, C.; Cardone, M.H.; Jones, J.A.; et al. Reduced occurrence of tumor flare with flavopiridol followed by combined flavopiridol and lenalidomide in patients with relapsed chronic lymphocytic leukemia (CLL). Am. J. Hematol. 2015, 90, 327–333. [Google Scholar] [CrossRef] [Green Version]
- Stephens, D.M.; Ruppert, A.S.; Maddocks, K.; Andritsos, L.; Baiocchi, R.; Jones, J.; Johnson, A.J.; Smith, L.L.; Zhao, Y.; Ling, Y.; et al. Cyclophosphamide, alvocidib (flavopiridol), and rituximab, a novel feasible chemoimmunotherapy regimen for patients with high-risk chronic lymphocytic leukemia. Leuk. Res. 2013, 37, 1195–1199. [Google Scholar] [CrossRef] [Green Version]
- Bologna, C.; Buonincontri, R.; Serra, S.; Vaisitti, T.; Audrito, V.; Brusa, D.; Pagnani, A.; Coscia, M.; D’Arena, G.; Mereu, E.; et al. SLAMF1 regulation of chemotaxis and autophagy determines CLL patient response. J. Clin. Investig. 2015, 126, 181–194. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.-L.; Huang, Y.; Wu, J.-Z.; Cao, X.; Liang, J.-H.; Xia, Y.; Wu, W.; Cao, L.; Zhu, H.-Y.; Wang, L.; et al. Expression of autophagy related genes in chronic lymphocytic leukemia is associated with disease course. Leuk. Res. 2018, 66, 8–14. [Google Scholar] [CrossRef]
- Arroyo, D.S.; Rodriguez, C.M.; Bussi, C.; Manzone-Rodriguez, C.; Sastre, D.; Heller, V.; Stanganelli, C.; Slavutsky, I.; Iribarren, P. Increased Expression of Autophagy Protein LC3 in Two Patients With Progressing Chronic Lymphocytic Leukemia. Front. Endocrinol. 2020, 11, 321. [Google Scholar] [CrossRef]
- Amrein, L.; Soulières, D.; Johnston, J.B.; Aloyz, R. p53 and autophagy contribute to dasatinib resistance in primary CLL lymphocytes. Leuk. Res. 2011, 35, 99–102. [Google Scholar] [CrossRef]
- Tasdemir, E.; Maiuri, M.C.; Morselli, E.; Criollo, A.; D’Amelio, M.; Djavaheri-Mergny, M.; Cecconi, F.; Tavernarakis, N.; Kroemer, G. A dual role of p53 in the control of autophagy. Autophagy 2008, 4, 810–814. [Google Scholar] [CrossRef] [Green Version]
- Pitini, V.V. The role of p53 and autophagy in Dasatinib resistance of CLL lymphocytes. Leuk. Res. 2011, 35, 32–33. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Zhang, W.; Yang, L.; Pelicano, H.; Zhou, K.; Yin, R.; Huang, R.; Zeng, J. Targeting the autophagy in bone marrow stromal cells overcomes resistance to vorinostat in chronic lymphocytic leukemia. Onco Targets Ther. 2018, 11, 5151–5170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bigas, A.; Rodriguez-Sevilla, J.J.; Espinosa, L.; Gallardo, F. Recent advances in T-cell lymphoid neoplasms. Exp. Hematol. 2022, 106, 3–18. [Google Scholar] [CrossRef]
- Terwilliger, T.; Abdul-Hay, M. Acute lymphoblastic leukemia: A comprehensive review and 2017 update. Blood Cancer J. 2017, 7, e577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evangelisti, C.; Evangelisti, C.; Chiarini, F.; Lonetti, A.; Buontempo, F.; Neri, L.M.; McCubrey, J.; Martelli, A.M. Autophagy in acute leukemias: A double-edged sword with important therapeutic implications. Biochim. Biophys. Acta 2015, 1853, 14–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catts, V.S.; Farnsworth, M.L.; Haber, M.; Norris, M.D.; Lutze-Mann, L.H.; Lock, R.B. High level resistance to glucocorticoids, associated with a dysfunctional glucocorticoid receptor, in childhood acute lymphoblastic leukemia cells selected for methotrexate resistance. Leukemia 2001, 15, 929–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarang, Z.; Gyurina, K.; Scholtz, B.; Kiss, C.; Szegedi, I. Altered expression of autophagy-related genes might contribute to glucocorticoid resistance in precursor B-cell-type acute lymphoblastic leukemia. Eur. J. Haematol. 2016, 97, 453–460. [Google Scholar] [CrossRef]
- Bonapace, L.; Bornhauser, B.C.; Schmitz, M.; Cario, G.; Ziegler, U.; Niggli, F.K.; Schafer, B.W.; Schrappe, M.; Stanulla, M.; Bourquin, J.P. Induction of autophagy-dependent necroptosis is required for childhood acute lymphoblastic leukemia cells to overcome glucocorticoid resistance. J. Clin. Investig. 2010, 120, 1310–1323. [Google Scholar] [CrossRef] [Green Version]
- Polak, A.; Kiliszek, P.; Sewastianik, T.; Szydłowski, M.; Jabłońska, E.; Białopiotrowicz, E.; Górniak, P.; Markowicz, S.; Nowak, E.; Grygorowicz, M.A.; et al. MEK Inhibition Sensitizes Precursor B-Cell Acute Lymphoblastic Leukemia (B-ALL) Cells to Dexamethasone through Modulation of mTOR Activity and Stimulation of Autophagy. PLoS ONE 2016, 11, e0155893. [Google Scholar] [CrossRef] [Green Version]
- Junk, S.; Cario, G.; Wittner, N.; Stanulla, M.; Scherer, R.; Schlegelberger, B.; Schrappe, M.; von Neuhoff, N.; Lauten, M. Bortezomib Treatment can Overcome Glucocorticoid Resistance in Childhood B-cell Precursor Acute Lymphoblastic Leukemia Cell Lines. Klin. Padiatr. 2015, 227, 123–130. [Google Scholar] [CrossRef]
- Wang, Z.; Zhu, S.; Zhang, G.; Liu, S. Inhibition of autophagy enhances the anticancer activity of bortezomib in B-cell acute lymphoblastic leukemia cells. Am. J. Cancer Res. 2015, 5, 639–650. [Google Scholar] [PubMed]
- Takahashi, H.; Inoue, J.; Sakaguchi, K.; Takagi, M.; Mizutani, S.; Inazawa, J. Autophagy is required for cell survival under L-asparaginase-induced metabolic stress in acute lymphoblastic leukemia cells. Oncogene 2017, 36, 4267–4276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, T.; Wang, J.; Wang, Y.; Wang, Y.; Cai, J.; Wang, M.; Chen, Q.; Song, J.; Yu, Z.; Huang, W.; et al. Inhibition of autophagy potentiates anticancer property of 20(S)-ginsenoside Rh2 by promoting mitochondria-dependent apoptosis in human acute lymphoblastic leukaemia cells. Oncotarget 2016, 7, 27336–27349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Dong, R.; Fan, W.W.; Zheng, X.C.; Li, A.M.; Wang, W.D. Timosaponin A-III induces autophagy of T-cell acute lymphoblastic leukemia Jurkat cells via inhibition of the PI3K/Akt/mTOR pathway. Oncol. Rep. 2019, 41, 2937–2944. [Google Scholar] [CrossRef]
- Pereira, J.K.N.; Machado-Neto, J.A.; Lopes, M.R.; Morini, B.C.; Traina, F.; Costa, F.F.; Saad, S.T.O.; Favaro, P. Molecular effects of the phosphatidylinositol-3-kinase inhibitor NVP-BKM120 on T and B-cell acute lymphoblastic leukaemia. Eur. J. Cancer 2015, 51, 2076–2085. [Google Scholar] [CrossRef]
- Zhou, D.; Dai, L.; Liu, X.; Que, F.; Xu, Y.; Luo, X.; Zhu, Y.; Liu, S.; Li, Y.; Yu, L. Bortezomib and obatoclax for dual blockade of protein degradation pathways show synergistic anti-tumor effect in human acute T lymphoblastic leukemia cells. Nan Fang Yi Ke Da Xue Xue Bao 2019, 39, 401–408. [Google Scholar] [CrossRef]
- Jing, B.; Jin, J.; Xiang, R.; Liu, M.; Yang, L.; Tong, Y.; Xiao, X.; Lei, H.; Liu, W.; Xu, H.; et al. Vorinostat and quinacrine have synergistic effects in T-cell acute lymphoblastic leukemia through reactive oxygen species increase and mitophagy inhibition. Cell Death Dis. 2018, 9, 589. [Google Scholar] [CrossRef]
- Holliday, M.W., Jr.; Cox, S.B.; Kang, M.H.; Maurer, B.J. C22:0- and C24:0-dihydroceramides Confer Mixed Cytotoxicity in T-Cell Acute Lymphoblastic Leukemia Cell Lines. PLoS ONE 2013, 8, e74768. [Google Scholar] [CrossRef] [Green Version]
- Bellodi, C.; Lidonnici, M.R.; Hamilton, A.; Helgason, G.V.; Soliera, A.R.; Ronchetti, M.; Galavotti, S.; Young, K.W.; Selmi, T.; Yacobi, R.; et al. Targeting autophagy potentiates tyrosine kinase inhibitor–induced cell death in Philadelphia chromosome–positive cells, including primary CML stem cells. J. Clin. Investig. 2009, 119, 1109–1123. [Google Scholar] [CrossRef]
- Sheng, Z.; Ma, L.; Sun, J.E.; Zhu, L.J.; Green, M.R. BCR-ABL suppresses autophagy through ATF5-mediated regulation of mTOR transcription. Blood 2011, 118, 2840–2848. [Google Scholar] [CrossRef] [Green Version]
- Colecchia, D.; Rossi, M.; Sasdelli, F.; Sanzone, S.; Strambi, A.; Chiariello, M. MAPK15 mediates BCR-ABL1-induced autophagy and regulates oncogene-dependent cell proliferation and tumor formation. Autophagy 2015, 11, 1790–1802. [Google Scholar] [CrossRef]
- Altman, B.J.; Jacobs, S.R.; Mason, E.F.; Michalek, R.D.; MacIntyre, A.N.; Coloff, J.L.; Ilkayeva, O.; Jia, W.; He, Y.-W.; Rathmell, J.C. Autophagy is essential to suppress cell stress and to allow BCR-Abl-mediated leukemogenesis. Oncogene 2011, 30, 1855–1867. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Medeiros, L.J.; Hu, S. Chronic Myeloid Leukemia: Beyond BCR-ABL1. Curr. Hematol. Malig. Rep. 2018, 13, 435–445. [Google Scholar] [CrossRef]
- Kayabasi, C.; Okcanoglu, T.B.; Yelken, B.O.; Asik, A.; Susluer, S.Y.; Avci, C.B.; Saydam, G.; Gunduz, C. Comparative effect of imatinib and ponatinib on autophagy and miRNome in chronic myeloid leukemia. Gene 2017, 637, 173–180. [Google Scholar] [CrossRef]
- Mitchell, R.; Hopcroft, L.E.M.; Baquero, P.; Allan, E.K.; Hewit, K.; James, D.; Hamilton, G.; Mukhopadhyay, A.; O’Prey, J.; Hair, A.; et al. Targeting BCR-ABL-Independent TKI Resistance in Chronic Myeloid Leukemia by mTOR and Autophagy Inhibition. J. Natl. Cancer Inst. 2018, 110, 467–478. [Google Scholar] [CrossRef] [Green Version]
- Baquero, P.; Dawson, A.; Mukhopadhyay, A.; Kuntz, E.M.; Mitchell, R.; Olivares, O.; Ianniciello, A.; Scott, M.T.; Dunn, K.; Nicastri, M.C.; et al. Targeting quiescent leukemic stem cells using second generation autophagy inhibitors. Leukemia 2019, 33, 981–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.; Fan, J.; Wang, Q.; Ju, D.; Feng, M.; Li, J.; Guan, Z.-B.; An, D.; Wang, X.; Ye, L. Diosgenin induces ROS-dependent autophagy and cytotoxicity via mTOR signaling pathway in chronic myeloid leukemia cells. Phytomedicine 2016, 23, 243–252. [Google Scholar] [CrossRef]
- Zhuang, J.; Yin, J.; Xu, C.; Mu, Y.; Lv, S. 20(S)-Ginsenoside Rh2 Induce the Apoptosis and Autophagy in U937 and K562 Cells. Nutrients 2018, 10, 328. [Google Scholar] [CrossRef] [Green Version]
- Short, N.J.; Rytting, M.E.; Cortes, J.E. Acute myeloid leukaemia. Lancet 2018, 392, 593–606. [Google Scholar] [CrossRef]
- Choi, E.-J.; Lee, J.-H.; Lee, J.-H.; Park, H.-S.; Ko, S.H.; Hur, E.-H.; Moon, J.; Goo, B.-K.; Kim, Y.; Seol, M.; et al. Comparison of anthracyclines used for induction chemotherapy in patients with FLT3 -ITD-mutated acute myeloid leukemia. Leuk. Res. 2018, 68, 51–56. [Google Scholar] [CrossRef]
- Levis, M. Quizartinib for the treatment of FLT3/ITD acute myeloid leukemia. Future Oncol. 2014, 10, 1571–1579. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Ge, Z.; Chen, B. Quizartinib (AC220): A promising option for acute myeloid leukemia. Drug Des. Dev. Ther. 2019, 13, 1117–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larrue, C.; Saland, E.; Boutzen, H.; Vergez, F.; David, M.; Joffre, C.; Hospital, M.-A.; Tamburini, J.; Delabesse, E.; Manenti, S.; et al. Proteasome inhibitors induce FLT3-ITD degradation through autophagy in AML cells. Blood 2016, 127, 882–892. [Google Scholar] [CrossRef] [Green Version]
- Heydt, Q.; Larrue, C.; Saland, E.; Bertoli, S.; Sarry, J.-E.; Besson, A.; Manenti, S.; Joffre, C.; Mansat-De Mas, V. Oncogenic FLT3-ITD supports autophagy via ATF4 in acute myeloid leukemia. Oncogene 2018, 37, 787–797. [Google Scholar] [CrossRef] [Green Version]
- Mohamadimaram, M.; Farsani, M.A.; Mirzaeian, A.; Shahsavan, S.; Hajifathali, A.; Parkhihdeh, S.; Mohammadi, M.H. Evaluation of ATG7 and Light Chain 3 (LC3) Autophagy Genes Expression in AML Patients. Iran. J. Pharm. Res. 2019, 18, 1060–1066. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Britschgi, A.; Schläfli, A.M.; Humbert, M.; Shan-Krauer, D.; Batliner, J.; Federzoni, E.A.; Ernst, M.; Torbett, B.E.; Yousefi, S.; et al. Low Autophagy (ATG) Gene Expression Is Associated with an Immature AML Blast Cell Phenotype and Can Be Restored during AML Differentiation Therapy. Oxidative Med. Cell. Longev. 2018, 2018, 1482795. [Google Scholar] [CrossRef]
- Wu, M.-Y.; Wang, S.-F.; Cai, C.-Z.; Tan, J.-Q.; Li, M.; Lu, J.-J.; Chen, X.-P.; Wang, Y.-T.; Zheng, W.; Lu, J.-H. Natural autophagy blockers, dauricine (DAC) and daurisoline (DAS), sensitize cancer cells to camptothecin-induced toxicity. Oncotarget 2017, 8, 77673–77684. [Google Scholar] [CrossRef]
- Wu, J.; Hu, G.; Dong, Y.; Ma, R.; Yu, Z.; Jiang, S.; Han, Y.; Yu, K.; Zhang, S. Matrine induces Akt/mTOR signalling inhibition-mediated autophagy and apoptosis in acute myeloid leukaemia cells. J. Cell. Mol. Med. 2017, 21, 1171–1181. [Google Scholar] [CrossRef]
- Zhu, H.-Y.; Huang, Z.-X.; Chen, G.-Q.; Sheng, F.; Zheng, Y.-S. Typhaneoside prevents acute myeloid leukemia (AML) through suppressing proliferation and inducing ferroptosis associated with autophagy. Biochem. Biophys. Res. Commun. 2019, 516, 1265–1271. [Google Scholar] [CrossRef]
- Piya, S.; Kornblau, S.M.; Ruvolo, V.R.; Mu, H.; Ruvolo, P.P.; McQueen, T.; Davis, R.E.; Hail, N., Jr.; Kantarjian, H.; Andreeff, M.; et al. Atg7 suppression enhances chemotherapeutic agent sensitivity and overcomes stroma-mediated chemoresistance in acute myeloid leukemia. Blood 2016, 128, 1260–1269. [Google Scholar] [CrossRef] [Green Version]
- Rothe, K.; Lin, H.; Lin, K.B.L.; Leung, A.; Wang, H.M.; Malekesmaeili, M.; Brinkman, R.R.; Forrest, D.L.; Gorski, S.; Jiang, X. The core autophagy protein ATG4B is a potential biomarker and therapeutic target in CML stem/progenitor cells. Blood 2014, 123, 3622–3634. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Mei, S.; Meng, W.; Xue, S.; Jiang, L.; Yang, Y.; Hui, L.; Chen, Y.; Guan, M.-X. CXCR4-mediated signaling regulates autophagy and influences acute myeloid leukemia cell survival and drug resistance. Cancer Lett. 2018, 425, 1–12. [Google Scholar] [CrossRef]
- Liu, S.; Lu, W.; Li, S.; Li, S.; Liu, J.; Xing, Y.; Zhang, S.; Zhou, J.Z.; Xing, H.; Xu, Y.; et al. Identification of JL1037 as a novel, specific, reversible lysine-specific demethylase 1 inhibitor that induce apoptosis and autophagy of AML cells. Oncotarget 2017, 8, 31901–31914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roman, E.; Smith, A.G. Epidemiology of lymphomas. Histopathology 2011, 58, 4–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mugnaini, E.N.; Ghosh, N. Lymphoma. Prim. Care 2016, 43, 661–675. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H. Targeting autophagy in lymphomas: A double-edged sword? Int. J. Hematol. 2018, 107, 502–512. [Google Scholar] [CrossRef]
- Steidl, C.; Connors, J.M.; Gascoyne, R.D. Molecular Pathogenesis of Hodgkin’s Lymphoma: Increasing Evidence of the Importance of the Microenvironment. J. Clin. Oncol. 2011, 29, 1812–1826. [Google Scholar] [CrossRef]
- Birkenmeier, K.; Moll, K.; Newrzela, S.; Hartmann, S.; Dröse, S.; Hansmann, M.-L. Basal autophagy is pivotal for Hodgkin and Reed-Sternberg cells’ survival and growth revealing a new strategy for Hodgkin lymphoma treatment. Oncotarget 2016, 7, 46579–46588. [Google Scholar] [CrossRef] [Green Version]
- Amaravadi, R.K.; Yu, D.; Lum, J.J.; Bui, T.; Christophorou, M.A.; Evan, G.I.; Thomas-Tikhonenko, A.; Thompson, C.B. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J. Clin. Investig. 2007, 117, 326–336. [Google Scholar] [CrossRef] [Green Version]
- Pierdominici, M.; Maselli, A.; Locatelli, S.L.; Ciarlo, L.; Careddu, G.; Patrizio, M.; Ascione, B.; Tinari, A.; Carlo-Stella, C.; Malorni, W.; et al. Estrogen receptor β ligation inhibits Hodgkin lymphoma growth by inducing autophagy. Oncotarget 2017, 8, 8522–8535. [Google Scholar] [CrossRef] [Green Version]
- Klein, J.M.; Henke, A.; Sauer, M.; Bessler, M.; Reiners, K.S.; Engert, A.; Hansen, H.P.; Von Strandmann, E.P. The Histone Deacetylase Inhibitor LBH589 (Panobinostat) Modulates the Crosstalk of Lymphocytes with Hodgkin Lymphoma Cell Lines. PLoS ONE 2013, 8, e79502. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; Lei, H.; He, M.; Gong, R.; Wang, Y.; He, X.; Li, G.; Pang, P.; Li, X.; Yu, S.; et al. Melatonin triggers autophagic cell death by regulating RORC in Hodgkin lymphoma. Biomed. Pharmacother. 2020, 123, 109811. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, S.G.; Fisher, R.I. The epidemiology of non-Hodgkin’s lymphoma. Oncogene 2004, 23, 6524–6534. [Google Scholar] [CrossRef] [Green Version]
- Nicotra, G.; Mercalli, F.; Peracchio, C.; Castino, R.; Follo, C.; Valente, G.; Isidoro, C. Autophagy-active beclin-1 correlates with favourable clinical outcome in non-Hodgkin lymphomas. Mod. Pathol. 2010, 23, 937–950. [Google Scholar] [CrossRef] [Green Version]
- Tan, P.; He, L.; Xing, C.; Mao, J.; Yu, X.; Zhu, M.; Diao, L.; Han, L.; Zhou, Y.; You, J.M.; et al. Myeloid loss of Beclin 1 promotes PD-L1hi precursor B cell lymphoma development. J. Clin. Investig. 2019, 129, 5261–5277. [Google Scholar] [CrossRef]
- Wang, Z.C.; Liu, Y.; Wang, H.; Han, Q.K.; Lu, C. Research on the relationship between artesunate and Raji cell autophagy and apoptosis of Burkitt’s lymphoma and its mechanism. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 2238–2243. [Google Scholar] [PubMed]
- Dong, X.; Qin, J.; Ma, J.; Zeng, Q.; Zhang, H.; Zhang, R.; Liu, C.; Xu, C.; Zhang, S.; Huang, S.; et al. BAFF inhibits autophagy promoting cell proliferation and survival by activating Ca2+-CaMKII-dependent Akt/mTOR signaling pathway in normal and neoplastic B-lymphoid cells. Cell. Signal. 2018, 53, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Wen, Y.; Zhou, M.; Li, J.; Wang, T.; Xu, P.; Ouyang, J. Ouabain induces apoptosis and autophagy in Burkitt’s lymphoma Raji cells. Biomed. Pharmacother. 2016, 84, 1841–1848. [Google Scholar] [CrossRef]
- Fan, J.; Zeng, X.; Li, Y.; Wang, S.; Wang, Z.; Sun, Y.; Gao, H.; Zhang, G.; Feng, M.; Ju, D. Autophagy Plays a Critical Role in ChLym-1-Induced Cytotoxicity of Non-Hodgkin’s Lymphoma Cells. PLoS ONE 2013, 8, e72478. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhang, X.; Fan, J.; Chen, W.; Luan, J.; Nan, Y.; Wang, S.; Chen, Q.; Zhang, Y.; Wu, Y.; et al. Activating Autophagy Enhanced the Antitumor Effect of Antibody Drug Conjugates Rituximab-Monomethyl Auristatin E. Front. Immunol. 2018, 9, 1799. [Google Scholar] [CrossRef] [PubMed]
- Oh, P.S.; Hwang, H.; Jeong, H.S.; Kwon, J.; Kim, H.S.; Kim, M.; Lim, S.; Sohn, M.H.; Jeong, H.J. Blue light emitting diode induces apoptosis in lymphoid cells by stimulating autophagy. Int. J. Biochem. Cell Biol. 2016, 70, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.H.; Cheng, S.; Zheng, Z.; Wang, L.; Shen, Y.; Shen, Z.X.; Chen, S.J.; Zhao, W.L. Histone deacetylase inhibitor potentiated the ability of MTOR inhibitor to induce autophagic cell death in Burkitt leukemia/lymphoma. J. Hematol. Oncol. 2013, 6, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, L.; Xie, L.; Zuo, C.; Ma, Z.; Zhang, Y.; Zhu, Y.; Gao, J. Targeting mTOR/p70S6K/glycolysis signaling pathway restores glucocorticoid sensitivity to 4E-BP1 null Burkitt Lymphoma. BMC Cancer 2015, 15, 529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Zeng, X.; Li, Y.; Wang, S.; Yang, P.; Cao, Z.; Wang, Z.; Song, P.; Mei, X.; Ju, D. A novel therapeutic approach against B-cell non-Hodgkin’s lymphoma through co-inhibition of Hedgehog signaling pathway and autophagy. Tumor Biol. 2015, 37, 7305–7314. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Li, Y.; Fan, J.; Zhao, H.; Xian, Z.; Sun, Y.; Wang, Z.; Wang, S.; Zhang, G.; Ju, D. Recombinant human arginase induced caspase-dependent apoptosis and autophagy in non-Hodgkin’s lymphoma cells. Cell Death Dis. 2013, 4, e840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campo, E.; Rule, S. Mantle cell lymphoma: Evolving management strategies. Blood 2015, 125, 48–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Chen, Z.; Miranda, R.N.; Medeiros, L.J.; McCarty, N. TG2 and NF-κB Signaling Coordinates the Survival of Mantle Cell Lymphoma Cells via IL6-Mediated Autophagy. Cancer Res. 2016, 76, 6410–6423. [Google Scholar] [CrossRef] [Green Version]
- Rosich, L.; Colomer, D.; Roué, G. Autophagy controls everolimus (RAD001) activity in mantle cell lymphoma. Autophagy 2013, 9, 115–117. [Google Scholar] [CrossRef] [Green Version]
- Alinari, L.; Baiocchi, R.A.; Prætorius-Ibba, M. FTY720-induced blockage of autophagy enhances anticancer efficacy of milatuzumab in mantle cell lymphoma: Is FTY720 the next autophagy-blocking agent in lymphoma treatment? Autophagy 2012, 8, 416–417. [Google Scholar] [CrossRef] [Green Version]
- Heine, S.; Kleih, M.; Giménez, N.; Böpple, K.; Ott, G.; Colomer, D.; Aulitzky, W.E.; van der Kuip, H.; Silkenstedt, E. Cyclin D1-CDK4 activity drives sensitivity to bortezomib in mantle cell lymphoma by blocking autophagy-mediated proteolysis of NOXA. J. Hematol. Oncol. 2018, 11, 112. [Google Scholar] [CrossRef] [PubMed]
- Yazbeck, V.Y.; Buglio, D.; Georgakis, G.V.; Li, Y.; Iwado, E.; Romaguera, J.E.; Kondo, S.; Younes, A. Temsirolimus downregulates p21 without altering cyclin D1 expression and induces autophagy and synergizes with vorinostat in mantle cell lymphoma. Exp. Hematol. 2008, 36, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-B.; Rahemtullah, A.; Hochberg, E. Primary Effusion Lymphoma. Oncologist 2007, 12, 569–576. [Google Scholar] [CrossRef] [Green Version]
- Narkhede, M.; Arora, S.; Ujjani, C. Primary effusion lymphoma: Current perspectives. OncoTargets Ther. 2018, 11, 3747–3754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alam, M.; Kariya, R.; Kawaguchi, A.; Matsuda, K.; Kudo, E.; Okada, S. Inhibition of autophagy by chloroquine induces apoptosis in primary effusion lymphoma in vitro and in vivo through induction of endoplasmic reticulum stress. Apoptosis 2016, 21, 1191–1201. [Google Scholar] [CrossRef] [PubMed]
- Granato, M.; Chiozzi, B.; Filardi, M.R.; Lotti, L.V.; Di Renzo, L.; Faggioni, A.; Cirone, M. Tyrosine kinase inhibitor tyrphostin AG490 triggers both apoptosis and autophagy by reducing HSF1 and Mcl-1 in PEL cells. Cancer Lett. 2015, 366, 191–197. [Google Scholar] [CrossRef]
- Granato, M.; Rizzello, C.; Gilardini Montani, M.S.; Cuomo, L.; Vitillo, M.; Santarelli, R.; Gonnella, R.; D’Orazi, G.; Faggioni, A.; Cirone, M. Quercetin induces apoptosis and autophagy in primary effusion lymphoma cells by inhibiting PI3K/AKT/mTOR and STAT3 signaling pathways. J. Nutr. Biochem. 2017, 41, 124–136. [Google Scholar] [CrossRef]
- Granato, M.; Santarelli, R.; Lotti, L.V.; Di Renzo, L.; Gonnella, R.; Garufi, A.; Trivedi, P.; Frati, L.; D’Orazi, G.; Faggioni, A.; et al. JNK and Macroautophagy Activation by Bortezomib Has a Pro-Survival Effect in Primary Effusion Lymphoma Cells. PLoS ONE 2013, 8, e75965. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Young, K.H.; Medeiros, L.J. Diffuse large B-cell lymphoma. Pathology 2018, 50, 74–87. [Google Scholar] [CrossRef] [Green Version]
- Friedberg, J.W. Relapsed/Refractory Diffuse Large B-Cell Lymphoma. Hematol. Am. Soc. Hematol. Educ. Program 2011, 2011, 498–505. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Chiang, Y.-L.; Lyssiotis, C.A.; Teater, M.R.; Hong, J.Y.; Shen, H.; Wang, L.; Hu, J.; Jing, H.; Chen, Z.; et al. Non-oncogene Addiction to SIRT3 Plays a Critical Role in Lymphomagenesis. Cancer Cell 2019, 35, 916–931.e9. [Google Scholar] [CrossRef] [PubMed]
- Li, L.-J.; Chai, Y.; Guo, X.-J.; Chu, S.-L.; Zhang, L.-S. The effects of the long non-coding RNA MALAT-1 regulated autophagy-related signaling pathway on chemotherapy resistance in diffuse large B-cell lymphoma. Biomed. Pharmacother. 2017, 89, 939–948. [Google Scholar] [CrossRef]
- Xiong, J.; Wang, L.; Fei, X.-C.; Jiang, X.-F.; Zheng, Z.; Zhao, Y.; Wang, C.-F.; Li, B.; Chen, S.-J.; Janin, A.; et al. MYC is a positive regulator of choline metabolism and impedes mitophagy-dependent necroptosis in diffuse large B-cell lymphoma. Blood Cancer J. 2017, 7, e582. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhou, X.; Zhang, Y.; Yang, J.; Xu, Y.; Zhao, Y.; Wang, X. CUL4B regulates autophagy via JNK signaling in diffuse large B-cell lymphoma. Cell Cycle 2019, 18, 379–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerhan, J.R. Epidemiology of Follicular Lymphoma. Hematol. Oncol. Clin. N. Am. 2020, 34, 631–646. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Roulland, S.; Gloghini, A.; Younes, A.; Von Keudell, G.; López-Guillermo, A.; FitzGibbon, J. Follicular lymphoma. Nat. Rev. Dis. Prim. 2019, 5, 83. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, A.; Marzec, J.; Clear, A.; Petty, R.D.; Coutinho, R.; Matthews, J.; Wilson, A.; Iqbal, S.; Calaminici, M.; Gribben, J.G.; et al. Dysregulation of autophagy in human follicular lymphoma is independent of overexpression of BCL-2. Oncotarget 2014, 5, 11653–11668. [Google Scholar] [CrossRef] [Green Version]
- Giatromanolaki, A.; Koukourakis, M.I.; Pouliliou, S.; Gatter, K.C.; Pezzella, F.; Harris, A.L.; Sivridis, E. Overexpression of LC3A autophagy protein in follicular and diffuse large B-cell lymphomas. Hematol. Oncol. Stem. Cell Ther. 2013, 6, 20–25. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Gatica, D.; Ying, Z.X.; Peterson, L.F.; Kim, P.K.; Bernard, D.; Saiya-Cork, K.; Wang, S.; Kaminski, M.S.; Chang, A.E.; et al. Follicular lymphoma–associated mutations in vacuolar ATPase ATP6V1B2 activate autophagic flux and mTOR. J. Clin. Investig. 2019, 129, 1626–1640. [Google Scholar] [CrossRef] [Green Version]
- Shah, U.A.; Shah, N.; Qiao, B.; Acuna-Villaorduna, A.; Pradhan, K.; Herrera, D.A.; Sica, R.A.; Shastri, A.; Mantzaris, I.; Derman, O.; et al. Epidemiology and survival trend of adult T-cell leukemia/lymphoma in the United States. Cancer 2020, 126, 567–574. [Google Scholar] [CrossRef]
- Phan, A.; Veldman, R.; Lechowicz, M.J. T-cell Lymphoma Epidemiology: The Known and Unknown. Curr. Hematol. Malign-Rep. 2016, 11, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Qiu, L.; Li, T.; Wang, X.; Deng, R.; Yi, H.; Su, Y.; Fan, F.Y. MiR-449a attenuates autophagy of T-cell lymphoma cells by downregulating ATG4B expression. BMB Rep. 2020, 53, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhang, J.; Zhang, X.; Li, X.; Li, L.; Li, Z.; Chen, R.; Zhang, L.; Wu, J.; Wang, X.; et al. AEG-1 is involved in hypoxia-induced autophagy and decreases chemosensitivity in T-cell lymphoma. Mol. Med. 2018, 24, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, K.-T.; Lin, M.-X.; Lin, S.-C.; Tung, Y.-T.; Lin, S.-H.; Lin, C.-C. Sinensetin induces apoptosis and autophagy in the treatment of human T-cell lymphoma. Anti-Cancer Drugs 2019, 30, 485–494. [Google Scholar] [CrossRef]
- Wang, L.; Yang, J.; Wang, H.-N.; Fu, R.-Y.; Liu, X.-D.; Piao, Y.-S.; Wei, L.-Q.; Wang, J.-W.; Zhang, L. LncRNA BCYRN1-induced autophagy enhances asparaginase resistance in extranodal NK/T-cell lymphoma. Theranostics 2021, 11, 925–940. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, E. Anaplastic Large-Cell Lymphoma, T-/Null-Cell Type. Oncologist 2006, 11, 831–840. [Google Scholar] [CrossRef] [Green Version]
- Mitou, G.; Frentzel, J.; Desquesnes, A.; Le Gonidec, S.; Alsaati, T.; Beau, I.; Lamant, L.; Meggetto, F.; Espinos, E.; Codogno, P.; et al. Targeting autophagy enhances the anti-tumoral action of crizotinib in ALK-positive anaplastic large cell lymphoma. Oncotarget 2015, 6, 30149–30164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, W.-Y.; Xiao, D.; Wang, L.; Dong, L.-H.; Yan, Z.-X.; Shen, Z.-X.; Chen, S.-J.; Chen, Y.; Zhao, W.-L. Therapeutic metformin/AMPK activation blocked lymphoma cell growth via inhibition of mTOR pathway and induction of autophagy. Cell Death Dis. 2012, 3, e275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Daghri, N.M.; Alokail, M.S.; Alkharfy, K.M.; Mohammed, A.K.; Abd-Alrahman, S.H.; Yakout, S.M.; E Amer, O.; Krishnaswamy, S. Fenugreek extract as an inducer of cellular death via autophagy in human T lymphoma Jurkat cells. BMC Complement. Altern. Med. 2012, 12, 202. [Google Scholar] [CrossRef] [Green Version]
- Maiuri, M.C.; Tasdemir, E.; Criollo, A.; Morselli, E.; Vicencio, J.M.; Carnuccio, R.; Kroemer, G. Control of autophagy by oncogenes and tumor suppressor genes. Cell Death Differ. 2009, 16, 87–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchihara, K.; Fujii, S.; Esumi, H. Autophagy and cancer: Dynamism of the metabolism of tumor cells and tissues. Cancer Lett. 2009, 278, 130–138. [Google Scholar] [CrossRef]
- Aita, V.M.; Liang, X.H.; Murty, V.; Pincus, D.L.; Yu, W.; Cayanis, E.; Kalachikov, S.; Gilliam, T.; Levine, B. Cloning and Genomic Organization of Beclin 1, a Candidate Tumor Suppressor Gene on Chromosome 17q21. Genomics 1999, 59, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Bekri, S.; Adélaïde, J.; Merscher, S.; Grosgeorge, J.; Caroli-Bosc, F.; Perucca-Lostanlen, D.; Kelley, P.; Pébusque, M.-J.; Theillet, C.; Birnbaum, D.; et al. Detailed map of a region commonly amplified at 11q13→q14 in human breast carcinoma. Cytogenet. Genome Res. 1997, 79, 125–131. [Google Scholar] [CrossRef]
- Liang, C.; Jung, J.U. Autophagy genes as tumor suppressors. Curr. Opin. Cell Biol. 2010, 22, 226–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebovitz, C.B.; Robertson, A.G.; Goya, R.; Jones, S.; Morin, R.; A Marra, M.; Gorski, S.M. Cross-cancer profiling of molecular alterations within the human autophagy interaction network. Autophagy 2015, 11, 1668–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothe, K.; Porter, V.; Jiang, X. Current Outlook on Autophagy in Human Leukemia: Foe in Cancer Stem Cells and Drug Resistance, Friend in New Therapeutic Interventions. Int. J. Mol. Sci. 2019, 20, 461. [Google Scholar] [CrossRef] [Green Version]
- Jaganathan, S.; Malek, E.; Vallabhapurapu, S.; Vallabhapurapu, S.; Driscoll, J.J. Bortezomib induces AMPK-dependent autophagosome formation uncoupled from apoptosis in drug resistant cells. Oncotarget 2014, 5, 12358–12370. [Google Scholar] [CrossRef] [Green Version]
- Massey, D.C.; Parkes, M. Genome-wide Association Scanning Highlights Two Autophagy Genes, ATG16L1 and IRGM, as Being Significantly Associated with Crohn’s Disease. Autophagy 2007, 3, 649–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rioux, J.D.; Xavier, R.J.; Taylor, K.D.; Silverberg, M.S.; Goyette, P.; Huett, A.; Green, T.; Kuballa, P.; Barmada, M.M.; Datta, L.W.; et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 2007, 39, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.-J.; Lu, X.-L.; Lv, J.-C.; Yang, H.-Z.; Qin, L.-X.; Zhao, M.-H.; Su, Y.; Li, Z.-G.; Zhang, H. Genetic association of PRDM1-ATG5 intergenic region and autophagy with systemic lupus erythematosus in a Chinese population. Ann. Rheum. Dis. 2011, 70, 1330–1337. [Google Scholar] [CrossRef] [PubMed]
- Verstockt, B.; Smith, K.; Lee, J.C. Genome-wide association studies in Crohn’s disease: Past, present and future. Clin. Transl. Immunol. 2018, 7, e1001. [Google Scholar] [CrossRef]
- Portilla-Fernandez, E.; Ghanbari, M.; Van Meurs, J.B.J.; Danser, A.H.J.; Franco, O.H.; Muka, T.; Roks, A.J.; Dehghan, A. Dissecting the association of autophagy-related genes with cardiovascular diseases and intermediate vascular traits: A population-based approach. PLoS ONE 2019, 14, e0214137. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.-H.; Ko, T.-M.; Chen, C.-H.; Chang, Y.-J.; Lu, L.-S.; Chang, C.-H.; Huang, K.-L.; Chang, T.-Y.; Lee, J.-D.; Chang, K.-C.; et al. A genome-wide association study links small-vessel ischemic stroke to autophagy. Sci. Rep. 2017, 7, 15229. [Google Scholar] [CrossRef] [Green Version]
- Park, H.; Kang, J.-H.; Lee, S. Autophagy in Neurodegenerative Diseases: A Hunter for Aggregates. Int. J. Mol. Sci. 2020, 21, 3369. [Google Scholar] [CrossRef] [PubMed]
- Gan-Or, Z.; Dion, P.A.; Rouleau, G.A. Genetic perspective on the role of the autophagy-lysosome pathway in Parkinson disease. Autophagy 2015, 11, 1443–1457. [Google Scholar] [CrossRef] [Green Version]
- Do, C.B.; Tung, J.Y.; Dorfman, E.; Kiefer, A.K.; Drabant, E.M.; Francke, U.; Mountain, J.L.; Goldman, S.M.; Tanner, C.M.; Langston, J.W.; et al. Web-Based Genome-Wide Association Study Identifies Two Novel Loci and a Substantial Genetic Component for Parkinson’s Disease. PLoS Genet. 2011, 7, e1002141. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamargo-Gómez, I.; Fernández, Á.F.; Mariño, G. Pathogenic Single Nucleotide Polymorphisms on Autophagy-Related Genes. Int. J. Mol. Sci. 2020, 21, 8196. [Google Scholar] [CrossRef] [PubMed]
- Fraiberg, M.; Elazar, Z. Genetic defects of autophagy linked to disease. Prog. Mol. Biol. Transl. Sci. 2020, 172, 293–323. [Google Scholar] [CrossRef] [PubMed]
- Went, M.; Sud, A.; Försti, A.; Halvarsson, B.-M.; Weinhold, N.; Kimber, S.; Van Duin, M.; Thorleifsson, G.; Holroyd, A.; Johnson, D.C.; et al. Identification of multiple risk loci and regulatory mechanisms influencing susceptibility to multiple myeloma. Nat. Commun. 2018, 9, 3707, Correction in Nat. Commun. 2019, 10, 213. [Google Scholar] [CrossRef]
- Castro, I.; Sampaio-Marques, B.; Areias, A.C.; Sousa, H.; Fernandes, Â.; Sanchez-Maldonado, J.; Cunha, C.; Carvalho, A.; Sainz, J.; Ludovico, P. Functional Genetic Variants in ATG10 Are Associated with Acute Myeloid Leukemia. Cancers 2021, 13, 1344. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.S.; Li, N.; Weinhold, N.; Försti, A.; Ali, M.; Van Duin, M.; Thorleifsson, G.; Johnson, D.C.; Chen, B.; Halvarsson, B.-M.; et al. Genome-wide association study identifies multiple susceptibility loci for multiple myeloma. Nat. Commun. 2016, 7, 12050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenberg, A.J.; Lee, A.M.; Serie, D.J.; McDonnell, S.K.; Cerhan, J.R.; Liebow, M.; Larson, D.R.; Colby, C.L.; Norman, A.D.; Kyle, R.A.; et al. Single-nucleotide polymorphism rs1052501 associated with monoclonal gammopathy of undetermined significance and multiple myeloma. Leukemia 2013, 27, 515–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pegliasco, J.; Hirsch, P.; Marzac, C.; Isnard, F.; Meniane, J.-C.; Deswarte, C.; Pellet, P.; Lemaitre, C.; Leroy, G.; Moraes, G.R.; et al. Germline ATG2B/GSKIP-containing 14q32 duplication predisposes to early clonal hematopoiesis leading to myeloid neoplasms. Leukemia 2022, 36, 126–137. [Google Scholar] [CrossRef]
- Saliba, J.; Saint-Martin, C.; Di Stefano, A.; Lenglet, G.; Marty, C.; Keren, B.; Pasquier, F.; Della Valle, V.; Secardin, L.; Leroy, G.; et al. Germline duplication of ATG2B and GSKIP predisposes to familial myeloid malignancies. Nat. Genet. 2015, 47, 1131–1140. [Google Scholar] [CrossRef] [PubMed]
- van der Velden, W.J.; Blijlevens, N.M.; Maas, F.M.; Schaap, N.P.; Jansen, J.H.; van der Reijden, B.A.; Feuth, T.; Dolstra, H.; Donnelly, J.P. NOD2 polymorphisms predict severe acute graft-versus-host and treatment-related mortality in T-cell-depleted haematopoietic stem cell transplantation. Bone Marrow Transplant. 2009, 44, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Varszegi, D.; Duga, B.; Melegh, B.I.; Sumegi, K.; Kisfali, P.; Maasz, A.; Melegh, B. Hodgkin Disease Therapy Induced Second Malignancy Susceptibility 6q21 Functional Variants in Roma and Hungarian Population Samples. Pathol. Oncol. Res. 2013, 20, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Best, T.; Li, D.; Skol, A.D.; Kirchhoff, T.; Jackson, S.A.; Yasui, Y.; Bhatia, S.; Strong, L.C.; Domchek, S.M.; Nathanson, K.; et al. Variants at 6q21 implicate PRDM1 in the etiology of therapy-induced second malignancies after Hodgkin’s lymphoma. Nat. Med. 2011, 17, 941–943. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García Ruiz, O.; Sánchez-Maldonado, J.M.; López-Nevot, M.Á.; García, P.; Macauda, A.; Hernández-Mohedo, F.; González-Sierra, P.A.; Martínez-Bueno, M.; Pérez, E.; Reyes-Zurita, F.J.; et al. Autophagy in Hematological Malignancies. Cancers 2022, 14, 5072. https://doi.org/10.3390/cancers14205072
García Ruiz O, Sánchez-Maldonado JM, López-Nevot MÁ, García P, Macauda A, Hernández-Mohedo F, González-Sierra PA, Martínez-Bueno M, Pérez E, Reyes-Zurita FJ, et al. Autophagy in Hematological Malignancies. Cancers. 2022; 14(20):5072. https://doi.org/10.3390/cancers14205072
Chicago/Turabian StyleGarcía Ruiz, Olga, José Manuel Sánchez-Maldonado, Miguel Ángel López-Nevot, Paloma García, Angelica Macauda, Francisca Hernández-Mohedo, Pedro Antonio González-Sierra, Manuel Martínez-Bueno, Eva Pérez, Fernando Jesús Reyes-Zurita, and et al. 2022. "Autophagy in Hematological Malignancies" Cancers 14, no. 20: 5072. https://doi.org/10.3390/cancers14205072
APA StyleGarcía Ruiz, O., Sánchez-Maldonado, J. M., López-Nevot, M. Á., García, P., Macauda, A., Hernández-Mohedo, F., González-Sierra, P. A., Martínez-Bueno, M., Pérez, E., Reyes-Zurita, F. J., Campa, D., Canzian, F., Jurado, M., Rodríguez-Sevilla, J. J., & Sainz, J. (2022). Autophagy in Hematological Malignancies. Cancers, 14(20), 5072. https://doi.org/10.3390/cancers14205072