
CLEFMA Induces the Apoptosis of Oral Squamous Carcinoma Cells through the Regulation of the P38/HO-1 Signalling Pathway



Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. MTT Cell Viability Assay
2.3. Colony Formation Assay
2.4. Sub-G1 Phase Ratio Analysis
2.5. Apoptotic Cell Death Assay
2.6. Proteome Profiler Human Apoptosis Array
2.7. Western Blot Analysis
2.8. Detection of Active Caspase-3
2.9. Small Interfering (Si) RNA Transfection
2.10. Tumour Growth in Nude Mice Model
2.11. Statistical Analysis
3. Results
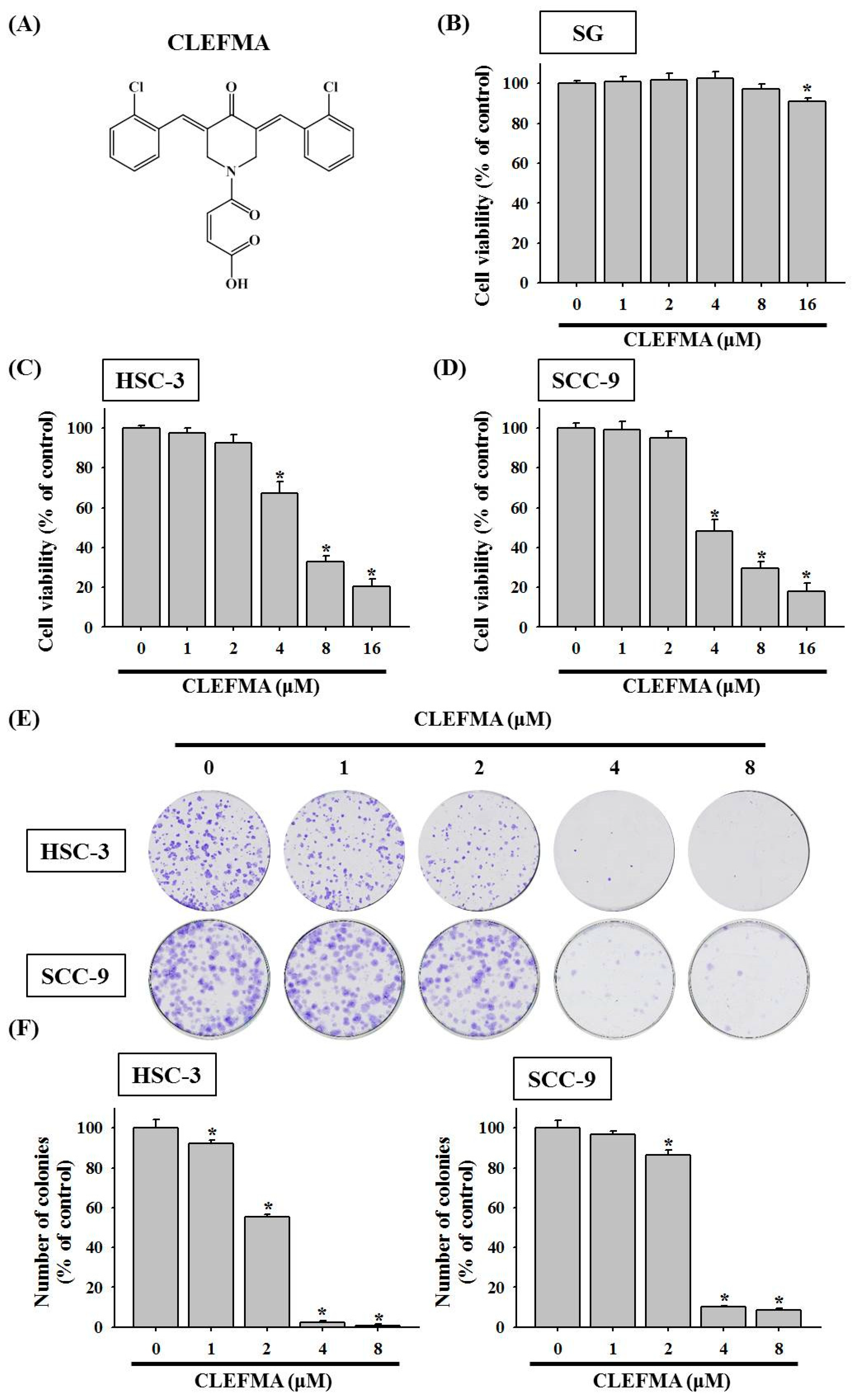
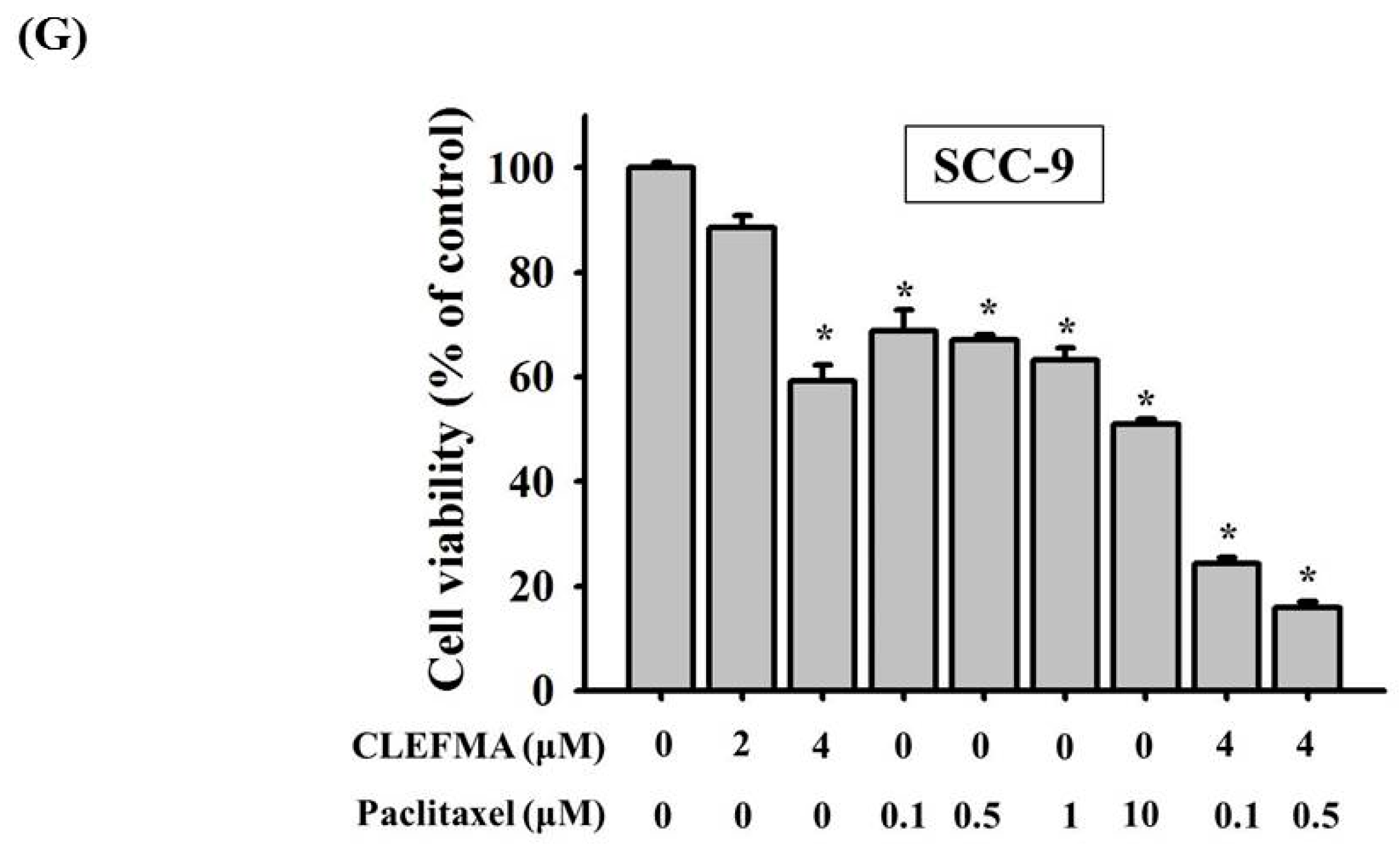
3.1. Decline in Cell Viability by CLEFMA in OSCC
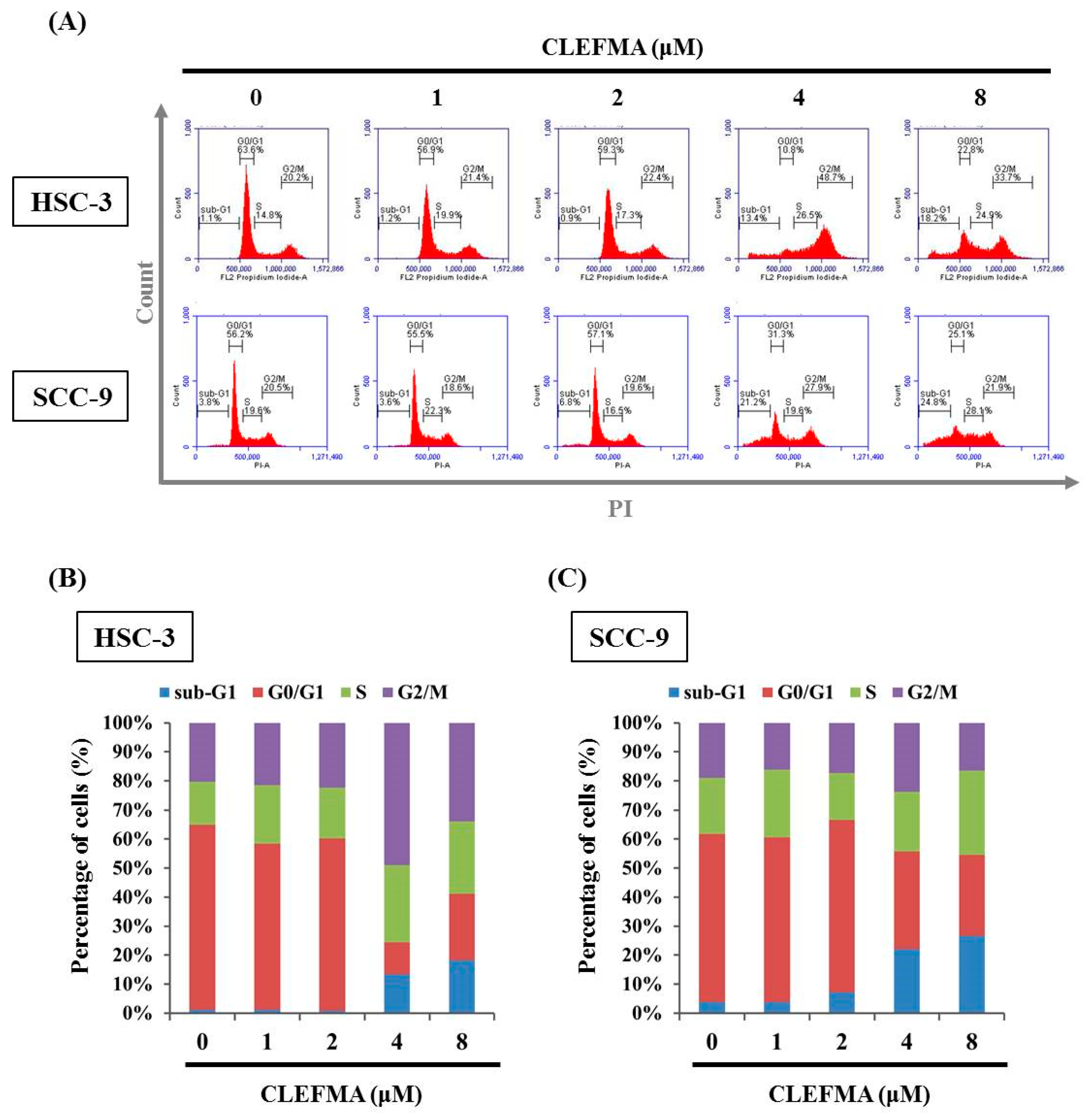
3.2. Increase in Apoptotic Cell Distribution by CLEFMA in OSCC
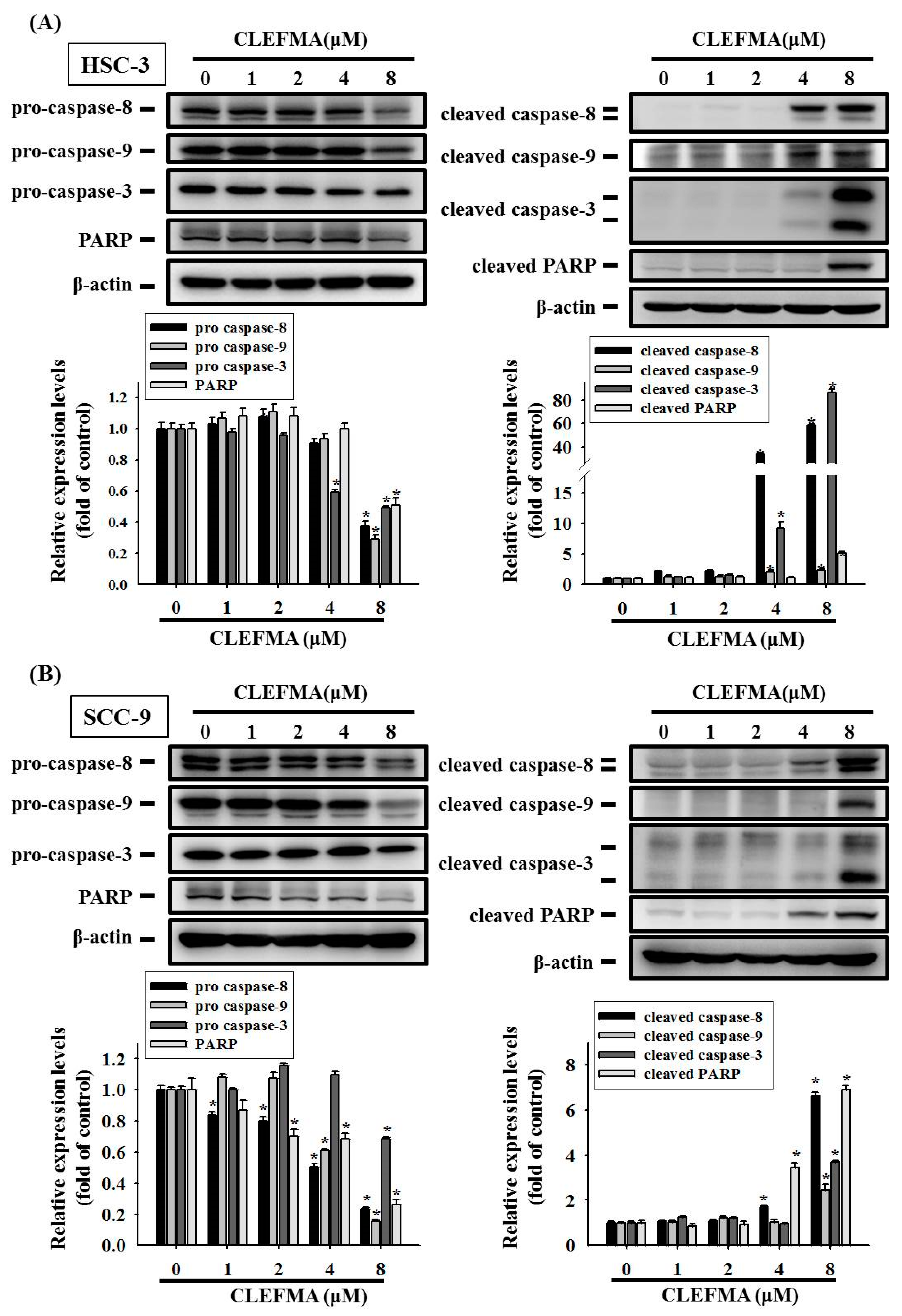
3.3. Apoptotic Effect of CLEFMA in OSCC
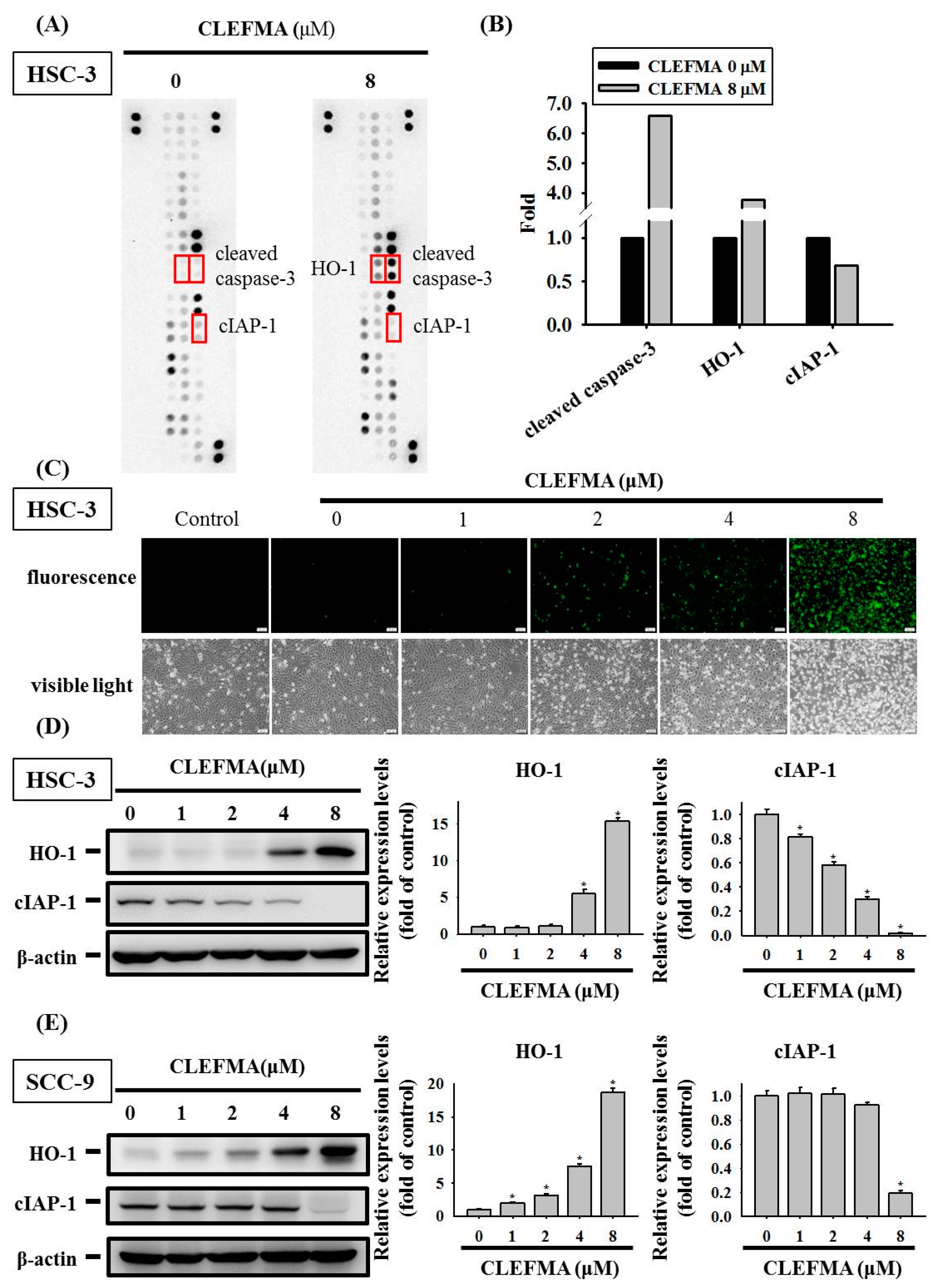
3.4. Regulation of cIAP-1 and HO-1 by CLEFMA-Induced Caspase-Mediated Apoptosis in OSCC
3.5. Activation of MAPK Signalling Cascades by CLEFMA in OSCC
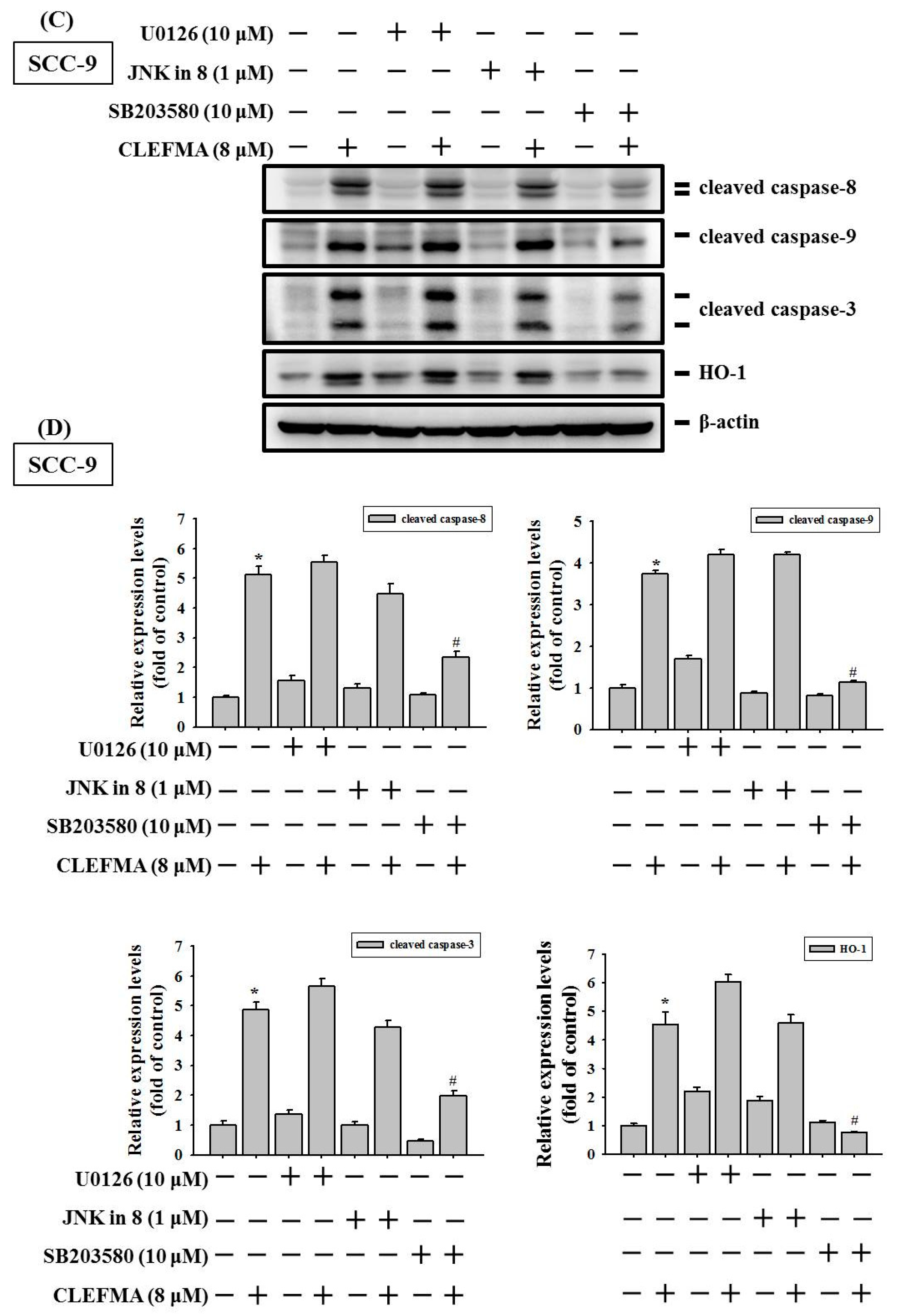
3.6. HO-1 Is Involved in Response to CLEFMA-Induced Caspase-Mediated Apoptosis in OSCC
3.7. Induction of Caspase-Mediated Apoptosis by CLEFMA Is Dependent on the Activation of p38 in OSCC
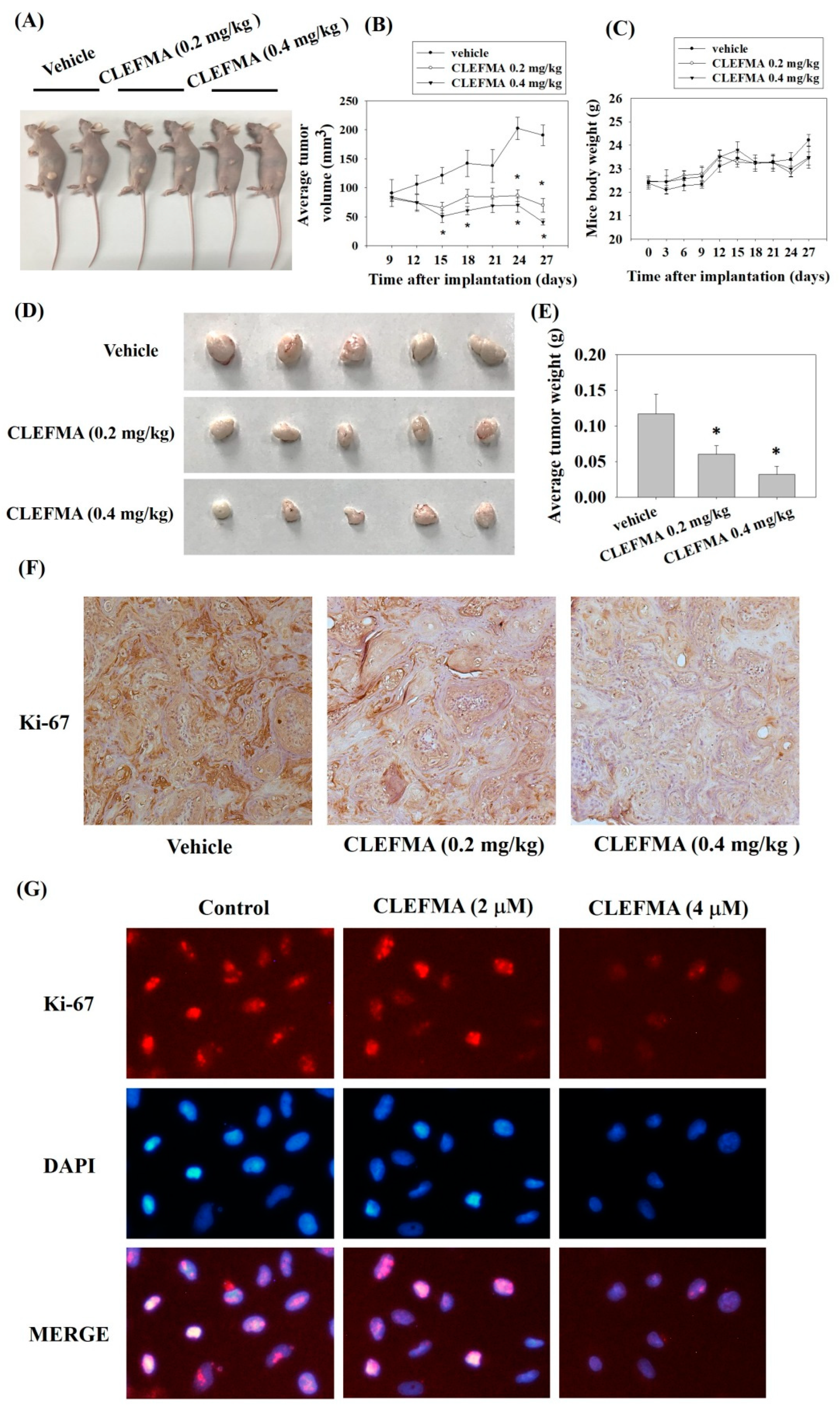
3.8. Inhibition of Tumour Growth by CLEFMA Treatment In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging 2016, 8, 603–619. [Google Scholar] [CrossRef] [Green Version]
- McComb, S.; Chan, P.K.; Guinot, A.; Hartmannsdottir, H.; Jenni, S.; Dobay, M.P.; Bourquin, J.P.; Bornhauser, B.C. Efficient apoptosis requires feedback amplification of upstream apoptotic signals by effector caspase-3 or -7. Sci. Adv. 2019, 5, eaau9433. [Google Scholar] [CrossRef] [Green Version]
- Bouraoui, Y.; Achour, M.; Royuela, M.; Oueslati, R. Immune profiling of human prostate epithelial cells determined by expression of p38/traf-6/erk map kinases pathways. Kaohsiung J. Med. Sci. 2018, 34, 125–133. [Google Scholar] [CrossRef]
- Jian, K.L.; Zhang, C.; Shang, Z.C.; Yang, L.; Kong, L.Y. Eucalrobusone c suppresses cell proliferation and induces ros-dependent mitochondrial apoptosis via the p38 mapk pathway in hepatocellular carcinoma cells. Phytomedicine 2017, 25, 71–82. [Google Scholar] [CrossRef]
- Chien, M.H.; Yang, W.E.; Yang, Y.C.; Ku, C.C.; Lee, W.J.; Tsai, M.Y.; Lin, C.W.; Yang, S.F. Dual targeting of the p38 mapk-ho-1 axis and ciap1/xiap by demethoxycurcumin triggers caspase-mediated apoptotic cell death in oral squamous cell carcinoma cells. Cancers 2020, 12, 703. [Google Scholar] [CrossRef] [Green Version]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef]
- Lee, G.; Park, J.S.; Lee, E.J.; Ahn, J.H.; Kim, H.S. Anti-inflammatory and antioxidant mechanisms of urolithin b in activated microglia. Phytomedicine 2018, 55, 50–57. [Google Scholar] [CrossRef]
- Al-Ishaq, R.K.; Overy, A.J.; Büsselberg, D. Phytochemicals and gastrointestinal cancer: Cellular mechanisms and effects to change cancer progression. Biomolecules 2020, 10, 105. [Google Scholar] [CrossRef] [Green Version]
- Yadav, V.R.; Sahoo, K.; Awasthi, V. Preclinical evaluation of 4-[3,5-bis(2-chlorobenzylidene)-4-oxo-piperidine-1-yl]-4-oxo-2-butenoic acid, in a mouse model of lung cancer xenograft. Br. J. Pharmacol. 2013, 170, 1436–1448. [Google Scholar] [CrossRef] [Green Version]
- Rao, G.; Houson, H.; Nkepang, G.; Yari, H.; Teng, C.; Awasthi, V. Induction of gut proteasome activity in hemorrhagic shock and its recovery by treatment with diphenyldihaloketones clefma and ef24. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G318–G327. [Google Scholar] [CrossRef]
- Yang, J.S.; Lin, R.C.; Hsieh, Y.H.; Wu, H.H.; Li, G.C.; Lin, Y.C.; Yang, S.F.; Lu, K.H. Clefma activates the extrinsic and intrinsic apoptotic processes through jnk1/2 and p38 pathways in human osteosarcoma cells. Molecules 2019, 24, 3280. [Google Scholar] [CrossRef] [Green Version]
- Monroe, J.D.; Hodzic, D.; Millay, M.H.; Patty, B.G.; Smith, M.E. Anti-cancer and ototoxicity characteristics of the curcuminoids, clefma and ef24, in combination with cisplatin. Molecules 2019, 24, 3889. [Google Scholar] [CrossRef] [Green Version]
- Lagisetty, P.; Vilekar, P.; Sahoo, K.; Anant, S.; Awasthi, V. Clefma-an anti-proliferative curcuminoid from structure-activity relationship studies on 3,5-bis(benzylidene)-4-piperidones. Bioorg. Med. Chem. 2010, 18, 6109–6120. [Google Scholar] [CrossRef] [Green Version]
- Smulow, J.B.; Glickman, I. An epithelial-like cell line in continuous culture from normal adult human gingiva. Proc. Soc. Exp. Biol. Med. 1966, 121, 1294–1296. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.H.; Chu, S.C.; Yang, S.F.; Hsieh, Y.S.; Lee, C.Y.; Chen, P.N. Induction of apoptotic but not autophagic cell death by cinnamomum cassia extracts on human oral cancer cells. J. Cell Physiol. 2019, 234, 5289–5303. [Google Scholar] [CrossRef]
- Yang, S.F.; Chen, Y.S.; Chien, H.W.; Wang, K.; Lin, C.L.; Chiou, H.L.; Lee, C.Y.; Chen, P.N.; Hsieh, Y.H. Melatonin attenuates epidermal growth factor-induced cathepsin s expression in arpe-19 cells: Implications for proliferative vitreoretinopathy. J. Pineal Res. 2020, 68, e12615. [Google Scholar] [CrossRef] [PubMed]
- Chien, M.H.; Shih, P.C.; Ding, Y.F.; Chen, L.H.; Hsieh, F.K.; Tsai, M.Y.; Li, P.Y.; Lin, C.W.; Yang, S.F. Curcumin analog, go-y078, induces ho-1 transactivation-mediated apoptotic cell death of oral cancer cells by triggering mapk pathways and ap-1 DNA-binding activity. Expert Opin. Ther. Targets 2022, 26, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Su, C.W.; Chuang, C.Y.; Chen, Y.T.; Yang, W.E.; Pan, Y.P.; Lin, C.W.; Yang, S.F. Flll32 triggers caspase-mediated apoptotic cell death in human oral cancer cells by regulating the p38 pathway. Int. J. Mol. Sci. 2021, 22, 11860. [Google Scholar] [CrossRef]
- Chen, C.W.; Hsieh, M.J.; Ju, P.C.; Hsieh, Y.H.; Su, C.W.; Chen, Y.L.; Yang, S.F.; Lin, C.W. Curcumin analog ho-3867 triggers apoptotic pathways through activating jnk1/2 signalling in human oral squamous cell carcinoma cells. J. Cell. Mol. Med. 2022, 26, 2273–2284. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Hsieh, M.J.; Hsieh, Y.S.; Chang, Y.C.; Chen, P.N.; Yang, S.F.; Ho, H.Y.; Chou, Y.E.; Lin, C.W. Antimetastatic effects of rheum palmatum l. Extract on oral cancer cells. Environ. Toxicol. 2017, 32, 2287–2294. [Google Scholar] [CrossRef]
- Chen, P.N.; Yang, S.F.; Yu, C.C.; Lin, C.Y.; Huang, S.H.; Chu, S.C.; Hsieh, Y.S. Duchesnea indica extract suppresses the migration of human lung adenocarcinoma cells by inhibiting epithelial-mesenchymal transition. Environ. Toxicol. 2017, 32, 2053–2063. [Google Scholar] [CrossRef] [PubMed]
- Derakhshan, A.; Chen, Z.; van Waes, C. Therapeutic small molecules target inhibitor of apoptosis proteins in cancers with deregulation of extrinsic and intrinsic cell death pathways. Clin. Cancer Res. 2017, 23, 1379–1387. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, Y.H.; Su, S.C.; Lin, C.W.; Chao, Y.H.; Yang, W.E.; Yang, S.F. Pathological and therapeutic aspects of matrix metalloproteinases: Implications in childhood leukemia. Cancer Metastasis Rev. 2019, 38, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.H.; Lin, C.W.; Hsieh, Y.H.; Su, S.C.; Reiter, R.J.; Yang, S.F. New insights into antimetastatic signaling pathways of melatonin in skeletomuscular sarcoma of childhood and adolescence. Cancer Metastasis Rev. 2020, 39, 303–320. [Google Scholar] [CrossRef]
- Lu, K.H.; Lu, E.W.; Lin, C.W.; Yang, J.S.; Yang, S.F. New insights into molecular and cellular mechanisms of zoledronate in human osteosarcoma. Pharmacol. Ther. 2020, 214, 107611. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A target for anticancer therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [Green Version]
- Wong, R.S. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, M.S.; Bishr, M.K.; Almutairi, F.M.; Ali, A.G. Inhibitors of apoptosis: Clinical implications in cancer. Apoptosis 2017, 22, 1487–1509. [Google Scholar] [CrossRef] [PubMed]
- Nagata, M.; Nakayama, H.; Tanaka, T.; Yoshida, R.; Yoshitake, Y.; Fukuma, D.; Kawahara, K.; Nakagawa, Y.; Ota, K.; Hiraki, A.; et al. Overexpression of ciap2 contributes to 5-fu resistance and a poor prognosis in oral squamous cell carcinoma. Br. J. Cancer 2011, 105, 1322–1330. [Google Scholar] [CrossRef]
- De Almagro, M.C.; Vucic, D. The inhibitor of apoptosis (iap) proteins are critical regulators of signaling pathways and targets for anti-cancer therapy. Exp. Oncol. 2012, 34, 200–211. [Google Scholar] [PubMed]
- Verma, A.K.; Ahmad, I.; Yadav, P.; Rahmani, A.H.; Khan, B.; Alsahli, M.A.; Joshi, P.C.; Ahmad, H.; Ali Beg, M.M. Expression and correlation of cell-free ciap-1 and ciap-2 mrna in breast cancer patients: A study from india. J. Oncol. 2020, 2020, 3634825. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Li, Z.; Bai, L.; Lin, Y. Nf-kappab in lung cancer, a carcinogenesis mediator and a prevention and therapy target. Front. Biosci. (Landmark Ed.) 2011, 16, 1172–1185. [Google Scholar] [CrossRef] [Green Version]
- Almeida, L.O.; Abrahao, A.C.; Rosselli-Murai, L.K.; Giudice, F.S.; Zagni, C.; Leopoldino, A.M.; Squarize, C.H.; Castilho, R.M. Nfκb mediates cisplatin resistance through histone modifications in head and neck squamous cell carcinoma (hnscc). FEBS Open Bio 2014, 4, 96–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.Y.; Chen, Y.C.; Shih, C.M.; Lin, C.M.; Cheng, C.H.; Chen, K.C.; Lin, C.W. The induction of heme oxygenase-1 suppresses heat shock protein 90 and the proliferation of human breast cancer cells through its byproduct carbon monoxide. Toxicol. Appl. Pharmacol. 2014, 274, 55–62. [Google Scholar] [CrossRef]
- Fang, J.; Islam, R.; Gao, S.; Zhang, C.; Kunisaki, R.; Sakaguchi, S.; Honda, N.; Zhou, J.R.; Yokomizo, K. Expression dynamics of heme oxygenase-1 in tumor cells and the host contributes to the progression of tumors. J. Pers. Med. 2021, 11, 1340. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Huang, L.Q.; He, H.S.; Yan, J.; Huang, C.; Wang, R.; Guan, Y.; Huang, D.P. Overexpression of heme oxygenase-1 in bone marrow stromal cells promotes multiple myeloma resistance through the jak2/stat3 pathway. Life Sci. 2020, 257, 118088. [Google Scholar] [CrossRef]
- Chiang, S.K.; Chen, S.E.; Chang, L.C. A dual role of heme oxygenase-1 in cancer cells. Int. J. Mol. Sci. 2018, 20, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagawa, T.; Omura, K.; Harada, H.; Nakaso, K.; Iwasa, S.; Koyama, Y.; Onizawa, K.; Yusa, H.; Yoshida, H. Heme oxygenase-1 expression predicts cervical lymph node metastasis of tongue squamous cell carcinomas. Oral Oncol. 2004, 40, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Liu, J.; Duan, H.; Li, R.; Peng, W.; Wu, C. Activation of nrf2/ho-1 signaling: An important molecular mechanism of herbal medicine in the treatment of atherosclerosis via the protection of vascular endothelial cells from oxidative stress. J. Adv. Res. 2021, 34, 43–63. [Google Scholar] [CrossRef]
- Liu, M.; Yang, X.; Liu, J.; Zhao, B.; Cai, W.; Li, Y.; Hu, D. Efficacy and safety of braf inhibition alone versus combined braf and mek inhibition in melanoma: A meta-analysis of randomized controlled trials. Oncotarget 2017, 8, 32258–32269. [Google Scholar] [CrossRef] [PubMed]
- Huynh, H.; Nguyen, T.T.; Chow, K.H.; Tan, P.H.; Soo, K.C.; Tran, E. Over-expression of the mitogen-activated protein kinase (mapk) kinase (mek)-mapk in hepatocellular carcinoma: Its role in tumor progression and apoptosis. BMC Gastroenterol. 2003, 3, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Jiang, S.; Zhou, L.; Yu, F.; Ding, H.; Li, P.; Zhou, M.; Wang, K. Potential mechanisms of action of curcumin for cancer prevention: Focus on cellular signaling pathways and mirnas. Int. J. Biol. Sci. 2019, 15, 1200–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teiten, M.H.; Dicato, M.; Diederich, M. Hybrid curcumin compounds: A new strategy for cancer treatment. Molecules 2014, 19, 20839–20863. [Google Scholar] [CrossRef] [PubMed]
- Raghuvanshi, D.; Nkepang, G.; Hussain, A.; Yari, H.; Awasthi, V. Stability study on an anti-cancer drug 4-(3,5-bis(2-chlorobenzylidene)-4-oxo-piperidine-1-yl)-4-oxo-2-butenoic acid (clefma) using a stability-indicating hplc method. J. Pharm. Anal. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Hussain, Z.; Thu, H.E.; Amjad, M.W.; Hussain, F.; Ahmed, T.A.; Khan, S. Exploring recent developments to improve antioxidant, anti-inflammatory and antimicrobial efficacy of curcumin: A review of new trends and future perspectives. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 77, 1316–1326. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, P.-N.; Lin, C.-W.; Yang, S.-F.; Chang, Y.-C. CLEFMA Induces the Apoptosis of Oral Squamous Carcinoma Cells through the Regulation of the P38/HO-1 Signalling Pathway. Cancers 2022, 14, 5519. https://doi.org/10.3390/cancers14225519
Chen P-N, Lin C-W, Yang S-F, Chang Y-C. CLEFMA Induces the Apoptosis of Oral Squamous Carcinoma Cells through the Regulation of the P38/HO-1 Signalling Pathway. Cancers. 2022; 14(22):5519. https://doi.org/10.3390/cancers14225519
Chicago/Turabian StyleChen, Pei-Ni, Chiao-Wen Lin, Shun-Fa Yang, and Yu-Chao Chang. 2022. "CLEFMA Induces the Apoptosis of Oral Squamous Carcinoma Cells through the Regulation of the P38/HO-1 Signalling Pathway" Cancers 14, no. 22: 5519. https://doi.org/10.3390/cancers14225519