
Extracellular Vesicles in Multiple Myeloma—Cracking the Code to a Better Understanding of the Disease
 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Stages of MM Progression
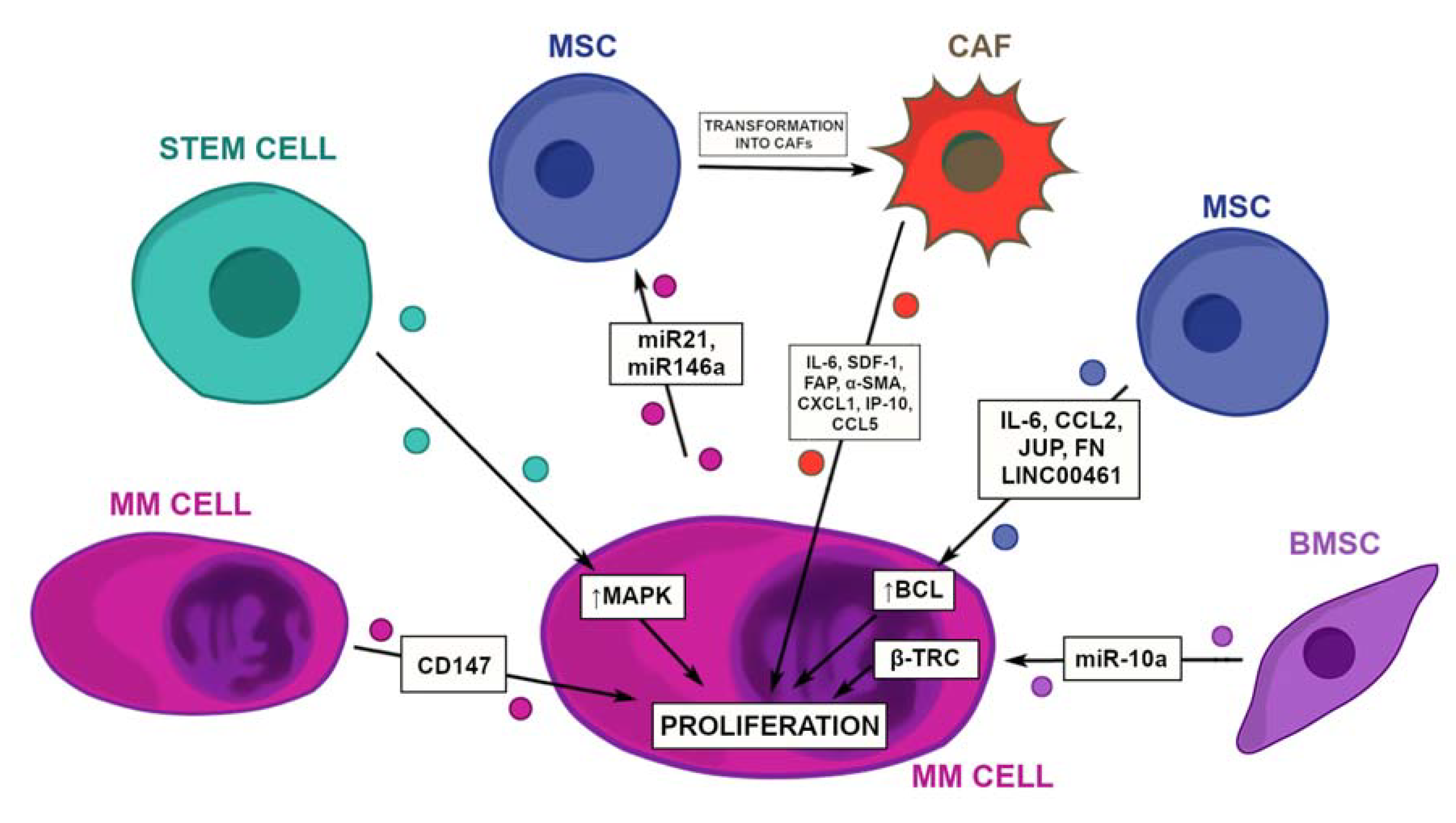
2.1. Tumor Growth and Proliferation
2.2. Progression
2.3. Angiogenesis
2.4. Matrix Remodeling and Osteolysis Induction
2.5. Immunosuppression
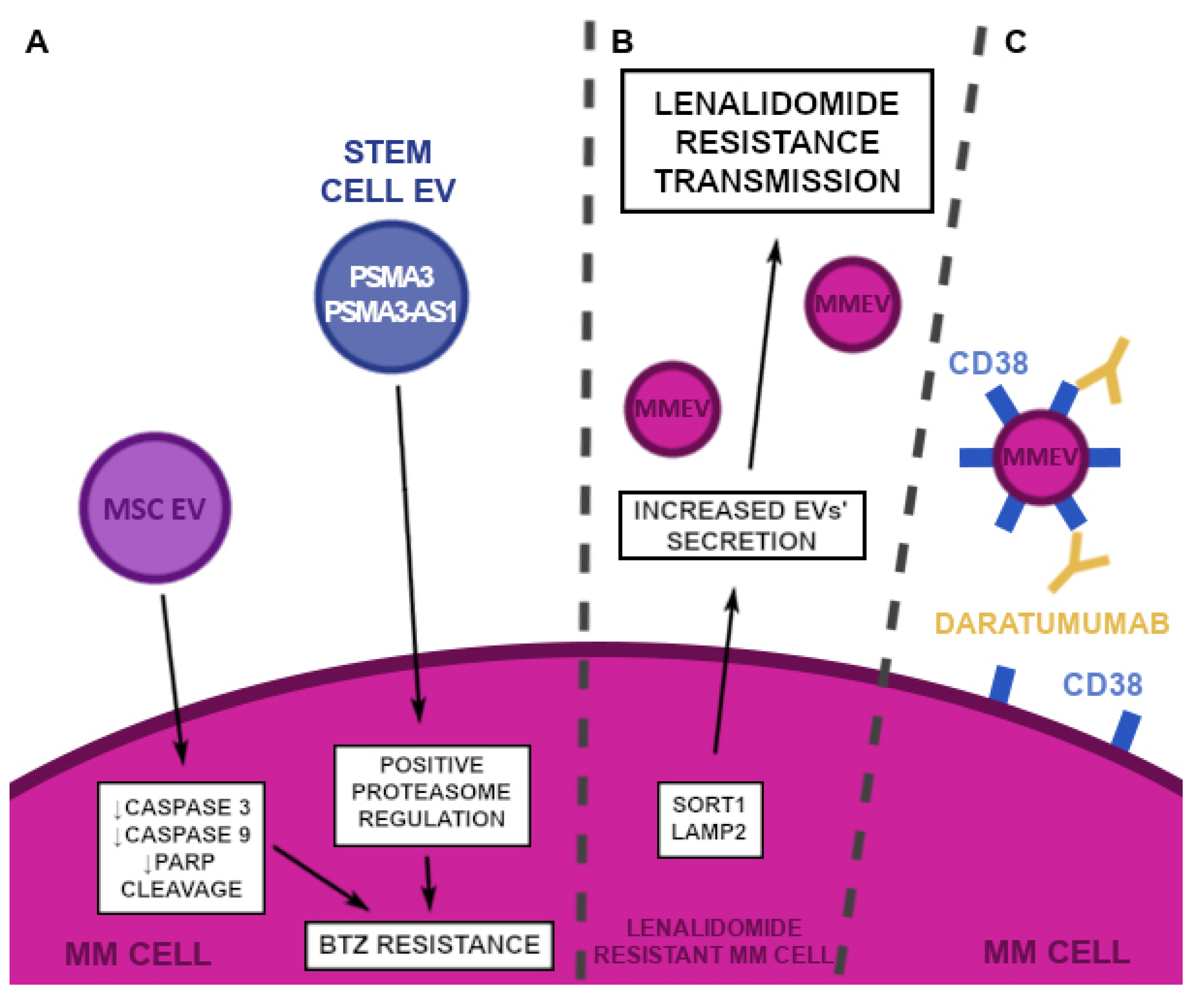
2.6. Drug Resistance
2.6.1. Proteasome Inhibitors (PI)
2.6.2. Immunomodulators
2.6.3. Monoclonal Antibodies
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADCC | antibody-dependent cell-mediated cytotoxicity |
| Akt | protein kinase B |
| ApoBDs | apoptotic bodies |
| ApoEVs | apoptotic extracellular vesicles |
| ApoExos | apoptotic exosomes |
| ApoMVs | apoptotic microvesicles |
| AREG | amphiregulin |
| ARMM | ARRDC1-mediated microvesicle |
| ARRDC1 | arrestin-domain-containing protein 1 |
| α-SMA | α-smooth muscle actin |
| BCL | B-cell lymphoma gene |
| Bcl-xL | B-cell lymphoma-extra large |
| Bi-EVs | MM-EVs pre-treated with bortezomib |
| BM | bone marrow |
| BM-MSC | bone marrow mesenchymal stromal cells |
| BMPs | bone morphogenetic proteins |
| β-TRC | β-transducin |
| CAFs | carcinoma-associated fibroblasts |
| CCL | CC chemokine ligand |
| CTSK | Cathepsin K |
| CXCL | CXC ligand |
| CXCR | CXC chemokine receptor |
| DC | dendritic cell |
| Del13 | chromosome 13 deletion |
| DKK1 | Dickkopf-related protein 1 |
| EC-EVs | endothelial cells-derived extracellular vesicles |
| ECs | endothelial cells |
| EGFR | epithelial growth factor receptor |
| eiF4GI | eukaryotic translation initiation factor 4GI |
| EMDR | environment-mediated drug resistance |
| EphA8 | ephrin receptor A8 kinase |
| ERK | extracellular signal-regulated kinases |
| ESCRT | endosomal sorting complexes required for transport |
| EVs | extracellular vesicles |
| FAP | fibroblast-activated protein |
| FIH-1 | factor-inhibiting hypoxia-inducible factor 1 |
| HER-2 | human epidermal growth factor receptor 2 |
| HR-MM cells | hypoxia-resistant multiple myeloma cells |
| Hsp-70 | heat shock protein 70 |
| IL | interleukin |
| IL-15Rα | interleukin 15 receptor α |
| ILVs | intraluminal vesicles |
| iNOS | inducible nitric oxide synthase |
| IP-10 | interferon-γ–inducible protein 10 |
| ISEV | International Society for Extracellular Vesicles |
| JNK | c-Jun N-terminal kinase |
| JUP | junction plakoglobin |
| LAMP2 | lysosomal associated membrane protein 2 |
| LINC00461 | long intergenic non-protein coding RNA 461 |
| lncRNAs | long-non-coding RNAs |
| MAPK | mitogen-activated protein kinase |
| MBD | myeloma-associated bone disease |
| Mcl-1 | myeloid cell leukemia-1 |
| MDSCs | myeloid-derived suppressor cells |
| MGUS | monoclonal gammopathy of undetermined significance |
| miR | microRNA |
| MM | multiple myeloma |
| MM MSC | multiple myeloma mesenchymal stromal cells |
| MM-EVs | multiple myeloma-derived extracellular vesicles |
| MMPs | metalloproteinases |
| MUC-1 | mucin 1 |
| MVB | multivesicular body |
| MVs | microvesicles |
| NFkB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NK | natural killer |
| NKG2DLs | natural-killer group 2, member D |
| OBs | osteoblasts |
| OCs | osteoclasts |
| OPG | osteoprotegerin |
| PDGF-BB | platelet-derived growth factor-BB |
| PGE2 | prostaglandin E2 |
| Pgp | P-glycoprotein |
| PI3K | phosphoinositide 3-kinase |
| piRNA | PIWI-interacting RNA |
| PM | plasma membrane |
| RANKL | receptor activator for nuclear factor κ B ligand |
| RHO GTPase | Ras homologous GTPase |
| RUNX2 | Runt-related transcription factor 2 |
| SDF-1 | stromal-derived factor 1 |
| SEMA5A | human Semaphorin-5A |
| SLAM | signaling lymphocytic activation molecule |
| SMAD5 | SMAD family member 5 |
| SMM | smoldering multiple myeloma |
| SORT1 | sortilin1 |
| STAT3 | signal transducer and activator of transcription 3 |
| TCR | T cell receptor |
| TF | tissue factor |
| TGF-β | tumor growth factor-β |
| TRAP | thrombospondin-related anonymous protein |
| uPAR | urokinase plasminogen activator receptor |
| UPP | ubiquitin-proteasome pathway |
| VEGF | vascular endothelial growth factor-1 |
References
- Brigle, K.; Rogers, B. Pathobiology and Diagnosis of Multiple Myeloma. Semin. Oncol. Nurs. 2017, 33, 225–236. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Surveillance, Epidemiology, and End Result Program. Cancer Stat Facts: Myeloma. Available online: https://seer.cancer.gov/statfacts/html/mulmy.html (accessed on 13 August 2022).
- Saltarella, I.; Lamanuzzi, A.; Apollonio, B.; Desantis, V.; Bartoli, G.; Vacca, A.; Frassanito, M.A. Role of Extracellular Vesicle-Based Cell-to-Cell Communication in Multiple Myeloma Progression. Cells 2021, 10, 3185. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.; Patel, A.; Goh, C.Y.; Moscvin, M.; Zhang, L.; Bianchi, G. Changing paradigms in diagnosis and treatment of monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM). Leukemia 2020, 34, 3111–3125. [Google Scholar] [CrossRef]
- Landgren, O.; Kyle, R.A.; Pfeiffer, R.M.; Katzmann, J.A.; Caporaso, N.E.; Hayes, R.; Dispenzieri, A.; Kumar, S.; Clark, R.J.; Baris, D.; et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: A prospective study. Blood 2009, 113, 5412–5417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.-V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Kyle, R.A.; Remstein, E.D.; Therneau, T.M.; Dispenzieri, A.; Kurtin, P.J.; Hodnefield, J.M.; Larson, D.R.; Plevak, M.F.; Jelinek, D.F.; Fonseca, R.; et al. Clinical Course and Prognosis of Smoldering (Asymptomatic) Multiple Myeloma. N. Engl. J. Med. 2007, 356, 2582–2590. [Google Scholar] [CrossRef] [PubMed]
- Brinton, L.T.; Sloane, H.S.; Kester, M.; Kelly, K.A. Formation and role of exosomes in cancer. Cell. Mol. Life Sci. 2014, 72, 659–671. [Google Scholar] [CrossRef] [Green Version]
- Bebelman, M.P.; Smit, M.J.; Pegtel, D.M.; Baglio, S.R. Biogenesis and function of extracellular vesicles in cancer. Pharmacol. Ther. 2018, 188, 1–11. [Google Scholar] [CrossRef]
- Rufino-Ramos, D.; Albuquerque, P.R.; Carmona, V.; Perfeito, R.; Nobre, R.J.; de Almeida, L.P. Extracellular vesicles: Novel promising delivery systems for therapy of brain diseases. J. Control. Release 2017, 262, 247–258. [Google Scholar] [CrossRef]
- Manna, I.; Quattrone, A.; De Benedittis, S.; Vescio, B.; Iaccino, E.; Quattrone, A. Exosomal miRNA as peripheral biomarkers in Parkinson’s disease and progressive supranuclear palsy: A pilot study. Park. Relat. Disord. 2021, 93, 77–84. [Google Scholar] [CrossRef]
- Tian, T.; Cao, L.; He, C.; Ye, Q.; Liang, R.; You, W.; Zhang, H.; Wu, J.; Ye, J.; Tannous, B.A.; et al. Targeted delivery of neural progenitor cell-derived extracellular vesicles for anti-inflammation after cerebral ischemia. Theranostics 2021, 11, 6507–6521. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Bayado, N.; He, D.; Li, J.; Chen, H.; Li, L.; Li, J.; Long, X.; Du, T.; Tang, J.; et al. Therapeutic Applications of Extracellular Vesicles for Myocardial Repair. Front. Cardiovasc. Med. 2021, 8, 758050. [Google Scholar] [CrossRef] [PubMed]
- Femminò, S.; Penna, C.; Margarita, S.; Comità, S.; Brizzi, M.F.; Pagliaro, P. Extracellular vesicles and cardiovascular system: Biomarkers and Cardioprotective Effectors. Vasc. Pharmacol. 2020, 135, 106790. [Google Scholar] [CrossRef] [PubMed]
- Kubo, H. Extracellular Vesicles in Lung Disease. Chest 2017, 153, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Dibavar, M.A.; Pourbagheri-Sigaroodi, A.; Asemani, Y.; Salari, S.; Bashash, D. Extracellular vesicles (EVs): What we know of the mesmerizing roles of these tiny vesicles in hematological malignancies? Life Sci. 2021, 271, 119177. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Caruso, S.; Poon, I.K.H. Apoptotic Cell-Derived Extracellular Vesicles: More Than Just Debris. Front. Immunol. 2018, 9, 1486. [Google Scholar] [CrossRef] [Green Version]
- Kakarla, R.; Hur, J.; Kim, Y.J.; Kim, J.; Chwae, Y.-J. Apoptotic cell-derived exosomes: Messages from dying cells. Exp. Mol. Med. 2020, 52, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Poon, I.K.H.; Parkes, M.A.F.; Jiang, L.; Atkin-Smith, G.K.; Tixeira, R.; Gregory, C.D.; Ozkocak, D.C.; Rutter, S.F.; Caruso, S.; Santavanond, J.P.; et al. Moving beyond size and phosphatidylserine exposure: Evidence for a diversity of apoptotic cell-derived extracellular vesicles in vitro. J. Extracell. Vesicles 2019, 8, 1608786. [Google Scholar] [CrossRef]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of Exosome Composition. Cell 2019, 177, 428–445.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolli, N.; Maura, F.; Minvielle, S.; Gloznik, D.; Szalat, R.; Fullam, A.; Martincorena, I.; Dawson, K.J.; Samur, M.K.; Zamora, J.; et al. Genomic patterns of progression in smoldering multiple myeloma. Nat. Commun. 2018, 9, 3363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roccaro, A.M.; Sacco, A.; Maiso, P.; Azab, A.K.; Tai, Y.T.; Reagan, M.; Azab, F.; Flores, L.M.; Campigotto, F.; Weller, E.; et al. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J. Clin. Investig. 2013, 123, 1542–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, M.; Yuan, H.; Liu, S.; Hu, Z.; Xiao, H. Exosome-transmitted LINC00461 promotes multiple myeloma cell proliferation and suppresses apoptosis by modulating microRNA/BCL-2 expression. Cytotherapy 2019, 21, 96–106. [Google Scholar] [CrossRef]
- Cimmino, A.; Calin, G.A.; Fabbri, M.; Iorio, M.V.; Ferracin, M.; Shimizu, M.; Wojcik, S.E.; Aqeilan, R.I.; Zupo, S.; Dono, M.; et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Natl. Acad. Sci. USA 2005, 102, 13944–13949. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Q.; Li, X.; Liu, J.; Ye, Q.; Chen, Y.; Tan, S.; Liu, J. Multiple Myeloma-Derived Exosomes Regulate the Functions of Mesenchymal Stem Cells Partially via Modulating miR-21 and miR-146a. Stem Cells Int. 2017, 2017, 9012152. [Google Scholar] [CrossRef]
- De Veirman, K.; Wang, J.; Xu, S.; Leleu, X.; Himpe, E.; Maes, K.; De Bruyne, E.; van Valckenborgh, E.; Vanderkerken, K.; Menu, E.; et al. Induction of miR-146a by multiple myeloma cells in mesenchymal stromal cells stimulates their pro-tumoral activity. Cancer Lett. 2016, 377, 17–24. [Google Scholar] [CrossRef]
- Umezu, T.; Imanishi, S.; Yoshizawa, S.; Kawana, C.; Ohyashiki, J.H.; Ohyashiki, K. Induction of multiple myeloma bone marrow stromal cell apoptosis by inhibiting extracellular vesicle miR-10a secretion. Blood Adv. 2019, 3, 3228–3240. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Zhang, P.; Gao, K.; Tang, Y.; Jin, X.; Zhang, Y.; Yi, Q.; Wang, C.; Yu, L. PLK1 and β-TrCP-dependent ubiquitination and degradation of Rap1GAP controls cell proliferation. PLoS ONE 2014, 9, e110296. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Song, X.; Lan, J.; Wang, X.; Wang, M. Bone marrow stromal cells derived exosomal miR-10a and miR-16 may be involved in progression of patients with multiple myeloma by regulating EPHA8 or IGF1R/CCND1. Medicine 2021, 100, e2344. [Google Scholar] [CrossRef]
- Dabbah, M.; Attar-Schneider, O.; Tartakover Matalon, S.; Shefler, I.; Jarchwsky Dolberg, O.; Lishner, M.; Drucker, L. Microvesicles derived from normal and multiple myeloma bone marrow mesenchymal stem cells differentially modulate myeloma cells’ phenotype and translation initiation. Carcinogenesis 2017, 38, 708–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arendt, B.K.; Walters, D.K.; Wu, X.; Tschumper, R.C.; Jelinek, D.F. Multiple myeloma dell-derived microvesicles are enriched in CD147 expression and enhance tumor cell proliferation. Oncotarget 2014, 5, 5686–5699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.J.; Mishima, Y.; Shi, J.; Sklavenitis-Pistofidis, R.; Redd, R.A.; Moschetta, M.; Manier, S.; Roccaro, A.M.; Sacco, A.; Tai, Y.-T.; et al. Progression signature underlies clonal evolution and dissemination of multiple myeloma. Blood 2021, 137, 2360–2372. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Lin, J.; Fang, H.; Fang, J.; Li, C.; Chen, W.; Liu, S.; Ondrejka, S.; Gong, Z.; Reu, F.; et al. Targeting the MALAT1/PARP1/LIG3 complex induces DNA damage and apoptosis in multiple myeloma. Leukemia 2018, 32, 2250–2262. [Google Scholar] [CrossRef] [PubMed]
- Ogiya, D.; Liu, J.; Ohguchi, H.; Kurata, K.; Samur, M.K.; Tai, Y.T.; Adamia, S.; Ando, K.; Hideshima, T.; Anderson, K.C. The JAK-STAT pathway regulates CD38 on myeloma cells in the bone marrow microenvironment: Therapeutic implications. Blood 2020, 136, 2334–2345. [Google Scholar] [CrossRef]
- Raje, N.S.; Bhatta, S.; Terpos, E. Role of the RANK/RANKL Pathway in Multiple Myeloma. Clin. Cancer Res. 2019, 25, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Henrich, S.E.; McMahon, K.M.; Plebanek, M.P.; Calvert, A.E.; Feliciano, T.J.; Parrish, S.; Tavora, F.; Mega, A.; De Souza, A.; Carneiro, B.A.; et al. Prostate cancer extracellular vesicles mediate intercellular communication with bone marrow cells and promote metastasis in a cholesterol-dependent manner. J. Extracell. Vesicles 2020, 10, e12042. [Google Scholar] [CrossRef]
- Tian, Y.; Ma, L.; Gong, M.; Su, G.; Zhu, S.; Zhang, W.; Wang, S.; Li, Z.; Chen, C.; Li, L.; et al. Protein Profiling and Sizing of Extracellular Vesicles from Colorectal Cancer Patients via Flow Cytometry. ACS Nano 2018, 12, 671–680. [Google Scholar] [CrossRef]
- Yang, N.; Li, S.; Li, G.; Zhang, S.; Tang, X.; Ni, S.; Jian, X.; Xu, C.; Zhu, J.; Lu, M. The role of extracellular vesicles in mediating progression, metastasis and potential treatment of hepatocellular carcinoma. Oncotarget 2017, 8, 3683–3695. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; De Veirman, K.; Faict, S.; Frassanito, M.A.; Ribatti, D.; Vacca, A.; Menu, E. Multiple myeloma exosomes establish a favourable bone marrow microenvironment with enhanced angiogenesis and immunosuppression. J. Pathol. 2016, 239, 162–173. [Google Scholar] [CrossRef]
- Colombo, M.; Galletti, S.; Bulfamante, G.; Falleni, M.; Tosi, D.; Todoerti, K.; Lazzari, E.; Crews, L.A.; Jamieson, C.H.; Ravaioli, S.; et al. Multiple myeloma-derived Jagged ligands increases autocrine and paracrine interleukin-6 expression in bone marrow niche. Oncotarget 2016, 7, 56013–56029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, M.; Giannandrea, D.; Lesma, E.; Basile, A.; Chiaramonte, R. Extracellular Vesicles Enhance Multiple Myeloma Metastatic Dissemination. Int. J. Mol. Sci. 2019, 20, 3236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Lei, Q.; Wang, H.; Xu, C.; Liu, T.; Kong, F.; Yang, C.; Yan, G.; Sun, L.; Zhao, A.; et al. Tumor-derived extracellular vesicles inhibit osteogenesis and exacerbate myeloma bone disease. Theranostics 2019, 9, 196–209. [Google Scholar] [CrossRef] [PubMed]
- Purushothaman, A.; Bandari, S.K.; Liu, J.; Mobley, J.A.; Brown, E.E.; Sanderson, R.D. Fibronectin on the Surface of Myeloma Cell-derived Exosomes Mediates Exosome-Cell Interactions. J. Biol. Chem. 2016, 291, 1652–1663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bochmann, I.; Ebstein, F.; Lehmann, A.; Wohlschlaeger, J.; Sixt, S.U.; Kloetzel, P.-M.; Dahlmann, B. T lymphocytes export proteasomes by way of microparticles: A possible mechanism for generation of extracellular proteasomes. J. Cell. Mol. Med. 2014, 18, 59–68. [Google Scholar] [CrossRef]
- Umezu, T.; Tadokoro, H.; Azuma, K.; Yoshizawa, S.; Ohyashiki, K.; Ohyashiki, J.H. Exosomal miR-135b shed from hypoxic multiple myeloma cells enhances angiogenesis by targeting factor-inhibiting HIF-1. Blood 2014, 124, 3748–3757. [Google Scholar] [CrossRef] [Green Version]
- Jakob, C.; Egerer, K.; Liebisch, P.; Türkmen, S.; Zavrski, I.; Kuckelkorn, U.; Heider, U.; Kaiser, M.; Fleissner, C.; Sterz, J.; et al. Circulating proteasome levels are an independent prognostic factor for survival in multiple myeloma. Blood 2007, 109, 2100–2105. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Zarfati, M.; Avivi, I.; Brenner, B.; Katz, T.; Aharon, A. Extracellular vesicles of multiple myeloma cells utilize the proteasome inhibitor mechanism to moderate endothelial angiogenesis. Angiogenesis 2018, 22, 185–196. [Google Scholar] [CrossRef]
- Chouaib, S.; Umansky, V.; Kieda, C. The role of hypoxia in shaping the recruitment of proangiogenic and immunosuppressive cells in the tumor microenvironment. Contemp. Oncol. 2018, 22, 7–13. [Google Scholar] [CrossRef]
- Gastelum, G.; Veena, M.; Lyons, K.; Lamb, C.; Jacobs, N.; Yamada, A.; Baibussinov, A.; Sarafyan, M.; Shamis, R.; Kraut, J.; et al. Can Targeting Hypoxia-Mediated Acidification of the Bone Marrow Microenvironment Kill Myeloma Tumor Cells? Front. Oncol. 2021, 11, 703878. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Hong, J.; Hong, M.; Wang, Y.; Yu, T.; Zang, S.; Wu, Q. piRNA-823 delivered by multiple myeloma-derived extracellular vesicles promoted tumorigenesis through re-educating endothelial cells in the tumor environment. Oncogene 2019, 38, 5227–5238. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.Y.; Moon, S.J.; Ratliff, B.B.; Ahn, S.H.; Jung, A.; Lee, M.; Lee, S.; Lim, B.J.; Kim, B.S.; Plotkin, M.D.; et al. Microparticles from Kidney-Derived Mesenchymal Stem Cells Act as Carriers of Proangiogenic Signals and Contribute to Recovery from Acute Kidney Injury. PLoS ONE 2014, 9, e87853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, X.; Gu, D.; Xing, X.; Cheng, Z.; Gong, D.; Zhang, G.; Zhu, Y. Human mesenchymal stromal cell-derived extracellular vesicles alleviate renal ischemic reperfusion injury and enhance angiogenesis in rats. Am. J. Transl. Res. 2016, 8, 4289–4299. [Google Scholar] [PubMed]
- Andersen, N.F.; Kristensen, I.B.; Preiss, B.S.; Christensen, J.H.; Abildgaard, N. Upregulation of Syndecan-1 in the bone marrow microenvironment in multiple myeloma is associated with angiogenesis. Eur. J. Haematol. 2015, 95, 211–217. [Google Scholar] [CrossRef]
- Taraboletti, G.; D’Ascenzo, S.; Borsotti, P.; Giavazzi, R.; Pavan, A.; Dolo, V. Shedding of the matrix metalloproteinases MMP-2, MMP-9, and MT1-MMP as membrane vesicle-associated components by endothelial cells. Am. J. Pathol. 2002, 160, 673–680. [Google Scholar] [CrossRef] [Green Version]
- Lombardo, G.; Dentelli, P.; Togliatto, G.; Rosso, A.; Gili, M.; Gallo, S.; Deregibus, M.C.; Camussi, G.; Brizzi, M.F. Activated Stat5 trafficking Via Endothelial Cell-derived Extracellular Vesicles Controls IL-3 Pro-angiogenic Paracrine Action. Sci. Rep. 2016, 6, 25689. [Google Scholar] [CrossRef] [Green Version]
- Theofilis, P.; Vogiatzi, G.; Oikonomou, E.; Gazouli, M.; Siasos, G.; Katifelis, H.; Perrea, D.; Vavuranakis, M.; Iliopoulos, D.C.; Tsioufis, C.; et al. The Effect of MicroRNA-126 Mimic Administration on Vascular Perfusion Recovery in an Animal Model of Hind Limb Ischemia. Front. Mol. Biosci. 2021, 8, 724465. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Abak, A.; Anamag, F.T.; Shoorei, H.; Majidpoor, J.; Taheri, M. The emerging role of non-coding RNAs in the regulation of PI3K/AKT pathway in the carcinogenesis process. Biomed. Pharmacother. 2021, 137, 111279. [Google Scholar] [CrossRef]
- Sleeman, J.P.; Christofori, G.; Fodde, R.; Collard, J.G.; Berx, G.; Decraene, C.; Rüegg, C. Concepts of metastasis in flux: The stromal progression model. Semin. Cancer Biol. 2012, 22, 174–186. [Google Scholar] [CrossRef]
- Migliorini, F.; Maffulli, N.; Trivellas, A.; Eschweiler, J.; Tingart, M.; Driessen, A. Bone metastases: A comprehensive review of the literature. Mol. Biol. Rep. 2020, 47, 6337–6345. [Google Scholar] [CrossRef] [PubMed]
- Rasch, S.; Lund, T.; Asmussen, J.; Nielsen, A.L.; Larsen, R.F.; Andersen, M.; Abildgaard, N. Multiple Myeloma Associated Bone Disease. Cancers 2020, 12, 2113. [Google Scholar] [CrossRef] [PubMed]
- Shupp, A.; Kolb, A.; Mukhopadhyay, D.; Bussard, K. Cancer Metastases to Bone: Concepts, Mechanisms, and Interactions with Bone Osteoblasts. Cancers 2018, 10, 182. [Google Scholar] [CrossRef] [Green Version]
- Faict, S.; Muller, J.; De Veirman, K.; De Bruyne, E.; Maes, K.; Vrancken, L.; Heusschen, R.; De Raeve, H.; Schots, R.; Vanderkerken, K.; et al. Exosomes play a role in multiple myeloma bone disease and tumor development by targeting osteoclasts and osteoblasts. Blood Cancer J. 2018, 8, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berasain, C.; Avila, M.A. Amphiregulin . Semin. Cell Dev. Biol. 2014, 28, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Raimondo, S.; Saieva, L.; Vicario, E.; Pucci, M.; Toscani, D.; Manno, M.; Raccosta, S.; Giuliani, N.; Alessandro, R. Multiple myeloma-derived exosomes are enriched of amphiregulin (AREG) and activate the epidermal growth factor pathway in the bone microenvironment leading to osteoclastogenesis. J. Hematol. Oncol. 2019, 12, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raimondi, L.; De Luca, A.; Amodio, N.; Manno, M.; Raccosta, S.; Taverna, S.; Bellavia, D.; Naselli, F.; Fontana, S.; Schillaci, O.; et al. Involvement of multiple myeloma cell-derived exosomes in osteoclast differentiation. Oncotarget 2015, 6, 13772–13789. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T. Realization of Osteolysis, Angiogenesis, Immunosuppression, and Drug Resistance by Extracellular Vesicles: Roles of RNAs and Proteins in Their Cargoes and of Ectonucleotidases of the Immunosuppressive Adenosinergic Noncanonical Pathway in the Bone Marrow Niche of Multiple Myeloma. Cancers 2021, 13, 2969. [Google Scholar]
- Löffler, D.; Brocke-Heidrich, K.; Pfeifer, G.; Stocsits, C.; Hackermüller, J.; Kretzschmar, A.K.; Burger, R.; Gramatzki, M.; Blumert, C.; Bauer, K.; et al. Interleukin-6–dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood 2007, 110, 1330–1333. [Google Scholar] [CrossRef] [Green Version]
- Pitari, M.R.; Rossi, M.; Amodio, N.; Botta, C.; Morelli, E.; Federico, C.; Gullà, A.; Caracciolo, D.; Di Martino, M.T.; Arbitrio, M.; et al. Inhibition of miR-21 restores RANKL/OPG ratio in multiple myeloma-derived bone marrow stromal cells and impairs the resorbing activity of mature osteoclasts. Oncotarget 2015, 6, 27343–27358. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Xu, H.; Han, H.; Song, S.; Zhang, X.; Ouyang, L.; Qian, C.; Hong, Y.; Qiu, Y.; Zhou, W.; et al. Exosome-mediated transfer of lncRUNX2-AS1 from multiple myeloma cells to MSCs contributes to osteogenesis. Oncogene 2018, 37, 5508–5519. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.; Kristensen, S.R.; Gregersen, H.; Teodorescu, E.M.; Christiansen, G.; Pedersen, S. Extracellular vesicle-associated procoagulant phospholipid and tissue factor activity in multiple myeloma. PLoS ONE 2019, 14, e0210835. [Google Scholar] [CrossRef] [PubMed]
- Labelle, M.; Begum, S.; Hynes, R.O. Platelets guide the formation of early metastatic niches. Proc. Natl. Acad. Sci. USA 2014, 111, E3053–E3061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takagi, S.; Tsukamoto, S.; Park, J.; Johnson, K.E.; Kawano, Y.; Moschetta, M.; Liu, C.-J.; Mishima, Y.; Kokubun, K.; Manier, S.; et al. Platelets Enhance Multiple Myeloma Progression via IL-1β Upregulation. Clin. Cancer Res. 2018, 24, 2430–2439. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, K.; Ramani, V.C.; Bandari, S.K.; Amin, R.; Brown, E.E.; Ritchie, J.P.; Stewart, M.D.; Sanderson, R.D. Heparanase promotes myeloma stemness and in vivo tumorigenesis. Matrix Biol. 2019, 88, 53–68. [Google Scholar] [CrossRef]
- Kawano, Y.; Moschetta, M.; Manier, S.; Glavey, S.; Görgün, G.T.; Roccaro, A.M.; Anderson, K.C.; Ghobrial, I.M. Targeting the bone marrow microenvironment in multiple myeloma. Immunol. Rev. 2015, 263, 160–172. [Google Scholar] [CrossRef]
- Wang, J.; De Veirman, K.; De Beule, N.; Maes, K.; De Bruyne, E.; van Valckenborgh, E.; Vanderkerken, K.; Menu, E. The bone marrow microenvironment enhances multiple myeloma progression by exosome-mediated activation of myeloid-derived suppressor cells. Oncotarget 2015, 6, 43992–44004. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Smyth, M.J.; Martinet, L. Cancer immunoediting and immune dysregulation in multiple myeloma. Blood 2020, 136, 2731–2740. [Google Scholar] [CrossRef]
- Chan, A.C.; Neeson, P.; Leeansyah, E.; Tainton, K.; Quach, H.; Prince, H.M.; Harrison, S.J.; Godfrey, I.D.; Ritchie, D.; Berzins, S.P. Natural killer T cell defects in multiple myeloma and the impact of lenalidomide therapy. Clin. Exp. Immunol. 2014, 175, 49–58. [Google Scholar] [CrossRef]
- Chung, D.J.; Pronschinske, K.B.; Shyer, J.A.; Sharma, S.; Leung, S.; Curran, S.A.; Lesokhin, A.M.; Devlin, S.M.; Giralt, S.A.; Young, J.W. T-cell Exhaustion in Multiple Myeloma Relapse after Autotransplant: Optimal Timing of Immunotherapy. Cancer Immunol. Res. 2016, 4, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Dhodapkar, M.V.; Krasovsky, J.; Osman, K.; Geller, M.D. Vigorous Premalignancy-specific Effector T Cell Response in the Bone Marrow of Patients with Monoclonal Gammopathy. J. Exp. Med. 2003, 198, 1753–1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillerey, C.; Ferrari de Andrade, L.; Vuckovic, S.; Miles, K.; Ngiow, S.F.; Yong, M.C.; Teng, M.W.; Colonna, M.; Ritchie, D.S.; Chesi, M. Immunosurveillance and therapy of multiple myeloma are CD226 dependent. J. Clin. Investig. 2015, 125, 2077–2089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurisic, V.; Srdic, T.; Konjevic, G.; Markovic, O.; Colovic, M. Clinical stage-depending decrease of NK cell activity in multiple myeloma patients. Med Oncol. 2007, 24, 312–317. [Google Scholar] [CrossRef]
- Malek, E.; de Lima, M.; Letterio, J.J.; Kim, B.-G.; Finke, J.H.; Driscoll, J.J.; Giralt, S.A. Myeloid-derived suppressor cells: The green light for myeloma immune escape. Blood Rev. 2016, 30, 341–348. [Google Scholar] [CrossRef]
- Xiang, X.; Poliakov, A.; Liu, C.; Liu, Y.; Deng, Z.-B.; Wang, J.; Cheng, Z.; Shah, S.V.; Wang, G.-J.; Zhang, L.; et al. Induction of myeloid-derived suppressor cells by tumor exosomes. Int. J. Cancer 2009, 124, 2621–2633. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Weber, R.; Groth, C.; Lasser, S.; Arkhypov, I.; Petrova, V.; Altevogt, P.; Utikal, J.; Umansky, V. IL-6 as a major regulator of MDSC activity and possible target for cancer immunotherapy. Cell. Immunol. 2020, 359, 104254. [Google Scholar] [CrossRef]
- Khalife, J.; Ghose, J.; Martella, M.; Viola, D.; Rocci, A.; Troadec, E.; Terrazas, C.; Satoskar, A.R.; Gunes, E.G.; Dona, A.; et al. MiR-16 regulates crosstalk in NF-κB tolerogenic inflammatory signaling between myeloma cells and bone marrow macrophages. JCI Insight 2019, 4, e129348. [Google Scholar] [CrossRef]
- Leaf, R.K.; Stroopinsky, D.; Pyzer, A.R.; Kruisbeek, A.M.; van Wetering, S.; Washington, A.; Ephraim, A.; Cole, L.; Morin, A.; Jain, S.; et al. DCOne as an Allogeneic Cell-based Vaccine for Multiple Myeloma. J. Immunother. 2017, 40, 315–322. [Google Scholar] [CrossRef]
- Vulpis, E.; Cecere, F.; Molfetta, R.; Soriani, A.; Fionda, C.; Peruzzi, G.; Caracciolo, G.; Palchetti, S.; Masuelli, L.; Simonelli, L.; et al. Genotoxic stress modulates the release of exosomes from multiple myeloma cells capable of activating NK cell cytokine production: Role of HSP70/TLR2/NF-kB axis. Oncoimmunology 2017, 6, e1279372. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, C.; Ricci, B.; Vulpis, E.; Fionda, C.; Ricciardi, M.R.; Petrucci, M.T.; Masuelli, L.; Peri, A.; Cippitelli, M.; Zingoni, A.; et al. Drug-Induced Senescent Multiple Myeloma Cells Elicit NK Cell Proliferation by Direct or Exosome-Mediated IL15 Trans-Presentation. Cancer Immunol. Res. 2018, 6, 860–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsner, L.; Muppala, V.; Gehrmann, M.; Lozano-Kuehne, J.; Malzahn, D.; Bickeböller, H.; Brunner, E.; Zientkowska, M.; Herrmann, T.; Walter, L.; et al. The Heat Shock Protein HSP70 Promotes Mouse NK Cell Activity against Tumors That Express Inducible NKG2D Ligands. J. Immunol. 2007, 179, 5523–5533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viaud, S.; Terme, M.; Flament, C.; Taieb, J.; André, F.; Novault, S.; Escudier, B.; Robert, C.; Caillat-Zucman, S.; Tursz, T.; et al. Dendritic cell-derived exosomes promote natural killer cell activation and proliferation: A role for NKG2D ligands and IL-15Ralpha. PLoS ONE 2009, 4, e4942. [Google Scholar] [CrossRef]
- Escudier, B.; Dorval, T.; Chaput, N.; André, F.; Caby, M.-P.; Novault, S.; Flament, C.; Leboulaire, C.; Borg, C.; Amigorena, S.; et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: Results of thefirst phase I clinical trial. J. Transl. Med. 2005, 3, 10. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, U.H.; Cornell, R.F.; Lakshman, A.; Gahvari, Z.J.; McGehee, E.; Jagosky, M.H.; Gupta, R.; Varnado, W.; Fiala, M.A.; Chhabra, S.; et al. Outcomes of patients with multiple myeloma refractory to CD38-targeted monoclonal antibody therapy. Leukemia 2019, 33, 2266–2275. [Google Scholar] [CrossRef]
- Schwarzenbach, H. Expression of MDR1/P-Glycoprotein, the Multidrug Resistance Protein MRP, and the Lung-Resistance Protein LRP in Multiple Myeloma. Med Oncol. 2002, 19, 87–104. [Google Scholar] [CrossRef]
- Pick, M.; Vainstein, V.; Goldschmidt, N.; Lavie, D.; Libster, D.; Gural, A.; Grisariu, S.; Avni, B.; Ben Yehuda, D.; Gatt, M.E. Daratumumab resistance is frequent in advanced-stage multiple myeloma patients irrespective of CD38 expression and is related to dismal prognosis. Eur. J. Haematol. 2018, 100, 494–501. [Google Scholar] [CrossRef]
- Meads, M.B.; Hazlehurst, L.A.; Dalton, W.S. The Bone Marrow Microenvironment as a Tumor Sanctuary and Contributor to Drug Resistance. Clin. Cancer Res. 2008, 14, 2519–2526. [Google Scholar] [CrossRef] [Green Version]
- Di Marzo, L.; Desantis, V.; Solimando, A.G.; Ruggieri, S.; Annese, T.; Nico, B.; Fumarulo, R.; Vacca, A.; Frassanito, M.A. Microenvironment drug resistance in multiple myeloma: Emerging new players. Oncotarget 2016, 7, 60698–60711. [Google Scholar] [CrossRef] [Green Version]
- Ribatti, D.; Nico, B.; Vacca, A. Multiple myeloma as a model for the role of bone marrow niches in the control of angiogenesis. Int. Rev. Cell Mol. Biol. 2015, 314, 259–282. [Google Scholar]
- Dimopoulos, M.A.; Moreau, P.; Terpos, E.; Mateos, M.V.; Zweegman, S.; Cook, G.; Delforge, M.; Hájek, R.; Schjesvold, F.; Cavo, M.; et al. Multiple myeloma: EHA-ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2021, 32, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Nunes, A.T.; Annunziata, C.M. Proteasome inhibitors: Structure and function. Semin. Oncol. 2017, 44, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Nefedova, Y.; l Landowski, T.H.; Dalton, W.S. Bone marrow stromal-derived soluble factors and direct cell contact contribute to de novo drug resistance of myeloma cells by distinct mechanisms. Leukemia 2003, 17, 1175–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Hendrix, A.; Hernot, S.; Lemaire, M.; De Bruyne, E.; van Valckenborgh, E.; Lahoutte, T.; De Wever, O.; Vanderkerken, K.; Menu, E. Bone marrow stromal cell–derived exosomes as communicators in drug resistance in multiple myeloma cells. Blood 2014, 124, 555–566. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Han, H.; Song, S.; Yi, N.; Qian, C.; Qiu, Y.; Zhou, W.; Hong, Y.; Zhuang, W.; Li, Z.; et al. Exosome-Transmitted PSMA3 and PSMA3-AS1 Promote Proteasome Inhibitor Resistance in Multiple Myeloma. Clin. Cancer Res. 2019, 25, 1923–1935. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Nakayama, J.; Yamamoto, Y.; Kuroda, M.; Hattori, Y.; Ochiya, T. SORT1/LAMP2-mediated Extracellular Vesicle Secretion and Cell Adhesion Are Linked to Lenalidomide Resistance in Multiple Myeloma. Blood Adv. 2022, 6, 2480–2495. [Google Scholar] [CrossRef]
- Mateos, M.-V.; Cavo, M.; Blade, J.; Dimopoulos, M.A.; Suzuki, K.; Jakubowiak, A.; Knop, S.; Doyen, C.; Lucio, P.; Nagy, Z.; et al. Overall survival with daratumumab, bortezomib, melphalan, and prednisone in newly diagnosed multiple myeloma (ALCYONE): A randomised, open-label, phase 3 trial. Lancet 2020, 395, 132–141. [Google Scholar] [CrossRef]
- Ashour, R.; Ri, M.; Aly, S.S.; Yoshida, T.; Tachita, T.; Kanamori, T.; Aoki, S.; Kinoshita, S.; Narita, T.; Totani, H.; et al. Expression analysis of two SLAM family receptors, SLAMF2 and SLAMF7, in patients with multiple myeloma. Int. J. Hematol. 2019, 110, 69–76. [Google Scholar] [CrossRef]
- Horenstein, A.L.; Bracci, C.; Morandi, F.; Malavasi, F. CD38 in Adenosinergic Pathways and Metabolic Re-programming in Human Multiple Myeloma Cells: In-tandem Insights from Basic Science to Therapy. Front. Immunol. 2019, 10, 760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morandi, F.; Marimpietri, D.; Horenstein, A.L.; Bolzoni, M.; Toscani, D.; Costa, F.; Castella, B.; Faini, A.C.; Massaia, M.; Pistoia, V.; et al. Microvesicles released from multiple myeloma cells are equipped with ectoenzymes belonging to canonical and non-canonical adenosinergic pathways and produce adenosine from ATP and NAD. Oncoimmunology 2018, 7, e1458809. [Google Scholar] [CrossRef] [PubMed]
- Röllig, C.; Schmidt, C.; Bornhäuser, M.; Ehninger, G.; Schmitz, M.; Auffermann-Gretzinger, S. Induction of Cellular Immune Responses in Patients with Stage-I Multiple Myeloma After Vaccination with Autologous Idiotype-pulsed Dendritic Cells. J. Immunother. 2011, 34, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Wolfers, J.; Lozier, A.; Raposo, G.; Regnault, A.; Théry, C.; Masurier, C.; Flament, C.; Pouzieux, S.; Faure, F.; Tursz, T.; et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat. Med. 2001, 7, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cao, Z.; Wang, P.; Zhang, X.; Tang, J.; He, Y.; Huang, Z.; Mao, X.; Shi, S.; Kou, X. Apoptotic Extracellular Vesicles Ameliorate Multiple Myeloma by Restoring Fas-Mediated Apoptosis. ACS Nano 2021, 15, 14360–14372. [Google Scholar] [CrossRef] [PubMed]
- Caby, M.-P.; Lankar, D.; Vincendeau-Scherrer, C.; Raposo, G.; Bonnerot, C. Exosomal-like vesicles are present in human blood plasma. Int. Immunol. 2005, 17, 879–887. [Google Scholar] [CrossRef] [Green Version]
- Palanisamy, V.; Sharma, S.; Deshpande, A.; Zhou, H.; Gimzewski, J.; Wong, D.T. Nanostructural and Transcriptomic Analyses of Human Saliva Derived Exosomes. PLoS ONE 2010, 5, e8577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paredes, P.T.; Esser, J.; Admyre, C.; Nord, M.; Rahman, Q.K.; Lukic, A.; Rådmark, O.; Grönneberg, R.; Grunewald, J.; Eklund, A.; et al. Bronchoalveolar lavage fluid exosomes contribute to cytokine and leukotriene production in allergic asthma. Allergy 2012, 67, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Muraoka, S.; Lin, W.; Chen, M.; Hersh, S.W.; Emili, A.; Xia, W.; Ikezu, T. Assessment of separation methods for extracellular vesicles from human and mouse brain tissues and human cerebrospinal fluids. Methods 2020, 177, 35–49. [Google Scholar] [CrossRef]
- Lin, L.-Y.; Yang, L.; Zeng, Q.; Wang, L.; Chen, M.-L.; Zhao, Z.-H.; Ye, G.-D.; Luo, Q.-C.; Lv, P.-Y.; Guo, Q.-W.; et al. Tumor-originated exosomal lncUEGC1 as a circulating biomarker for early-stage gastric cancer. Mol. Cancer 2018, 17, 84. [Google Scholar] [CrossRef] [Green Version]
- Logozzi, M.; De Milito, A.; Lugini, L.; Borghi, M.; Calabrò, L.; Spada, M.; Perdicchio, M.; Marino, M.L.; Federici, C.; Iessi, E.; et al. High Levels of Exosomes Expressing CD63 and Caveolin-1 in Plasma of Melanoma Patients. PLoS ONE 2009, 4, e5219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maisano, D.; Mimmi, S.; Dattilo, V.; Marino, F.; Gentile, M.; Vecchio, E.; Fiume, G.; Nisticò, N.; Aloisio, A.; de Santo, M.P.; et al. A novel phage display based platform for exosome diversity characterization. Nanoscale 2022, 14, 2998–3003. [Google Scholar] [CrossRef] [PubMed]
- Sedlarikova, L.; Bollova, B.; Radova, L.; Brozova, L.; Jarkovsky, J.; Almasi, M.; Penka, M.; Kuglík, P.; Sandecká, V.; Stork, M.; et al. Circulating exosomal long noncoding RNA PRINS—First findings in monoclonal gammopathies. Hematol. Oncol. 2018, 36, 786–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Noto, G.; Chiarini, M.; Paolini, L.; Mazzoldi, E.L.; Giustini, V.; Radeghieri, A.; Caimi, L.; Ricotta, D. Immunoglobulin Free Light Chains and GAGs Mediate Multiple Myeloma Extracellular Vesicles Uptake and Secondary NfκB Nuclear Translocation. Front. Immunol. 2014, 5, 517. [Google Scholar] [CrossRef] [Green Version]
- Manier, S.; Liu, C.-J.; Avet-Loiseau, H.; Park, J.; Shi, J.; Campigotto, F.; Salem, K.Z.; Huynh, D.; Glavey, S.V.; Rivotto, B.; et al. Prognostic role of circulating exosomal miRNAs in multiple myeloma. Blood 2017, 129, 2429–2436. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Classical Exosomes | Non-Classical Exosomes | Classical Microvesicles | Large Oncosomes | ARMM | Apoptotic Exosomes | Apoptotic Microvesicles | Apoptotic Bodies | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Category | Exosomes | Microvesicles | Apoptotic Extracellular Vesicles | [17,18,19,20,21,22] | |||||
| Size | 40–150 nm | 150–1000 nm | 1–10 μm | 40–100 nm | <150 nm | 100–1000 nm | 1–5 μm | ||
| EV class | Small EV | Small EV | Large EV | Large EV | Small EV | Small EV | Small to Large EV | Large EV | |
| Biogenesis | Exocytosis of MVBs | Direct budding from PM | Apoptosis | ||||||
| dependent on RHO GTPases | dependent on ARRDC1 and ESCRT | caspase 3-dependent formation of MVBs and its exocytosis | possibly via direct budding | apoptotic membrane blebbing | |||||
| Markers | CD63, CD81, CD9 | (CD63-, CD81- and CD9-negative) a | Annexin A1, annexin A2 | Annexin A1, annexin A2 | ARRDC1 | Annexin V | |||
| CD63, LAMP1, HSP70, S1PR1 and 3 | |||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iskrzak, J.; Zygmunciak, P.; Misiewicz-Krzemińska, I.; Puła, B. Extracellular Vesicles in Multiple Myeloma—Cracking the Code to a Better Understanding of the Disease. Cancers 2022, 14, 5575. https://doi.org/10.3390/cancers14225575
Iskrzak J, Zygmunciak P, Misiewicz-Krzemińska I, Puła B. Extracellular Vesicles in Multiple Myeloma—Cracking the Code to a Better Understanding of the Disease. Cancers. 2022; 14(22):5575. https://doi.org/10.3390/cancers14225575
Chicago/Turabian StyleIskrzak, Justyna, Przemysław Zygmunciak, Irena Misiewicz-Krzemińska, and Bartosz Puła. 2022. "Extracellular Vesicles in Multiple Myeloma—Cracking the Code to a Better Understanding of the Disease" Cancers 14, no. 22: 5575. https://doi.org/10.3390/cancers14225575
APA StyleIskrzak, J., Zygmunciak, P., Misiewicz-Krzemińska, I., & Puła, B. (2022). Extracellular Vesicles in Multiple Myeloma—Cracking the Code to a Better Understanding of the Disease. Cancers, 14(22), 5575. https://doi.org/10.3390/cancers14225575