
The TLK1–MK5 Axis Regulates Motility, Invasion, and Metastasis of Prostate Cancer Cells



Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmids and Antibodies
2.2. Cell Culture
2.3. Lentiviral Transduction
2.4. Cell Treatment
2.5. Scratch Wound Repair Assay
2.6. Trans-Well Invasion Assay
2.7. Immunocytochemistry (ICC) and Actin Staining
2.8. Western Blotting
2.9. Real-Time Quantitative PCR (RT-qPCR)
2.10. Tail-Vein Injection
2.11. Immunohistochemistry (IHC)
2.12. Statistical Analysis
3. Results
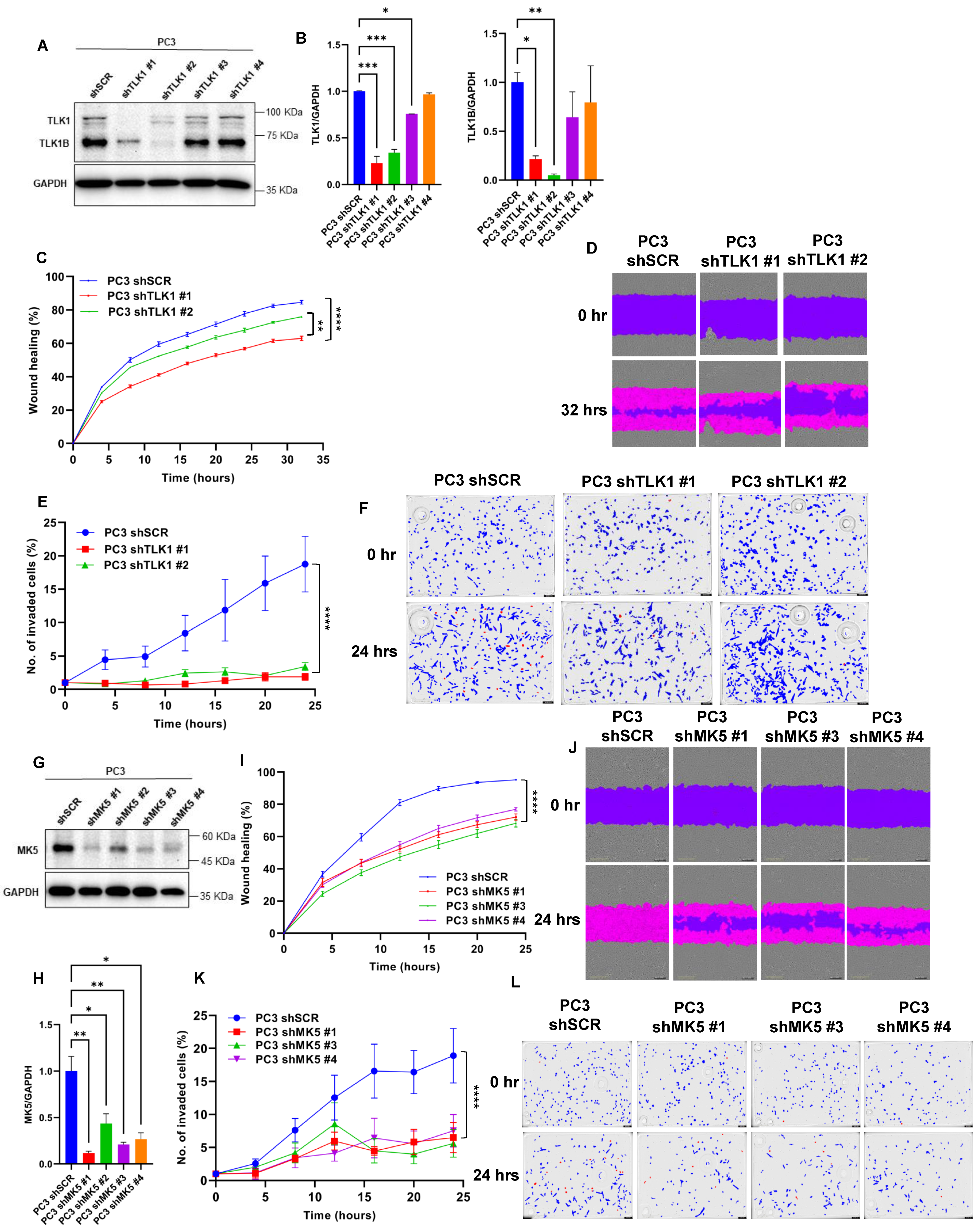
3.1. Knockdown of TLK1 or MK5 Results in Reduced Motility and Invasive Capacity of PC3 Cells
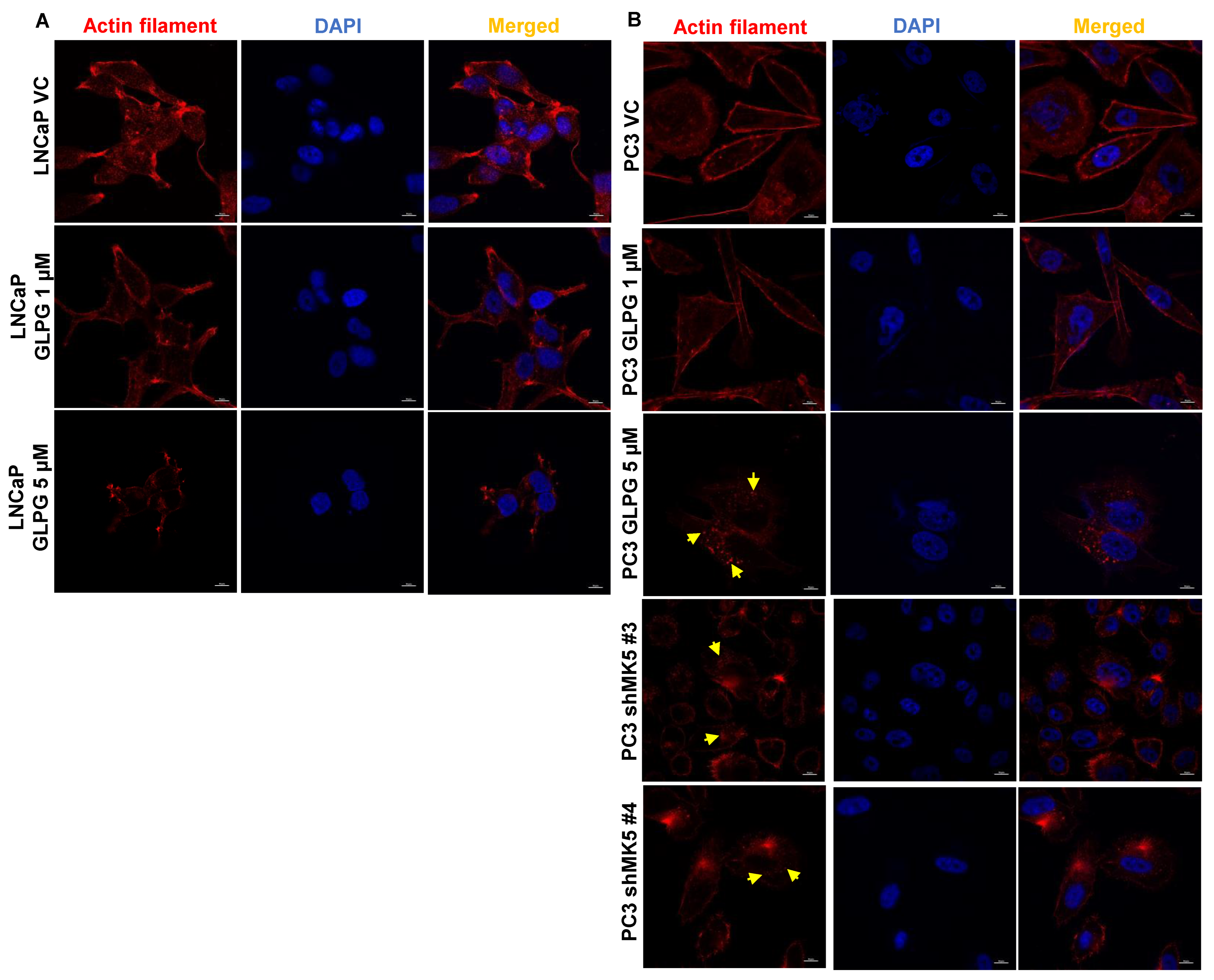
3.2. Treatment of LNCaP and PC3 Cells with GLPG Results in Actin Fibers’ Relocalization, Which Likely Affects Motility
3.3. The Motility Properties of MK5 Can Be Explained via Alterations of Focal Adhesion Complex
3.4. MK5 Could Promote Stress Fibers Formation through Its Effect on HSP27 Phosphorylation and Suppressing Its Actin Fibers’ “Capping Activity”
3.5. Relocalization of Paxillin to Focal Contacts of the Basal Surface Area of the Cells in PC3 Cells Treated with GLPG
3.6. The Loss of Invasive Properties of the Cells upon GLPG Treatment Correlate with Reduced MMPs Expression
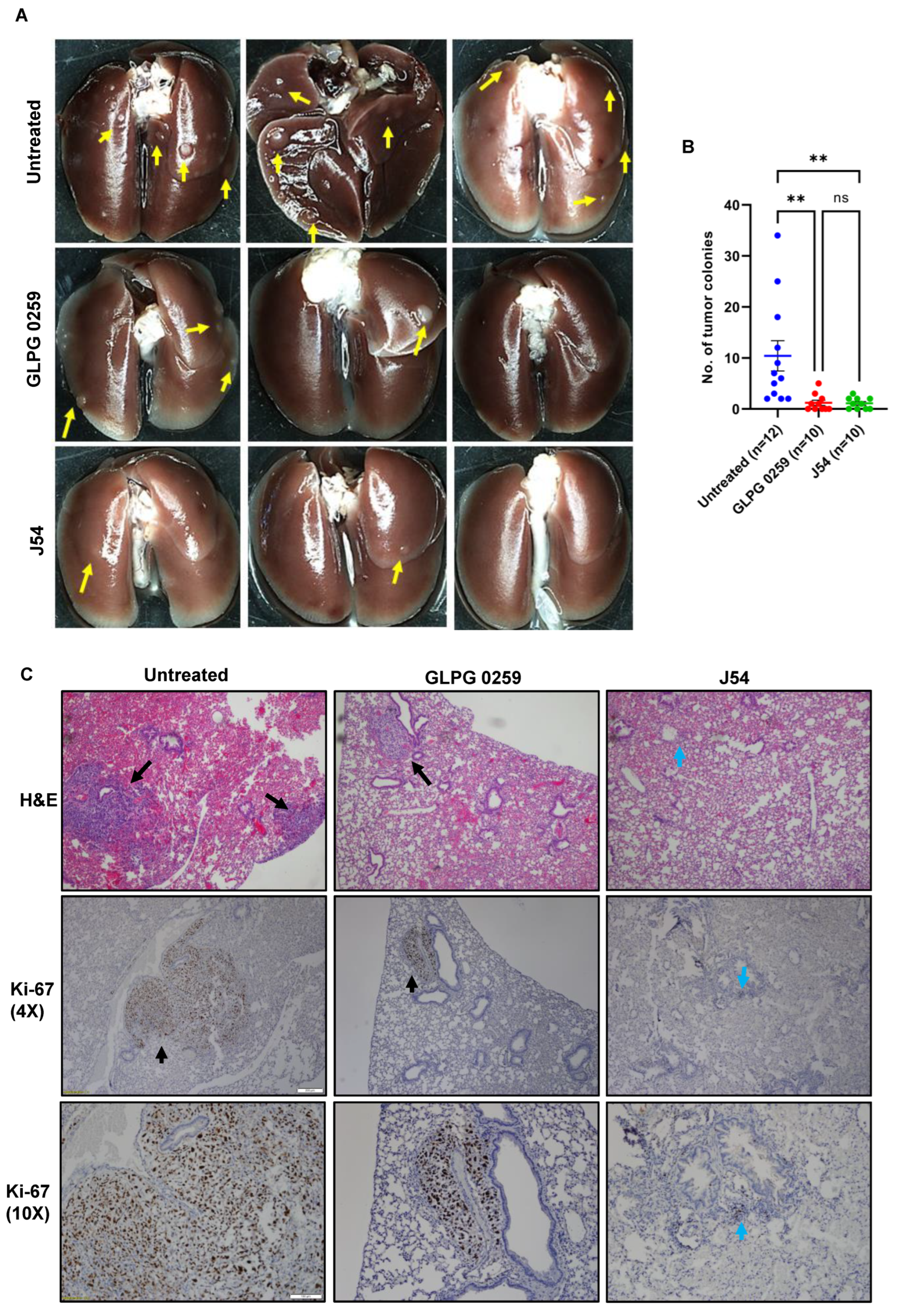
3.7. Treatment with GLPG or J54 Reduces Experimental Metastases
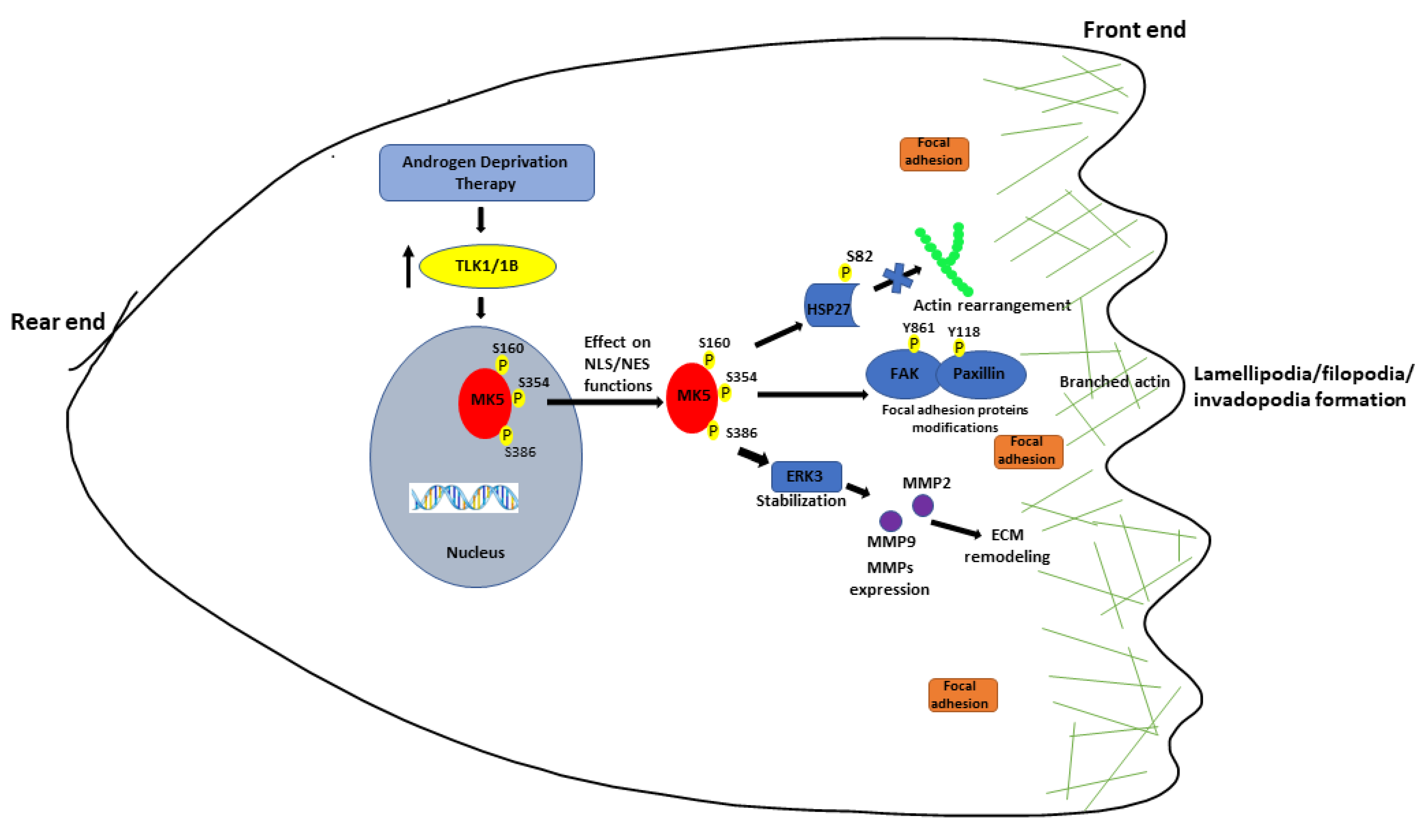
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA A Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2021. CA A Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Giamas, G.; Man, Y.L.; Hirner, H.; Bischof, J.; Kramer, K.; Khan, K.; Ahmed, S.S.L.; Stebbing, J.; Knippschild, U. Kinases as targets in the treatment of solid tumors. Cell. Signal. 2010, 22, 984–1002. [Google Scholar] [CrossRef]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Khalil, M.I.; Singh, V.; King, J.; De Benedetti, A. TLK1-mediated MK5-S354 phosphorylation drives prostate cancer cell motility and may signify distinct pathologies. Mol. Oncol. 2022, 16, 2537–2557. [Google Scholar] [CrossRef]
- Khalil, M.I.; Singh, V.; King, J.; De Benedetti, A. TLK1-MK5 axis drives prostate cancer cell motility and pathologic features of aggressiveness. Res. Sq. 2021. [Google Scholar] [CrossRef]
- Khalil, M.I.; De Benedetti, A. Tousled-like kinase 1: A novel factor with multifaceted role in mCRPC progression and development of therapy resistance. Cancer Drug Resist. 2022, 5, 93–101. [Google Scholar] [CrossRef]
- De Benedetti, A. The Tousled-Like-Kinases as guardians of genome integrity. ISRN Mol. Biol. 2012, 2012, 1–9. [Google Scholar] [CrossRef]
- Segura-Bayona, S.; Stracker, T.H. The Tousled-like kinases regulate genome and epigenome stability: Implications in development and disease. Cell Mol Life Sci. 2019, 76, 3827–3841. [Google Scholar] [CrossRef]
- Sunavala-Dossabhoy, G. Preserving salivary gland physiology against genotoxic damage—The Tousled way. Oral Dis. 2018, 24, 1390–1398. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.; Connelly, Z.M.; Shen, X.; De Benedetti, A. Identification of the proteome complement of humanTLK1 reveals it binds and phosphorylates NEK1 regulating its activity. Cell Cycle 2017, 16, 915–926. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.; Jaiswal, P.; Ghosh, I.; Koul, H.K.; Yu, X.; De Benedetti, A. Targeting the TLK1/NEK1 DDR axis with Thioridazine suppresses outgrowth of Androgen Independent Prostate tumors. Int. J. Cancer 2019, 145, 1055–1067. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.; Jaiswal, P.K.; Ghosh, I.; Koul, H.K.; Yu, X.; De Benedetti, A. The TLK1-Nek1 axis promotes prostate cancer progression. Cancer Lett. 2019, 453, 131–141. [Google Scholar] [CrossRef]
- Singh, V.; Khalil, M.I.; De Benedetti, A. The TLK1/Nek1 axis contributes to mitochondrial integrity and apoptosis prevention via phosphorylation of VDAC1. Cell Cycle 2020, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Khalil, M.I.; Ghosh, I.; Singh, V.; Chen, J.; Zhu, H.; De Benedetti, A. NEK1 Phosphorylation of YAP Promotes Its Stabilization and Transcriptional Output. Cancers 2020, 12, 3666. [Google Scholar] [CrossRef]
- Khalil, M.I.; Madere, C.; Ghosh, I.; Adam, R.M.; De Benedetti, A. Interaction of TLK1 and AKTIP as a Potential Regulator of AKT Activation in Castration-Resistant Prostate Cancer Progression. Pathophysiology 2021, 28, 339–354. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-A.; Tan, Y.; Wang, X.; Cao, X.; Veeraraghavan, J.; Liang, Y.; Edwards, D.P.; Huang, S.; Pan, X.; Li, K.; et al. Comprehensive functional analysis of the tousled-like kinase 2 frequently amplified in aggressive luminal breast cancers. Nat. Commun. 2016, 7, 12991. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.; Yao, Z.; Zhao, N.; Zhang, C. TLK2 enhances aggressive phenotypes of glioblastoma cells through the activation of SRC signaling pathway. Cancer Biol Ther. 2019, 20, 101–108. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, W.; Wu, H.; Zhou, Y.; Qin, X.; Wang, Y.; Wu, J.; Sun, X.-Y.; Yang, Y.; Xu, H.; et al. The essential role of PRAK in tumor metastasis and its therapeutic potential. Nat. Commun. 2021, 12, 1736. [Google Scholar] [CrossRef]
- Yoshizuka, N.; Chen, R.M.; Xu, Z.; Liao, R.; Hong, L.; Hu, W.-Y.; Yu, G.; Han, J.; Chen, L.; Sun, P. A novel function of p38-regulated/activated kinase in endothelial cell migration and tumor angiogenesis. Mol. Cell Biol. 2012, 32, 606–618. [Google Scholar] [CrossRef] [Green Version]
- Gerits, N.; Mikalsen, T.; Kostenko, S.; Shiryaev, A.; Johannessen, M.; Moens, U. Modulation of F-actin rearrangement by the cyclic AMP/cAMP-dependent protein kinase (PKA) pathway is mediated by MAPK-activated protein kinase 5 and requires PKA-induced nuclear export of MK5. J. Biol. Chem. 2007, 282, 37232–37243. [Google Scholar] [CrossRef] [Green Version]
- Kostenko, S.; Johannessen, M.; Moens, U. PKA-induced F-actin rearrangement requires phosphorylation of Hsp27 by the MAPKAP kinase MK5. Cell. Signal. 2009, 21, 712–718. [Google Scholar] [CrossRef]
- Tak, H.; Jang, E.; Kim, S.B.; Park, J.; Suk, J.; Yoon, Y.S.; Ahn, J.K.; Lee, J.-H.; Joe, C.O. 14-3-3epsilon inhibits MK5-mediated cell migration by disrupting F-actin polymerization. Cell. Signal. 2007, 19, 2379–2387. [Google Scholar] [CrossRef]
- Dwyer, S.F.; Gelman, I.H. Cross-phosphorylation and interaction between Src/FAK and MAPKAP5/PRAK in early focal adhesions controls cell motility. J. Cancer Biol. Res. 2014, 2, 1045. [Google Scholar]
- Stöhr, N.; Köhn, M.; Lederer, M.; Glaß, M.; Reinke, C.; Singer, R.H.; Hüttelmaier, S. IGF2BP1 promotes cell migration by regulating MK5 and PTEN signaling. Genes Dev. 2012, 26, 176–189. [Google Scholar] [CrossRef] [Green Version]
- Schlaepfer, D.D.; Hauck, C.R.; Sieg, D.J. Signaling through focal adhesion kinase. Prog. Biophys. Mol. Biol. 1999, 71, 435–478. [Google Scholar] [CrossRef] [Green Version]
- Brunton, V.G.; Avizienyte, E.; Fincham, V.J.; Serrels, B.; Metcalf, C.A.; Sawyer, T.K.; Frame, M.C. Identification of Src-specific phosphorylation site on focal adhesion kinase: Dissection of the role of Src SH2 and catalytic functions and their consequences for tumor cell behavior. Cancer Res. 2005, 65, 1335–1342. [Google Scholar] [CrossRef] [Green Version]
- Slack, J.K.; Adams, R.B.; Rovin, J.D.; Bissonette, E.A.; Stoker, C.E.; Parsons, J.T. Alterations in the focal adhesion kinase/Src signal transduction pathway correlate with increased migratory capacity of prostate carcinoma cells. Oncogene 2001, 20, 1152–1163. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Gallo, K.A. MLK3 regulates paxillin phosphorylation in chemokine-mediated breast cancer cell migration and invasion to drive metastasis. Cancer Res. 2012, 72, 4130–4140. [Google Scholar] [CrossRef]
- Tsubouchi, A.; Sakakura, J.; Yagi, R.; Mazaki, Y.; Schaefer, E.; Yano, H.; Sabe, H. Localized suppression of RhoA activity by Tyr31/118-phosphorylated paxillin in cell adhesion and migration. J. Cell Biol. 2002, 159, 673–683. [Google Scholar] [CrossRef] [Green Version]
- Lavoie, J.N.; Hickey, E.; Weber, L.A.; Landry, J. Modulation of actin microfilament dynamics and fluid phase pinocytosis by phosphorylation of heat shock protein 27. J. Biol. Chem. 1993, 268, 24210–24214. [Google Scholar] [CrossRef]
- Huot, J.; Houle, F.; Spitz, D.R.; Landry, J. HSP27 phosphorylation-mediated resistance against actin fragmentation and cell death induced by oxidative stress. Cancer Res. 1996, 56, 273–279. [Google Scholar]
- Song, I.S.; Kang, S.-S.; Kim, E.-S.; Park, H.-M.; Choi, C.Y.; Tchah, H.; Kim, J.Y. Heat shock protein 27 phosphorylation is involved in epithelial cell apoptosis as well as epithelial migration during corneal epithelial wound healing. Exp. Eye Res. 2014, 118, 36–41. [Google Scholar] [CrossRef]
- Wolfson, R.K.; Chiang, E.T.; Garcia, J.G. HMGB1 induces human lung endothelial cell cytoskeletal rearrangement and barrier disruption. Microvasc. Res. 2011, 81, 189–197. [Google Scholar] [CrossRef] [Green Version]
- Huot, J.; Lambert, H.; Lavoie, J.N.; Guimond, A.; Houle, F.; Landry, J. Characterization of 45-kDa/54-kDa HSP27 kinase, a stress-sensitive kinase which may activate the phosphorylation-dependent protective function of mammalian 27-kDa heat-shock protein HSP27. Eur. J. Biochem. 1995, 227, 416–427. [Google Scholar] [CrossRef]
- Katsogiannou, M.; Andrieu, C.; Rocchi, P. Heat shock protein 27 phosphorylation state is associated with cancer progression. Front Genet. 2014, 5, 346. [Google Scholar] [CrossRef] [Green Version]
- New, L.; Jiang, Y.; Zhao, M.; Liu, K.; Zhu, W.; Flood, L.J.; Kato, Y.; Parry, G.C.; Han, J. PRAK, a novel protein kinase regulated by the p38 MAP kinase. EMBO J. 1998, 17, 3372–3384. [Google Scholar] [CrossRef] [Green Version]
- Kostenko, S.; Moens, U. Heat shock protein 27 phosphorylation: Kinases, phosphatases, functions and pathology. Cell. Mol. Life Sci. 2009, 66, 3289–3307. [Google Scholar] [CrossRef]
- Shiryaev, A.; Dumitriu, G.; Moens, U. Distinct roles of MK2 and MK5 in cAMP/PKA- and stress/p38MAPK-induced heat shock protein 27 phosphorylation. J. Mol. Signal. 2011, 6, 4. [Google Scholar] [CrossRef] [Green Version]
- Shiryaev, A.; Moens, U. Mitogen-activated protein kinase p38 and MK2, MK3 and MK5: Ménage à trois or ménage à quatre? Cell Signal. 2010, 22, 1185–1192. [Google Scholar] [CrossRef]
- Coulombe, P.; Rodier, G.; Bonneil, E.; Thibault, P.; Meloche, S. N-Terminal ubiquitination of extracellular signal-regulated kinase 3 and p21 directs their degradation by the proteasome. Mol. Cell. Biol. 2004, 24, 6140–6150. [Google Scholar] [CrossRef] [Green Version]
- Coulombe, P.; Rodier, G.; Pelletier, S.; Pellerin, J.; Meloche, S. Rapid turnover of extracellular signal-regulated kinase 3 by the ubiquitin-proteasome pathway defines a novel paradigm of mitogen-activated protein kinase regulation during cellular differentiation. Mol. Cell. Biol. 2003, 23, 4542–4558. [Google Scholar] [CrossRef] [Green Version]
- Seternes, O.M.; Mikalsen, T.; Johansen, B.; Michaelsen, E.; Armstrong, C.G.; Morrice, N.A.; Turgeon, B.; Meloche, S.; Moens, U.; Keyse, M.S. Activation of MK5/PRAK by the atypical MAP kinase ERK3 defines a novel signal transduction pathway. Embo J. 2004, 23, 4780–4791. [Google Scholar] [CrossRef]
- Al-Mahdi, R.; Babteen, N.; Thillai, K.; Holt, M.; Johansen, B.; Wetting, H.L.; Seternes, O.-M.; Wells, C.M. A novel role for atypical MAPK kinase ERK3 in regulating breast cancer cell morphology and migration. Cell Adhes. Migr. 2015, 9, 483–494. [Google Scholar] [CrossRef] [Green Version]
- Long, W.; Foulds, C.; Qin, J.; Liu, J.; Ding, C.; Lonard, D.M.; Solis, L.M.; Wistuba, I.I.; Tsai, S.Y.; Tsai, M.-J.; et al. ERK3 signals through SRC-3 coactivator to promote human lung cancer cell invasion. J. Clin. Investig. 2012, 122, 1869–1880. [Google Scholar] [CrossRef]
- Elkin, M.; Vlodavsky, I. Tail vein assay of cancer metastasis. Curr. Protoc. Cell Biol. 2001, 12, 19.2.1–19.2.7. [Google Scholar] [CrossRef]
- Wu, X.; Gong, S.; Roy-Burman, P.; Lee, P.; Culig, Z. Current mouse and cell models in prostate cancer research. Endocr. Relat. Cancer 2013, 20, R155–R170. [Google Scholar] [CrossRef] [Green Version]
- Stopsack, K.H. Efficacy of PARP Inhibition in Metastatic Castration-resistant Prostate Cancer is Very Different with Non-BRCA DNA Repair Alterations: Reconstructing Prespecified Endpoints for Cohort B from the Phase 3 PROfound Trial of Olaparib. Eur. Urol. 2021, 79, 442–445. [Google Scholar] [CrossRef]
- Schaller, M.D.; Parsons, J.T. pp125FAK-dependent tyrosine phosphorylation of paxillin creates a high-affinity binding site for Crk. Mol. Cell. Biol. 1995, 15, 2635–2645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, D.J.; Schroeder, M.J.; Brame, C.J.; Whitmore, L.; Shabanowitz, J.; Hunt, D.F.; Horwitz, A.R. Paxillin phosphorylation sites mapped by mass spectrometry. J. Cell Sci. 2005, 118, 4925–4929. [Google Scholar] [CrossRef] [PubMed]
- Rajah, A.; Boudreau, C.G.; Ilie, A.; Wee, T.-L.; Tang, K.; Borisov, A.Z.; Orlowski, J.; Brown, C.M. Paxillin S273 Phosphorylation Regulates Adhesion Dynamics and Cell Migration through a Common Protein Complex with PAK1 and βPIX. Sci. Rep. 2019, 9, 11430. [Google Scholar] [CrossRef] [Green Version]
- Roof, D.J.; Hayes, A.; Adamian, M.; Chishti, A.H.; Li, T. Molecular Characterization of abLIM, a Novel Actin-binding and Double Zinc Finger Protein. J. Cell Biol. 1997, 138, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Wang, Y.-H.; Wu, X.-N.; Wu, S.-Q.; Lu, B.-J.; Dong, M.-Q.; Zhang, H.; Sun, P.; Lin, S.-C.; Guan, K.-L.; et al. Inactivation of Rheb by PRAK-mediated phosphorylation is essential for energy-depletion-induced suppression of mTORC1. Nat. Cell Biol. 2011, 13, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence-5′-3′ |
|---|---|
| MMP2-F | 5′-TCCACCACCTACAACTTTGAG-3′ |
| MMP2-R | 5′-GTGCAGCTGTCATAGGATGT-3′ |
| MMP9-F | 5′-ACATCGTCATCCAGTTTGGTG-3′ |
| MMP9-R | 5′-CGTCGAAATGGGCGTCT-3′ |
| MMP10-F | 5′-GGAGACTTTTACTCTTTTGATGGC-3′ |
| MMP10-R | 5′-AGCAACGAGGAATAAATTGGTG-3′ |
| GAPDH-F | 5′-ACATCGCTCAGACACCATG-3′ |
| GAPDH-R | 5′-TGTAGTTGAGGTCAATGAAGGG-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalil, M.I.; De Benedetti, A. The TLK1–MK5 Axis Regulates Motility, Invasion, and Metastasis of Prostate Cancer Cells. Cancers 2022, 14, 5728. https://doi.org/10.3390/cancers14235728
Khalil MI, De Benedetti A. The TLK1–MK5 Axis Regulates Motility, Invasion, and Metastasis of Prostate Cancer Cells. Cancers. 2022; 14(23):5728. https://doi.org/10.3390/cancers14235728
Chicago/Turabian StyleKhalil, Md Imtiaz, and Arrigo De Benedetti. 2022. "The TLK1–MK5 Axis Regulates Motility, Invasion, and Metastasis of Prostate Cancer Cells" Cancers 14, no. 23: 5728. https://doi.org/10.3390/cancers14235728
APA StyleKhalil, M. I., & De Benedetti, A. (2022). The TLK1–MK5 Axis Regulates Motility, Invasion, and Metastasis of Prostate Cancer Cells. Cancers, 14(23), 5728. https://doi.org/10.3390/cancers14235728