
Ten Years of CRISPRing Cancers In Vitro

Abstract
:Simple Summary
Abstract
1. Introduction
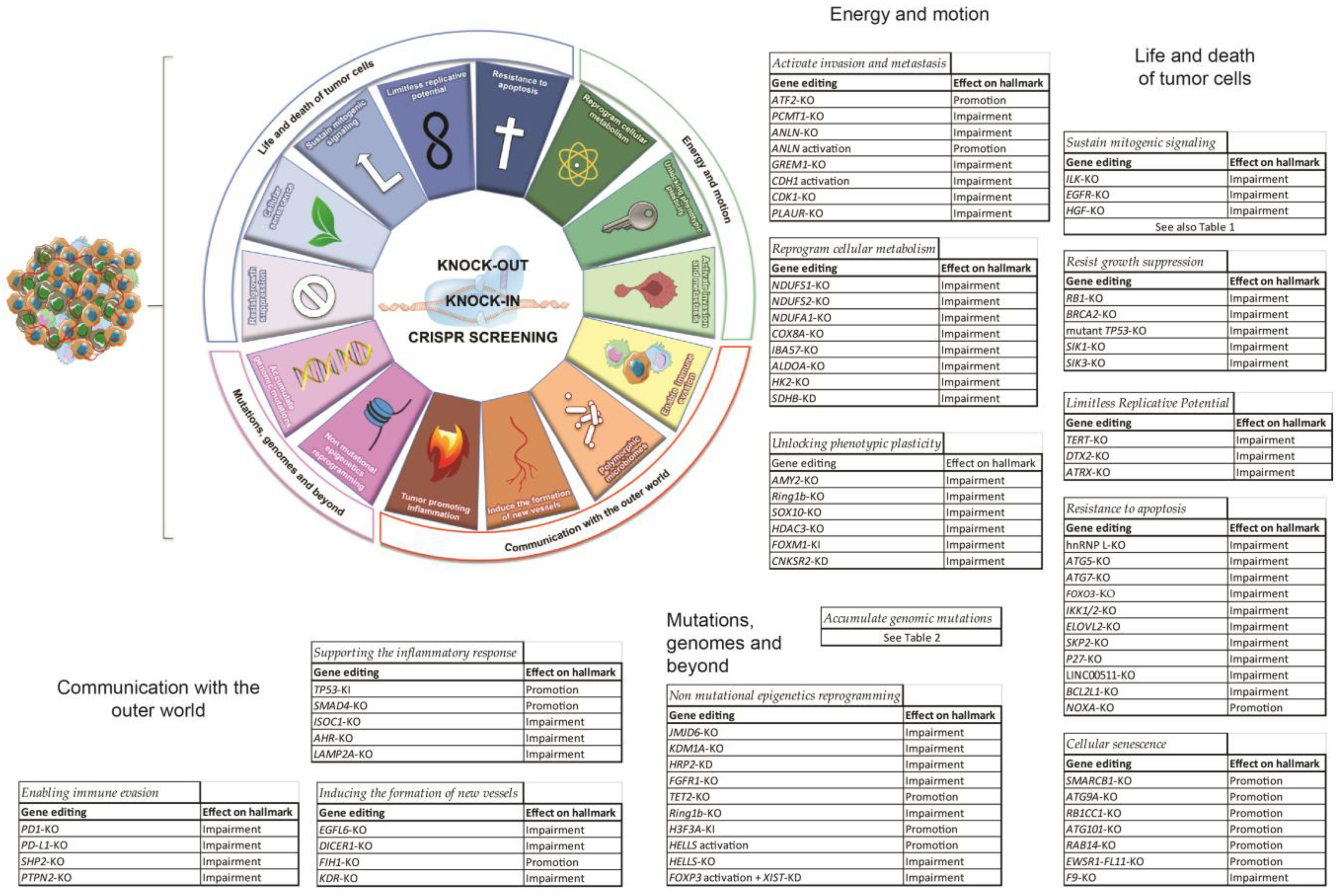
2. CRISPR and Cancer
2.1. Life and Death of Tumor Cells
2.1.1. Sustain Mitogenic Signaling
2.1.2. Resist Growth Suppression
2.1.3. Limitless Replicative Potential
2.1.4. Resistance to Apoptosis
2.1.5. Cellular Senescence
2.2. Mutations, Genomes and Beyond
2.2.1. Accumulate Genomic Mutations
2.2.2. Non-Mutational Epigenetics Reprogramming
2.3. Energy and Motion
2.3.1. Reprogram Cellular Metabolism
2.3.2. Unlocking Phenotypic Plasticity
2.3.3. Activate Invasion and Metastasis
2.4. Communication with the Outer World
2.4.1. Supporting the Inflammatory Response
2.4.2. Enabling Immune Evasion
2.4.3. Inducing the Formation of New Vessels
2.4.4. Polymorphic Microbiomes
3. Discussion and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakata, A. Nucleotide Sequence of the iap Gene, Responsible for Alkaline Phosphatase Isozyme Conversion in Escherichia coli, and Identification of the Gene Product. J. Bacteriol. 1987, 169, 5429–5433. [Google Scholar] [CrossRef] [Green Version]
- Mojica, F.J.M.; Díez-Villaseñor, C.; García-Martínez, J.; Soria, E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 2005, 60, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Wiedenheft, B.; Sternberg, S.H.; Doudna, J.A. RNA-guided genetic silencing systems in bacteria and archaea. Nature 2012, 482, 331–338. [Google Scholar] [CrossRef]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR Provides Acquired Resistance Against Viruses in Prokaryotes. Science 2007, 315, 170–172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Qin, C.; An, C.; Zheng, X.; Wen, S.; Chen, W.; Liu, X.; Lv, Z.; Yang, P.; Xu, W.; et al. Application of the CRISPR/Cas9-based gene editing technique in basic research, diagnosis, and therapy of cancer. Mol. Cancer 2021, 20, 126. [Google Scholar] [CrossRef] [PubMed]
- Moses, C.; Garcia-Bloj, B.; Harvey, A.R.; Blancafort, P. Hallmarks of cancer: The CRISPR generation. Eur. J. Cancer 2018, 93, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Kannan, R.; Ventura, A. The CRISPR revolution and its impact on cancer research. Swiss Med. Wkly. 2015, 145, w14230. [Google Scholar] [CrossRef]
- Wild, C.; Weiderpass, E.; Stewart, B. World Cancer Report: Cancer Research for Cancer Development; IARC: Cedex, France, 2020.
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer Review evolve progressively from normalcy via a series of pre. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Shan, B.-Q.; Wang, X.-M.; Zheng, L.; Han, Y.; Gao, J.; Lv, M.; Zhang, Y.; Liu, Y.-X.; Zhang, H.; Chen, H.-S.; et al. DCAF13 promotes breast cancer cell proliferation by ubiquitin inhibiting PERP expression. Cancer Sci. 2022, 113, 1587–1600. [Google Scholar] [CrossRef] [PubMed]
- Tzelepis, K.; Koike-Yusa, H.; De Braekeleer, E.; Li, Y.; Metzakopian, E.; Dovey, O.M.; Mupo, A.; Grinkevich, V.; Li, M.; Mazan, M.; et al. A CRISPR Dropout Screen Identifies Genetic Vulnerabilities and Therapeutic Targets in Acute Myeloid Leukemia. Cell Rep. 2016, 17, 1193–1205. [Google Scholar] [CrossRef] [Green Version]
- Singhal, J.; Chikara, S.; Horne, D.; Awasthi, S.; Salgia, R.; Singhal, S.S. Targeting RLIP with CRISPR/Cas9 controls tumor growth. Carcinogenesis 2020, 42, 48–57. [Google Scholar] [CrossRef]
- Wang, Q.; Li, J.; Zhu, J.; Mao, J.; Duan, C.; Liang, X.; Zhu, L.; Zhu, M.; Zhang, Z.; Lin, F.; et al. Genome-wide CRISPR/Cas9 screening for therapeutic targets in NSCLC carrying wild-type TP53 and receptor tyrosine kinase genes. Clin. Transl. Med. 2022, 12, e882. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Cao, C.; Gan, Y.; Wang, X.; Wu, T.; Zhang, Q.; Liu, Y.; Yao, L. ZNF677 suppresses renal cell carcinoma progression through N6-methyladenosine and transcriptional repression of CDKN3. Clin. Transl. Med. 2022, 12, e906. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, B.; Murugan, S.; Palagani, A.; Sarkar, D.K. Beta 2 adrenergic receptor and mu opioid receptor interact to potentiate the aggressiveness of human breast cancer cell by activating the glycogen synthase kinase 3 signaling. Breast Cancer Res. 2022, 24, 33. [Google Scholar] [CrossRef]
- Quan, M.; Oh, Y.; Cho, S.-Y.; Kim, J.H.; Moon, H.-G. Polo-Like Kinase 1 Regulates Chromosomal Instability and Paclitaxel Resistance in Breast Cancer Cells. J. Breast Cancer 2022, 25, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Bakhshi, S.E.; Roushandeh, A.M.; Roudkenar, M.H.; Shekarchi, S.; Bahadori, M.H. CRISPR/Cas9-mediated knockout of HO-1 decreased the proliferation and migration of T47D cells and increased cisplatin-induced apoptosis: An in vitro study. Med. Oncol. 2022, 39, 175. [Google Scholar] [CrossRef]
- Liu, Y.; Kong, X.-X.; He, J.-J.; Xu, Y.-B.; Zhang, J.-K.; Zou, L.-Y.; Ding, K.-F.; Xu, D. OLA1 promotes colorectal cancer tumorigenesis by activation of HIF1α/CA9 axis. BMC Cancer 2022, 22, 424. [Google Scholar] [CrossRef]
- He, W.; Lin, S.; Guo, Y.; Wu, Y.; Zhang, L.-L.; Deng, Q.; Du, Z.-M.; Wei, M.; Zhu, W.; Chen, W.-J.; et al. Targeted demethylation at ZNF154 promotor upregulates ZNF154 expression and inhibits the proliferation and migration of Esophageal Squamous Carcinoma cells. Oncogene 2022, 41, 4537–4546. [Google Scholar] [CrossRef]
- Nagarajan, S.; Rao, S.V.; Sutton, J.; Cheeseman, D.; Dunn, S.; Papachristou, E.K.; Prada, J.-E.G.; Couturier, D.-L.; Kumar, S.; Kishore, K.; et al. ARID1A influences HDAC1/BRD4 activity, intrinsic proliferative capacity and breast cancer treatment response. Nat. Genet. 2020, 52, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Körner, S.; Pick, T.; Bochen, F.; Wemmert, S.; Körbel, C.; Menger, M.D.; Cavalié, A.; Kühn, J.-P.; Schick, B.; Linxweiler, M. Antagonizing Sec62 function in intracellular Ca2+ homeostasis represents a novel therapeutic strategy for head and neck cancer. Front. Physiol. 2022, 13, 880004. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, H.; Cingoz, H.; Tufan, T.; Yang, J.; Adair, S.J.; Tummala, K.S.; Kuscu, C.; Kinali, M.; Comertpay, G.; Nagdas, S.; et al. ISL2 is a putative tumor suppressor whose epigenetic silencing reprograms the metabolism of pancreatic cancer. Dev. Cell 2022, 57, 1331–1346.e9. [Google Scholar] [CrossRef] [PubMed]
- Huebner, K.; Erlenbach-Wuensch, K.; Prochazka, J.; Sheraj, I.; Hampel, C.; Mrazkova, B.; Michalcikova, T.; Tureckova, J.; Iatsiuk, V.; Weissmann, A.; et al. ATF2 loss promotes tumor invasion in colorectal cancer cells via upregulation of cancer driver TROP2. Cell Mol. Life Sci. 2022, 79, 423. [Google Scholar] [CrossRef]
- Shi, S.; Chen, H.; Wang, H.; Wan, J.; Shi, Y.; Li, J.; Wang, S.; Shi, J.; Lv, J.; Wu, T.; et al. Genome-wide CRISPR knockout screening identified G protein pathway suppressor 2 as a novel tumor suppressor for uveal melanoma metastasis. J. Cancer Res. Clin. Oncol. 2022; ahead of print. [Google Scholar] [CrossRef]
- Peng, W.-X.; Huang, J.; Yang, L.; Gong, A.-H.; Mo, Y.-Y. Linc-RoR promotes MAPK/ERK signaling and confers estrogen-independent growth of breast cancer. Mol. Cancer 2017, 16, 161. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Z.; Zhang, X.; Jiang, C.-Q.; Zhang, Y.-G.; Wu, X.; Li, J.; Tang, S.; Li, L.; Gu, L.-J.; Xie, X.-Y.; et al. Identifying novel therapeutic targets in gastric cancer using genome-wide CRISPR-Cas9 screening. Oncogene 2022, 41, 2069–2078. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, W.; Cheng, X.; Wang, F.; Gao, C.; Song, F.; Song, F.; Liang, X.; Fang, W.; Chen, Z. Sphingomyelin Phodiesterase Acid-Like 3A Promotes Hepatocellular Carcinoma Growth Through the Enhancer of Rudimentary Homolog. Front. Oncol. 2022, 12, 852765. [Google Scholar] [CrossRef]
- Shmakova, A.A.; Klimovich, P.S.; Rysenkova, K.D.; Popov, V.S.; Gorbunova, A.S.; Karpukhina, A.A.; Karagyaur, M.N.; Rubina, K.A.; Tkachuk, V.A.; Semina, E.V. Urokinase Receptor uPAR Downregulation in Neuroblastoma Leads to Dormancy, Chemoresistance and Metastasis. Cancers 2022, 14, 994. [Google Scholar] [CrossRef]
- Quijano-Rubio, C.; Silginer, M.; Weller, M. CD95 gene deletion may reduce clonogenic growth and invasiveness of human glioblastoma cells in a CD95 ligand-independent manner. Cell Death Discov. 2022, 8, 341. [Google Scholar] [CrossRef]
- JChen, J.; Bell, J.; Lau, B.T.; Whittaker, T.; Stapleton, D.; Ji, H.P. A functional CRISPR/Cas9 screen identifies kinases that modulate FGFR inhibitor response in gastric cancer. Oncogenesis 2019, 8, 33. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Arguello, O.A.D.; Chen, D.; Chen, S.; Saber, A.; Haisma, H.J. CRISPR-mediated ablation of overexpressed EGFR in combination with sunitinib significantly suppresses renal cell carcinoma proliferation. PLoS ONE 2020, 15, e0232985. [Google Scholar] [CrossRef]
- Lee, H.K.; Lim, H.M.; Park, S.-H.; Nam, M.J. Knockout of hepatocyte growth factor by CRISPR/Cas9 system induces apoptosis in hepatocellular carcinoma cells. J. Pers. Med. 2021, 11, 983. [Google Scholar] [CrossRef]
- Tejero, R.; Huang, Y.; Katsyv, I.; Kluge, M.; Lin, J.-Y.; Tome-Garcia, J.; Daviaud, N.; Wang, Y.; Zhang, B.; Tsankova, N.M.; et al. Gene signatures of quiescent glioblastoma cells reveal mesenchymal shift and interactions with niche microenvironment. EBioMedicine 2019, 42, 252–269. [Google Scholar] [CrossRef] [Green Version]
- Tang, N.; Cheng, C.; Zhang, X.; Qiao, M.; Li, N.; Mu, W.; Wei, X.-F.; Han, W.; Wang, H. TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight 2020, 5, e133977. [Google Scholar] [CrossRef]
- Jiang, Y.; Chu, W.K. Potential Roles of the Retinoblastoma Protein in Regulating Genome Editing. Front. Cell Dev. Biol. 2018, 6, 81. [Google Scholar] [CrossRef]
- Kanber, D.; Woestefeld, J.; Döpper, H.; Bozet, M.; Brenzel, A.; Altmüller, J.; Kilpert, F.; Lohmann, D.; Pommerenke, C.; Steenpass, L. RB1-Negative Retinal Organoids Display Proliferation of Cone Photoreceptors and Loss of Retinal Differentiation. Cancers 2022, 14, 2166. [Google Scholar] [CrossRef]
- Marshall, A.E.; Roes, M.V.; Passos, D.T.; DeWeerd, M.C.; Chaikovsky, A.C.; Sage, J.; Howlett, C.J.; Dick, F.A. RB1 Deletion in Retinoblastoma Protein Pathway-Disrupted Cells Results in DNA Damage and Cancer Progression. Mol. Cell Biol. 2019, 39, e00105-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oser, M.G.; Fonseca, R.; Chakraborty, A.A.; Brough, R.; Spektor, A.; Jennings, R.B.; Flaifel, A.; Novak, J.S.; Gulati, A.; Buss, E.; et al. Cells lacking the RB1 tumor suppressor gene are hyperdependent on aurora B kinase for survival. Cancer Discov. 2019, 9, 230–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, G.; Armenia, J.; Mazzu, Y.Z.; Nandakumar, S.; Stopsack, K.H.; Atiq, M.O.; Komura, K.; Jehane, L.; Hirani, R.; Chadalavada, K.; et al. Significance of BRCA2 and RB1 co-loss in aggressive prostate cancer progression. Clin. Cancer Res. 2020, 26, 2047–2064. [Google Scholar] [CrossRef]
- Tang, F.; Min, L.; Seebacher, N.A.; Li, X.; Zhou, Y.; Hornicek, F.J.; Wei, Y.; Tu, C.; Duan, Z. Targeting mutant TP53 as a potential therapeutic strategy for the treatment of osteosarcoma. J. Orthop. Res. 2019, 37, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Hollstein, P.E.; Eichner, L.J.; Brun, S.N.; Kamireddy, A.; Svensson, R.U.; Vera, L.I.; Ross, D.S.; Rymoff, T.J.; Hutchins, A.; Galvez, H.M.; et al. The AMPK-related kinases SIK1 and SIK3 mediate key tumor-suppressive effects of LKB1 in NSCLC. Cancer Discov. 2019, 9, 1606–1627. [Google Scholar] [CrossRef] [Green Version]
- Hayflick, L. The illusion of cell immortality. Br. J. Cancer 2000, 83, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Saraswati, A.P.; Relitti, N.; Brindisi, M.; Gemma, S.; Zisterer, D.; Butini, S.; Campiani, G. Raising the bar in anticancer therapy: Recent advances in, and perspectives on, telomerase inhibitors. Drug Discov. Today 2019, 24, 1370–1388. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Bacchetti, S. A Survey of Telomerase Activity in Human Cancer. Eur. J. Cancer 1997, 33, 787–791. [Google Scholar] [CrossRef]
- Wen, L.; Zhao, C.; Song, J.; Ma, L.; Ruan, J.; Xia, X.; Chen, Y.E.; Zhang, J.; Ma, P.X.; Xu, J. CRISPR/Cas9-mediated TERT disruption in cancer cells. Int. J. Mol. Sci. 2020, 21, 653. [Google Scholar] [CrossRef] [Green Version]
- Chiba, K.; Johnson, J.Z.; Vogan, J.M.; Wagner, T.; Boyle, J.M.; Hockemeyer, D. Cancer-associated TERT promoter mutations abrogate telomerase silencing. Elife 2015, 4, e07918. [Google Scholar] [CrossRef]
- Xi, L.; Schmidt, J.C.; Zaug, A.J.; Ascarrunz, D.R.; Cech, T.R. A novel two-step genome editing strategy with CRISPR-Cas9 provides new insights into telomerase action and TERT gene expression. Genome Biol. 2015, 16, 231. [Google Scholar] [CrossRef] [Green Version]
- Dai, W.; Xu, X.; Wang, D.; Wu, J.; Wang, J. Cancer therapy with a CRISPR-assisted telomerase-activating gene expression system. Oncogene 2019, 38, 4110–4124. [Google Scholar] [CrossRef]
- Zhou, Z.; Li, Y.; Xu, H.; Xie, X.; He, Z.; Lin, S.; Li, R.; Jin, S.; Cui, J.; Hu, H.; et al. An inducible CRISPR/Cas9 screen identifies DTX2 as a transcriptional regulator of human telomerase. iScience 2022, 25, 103813. [Google Scholar] [CrossRef]
- Shay, J.; Reddel, R.; Wright, W. Cancer and Telomeres—An ALTernative to Telomerase. Science 2012, 336, 1387–1388. [Google Scholar] [CrossRef]
- Graham, M.K.; Kim, J.; Da, J.; Brosnan-Cashman, J.A.; Rizzo, A.; Del Valle, J.A.B.; Chia, L.; Rubenstein, M.; Davis, C.; Zheng, Q.; et al. Functional loss of ATRX and TERC activates Alternative Lengthening of Telomeres (ALT) in LAPC4 prostate cancer cells. Mol. Cancer Res. 2019, 17, 2480–2491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishor, A.; Ge, Z.; Hogg, J.R. hnRNP L-dependent protection of normal mRNAs from NMD subverts quality control in B cell lymphoma. EMBO J. 2018, 38, e99128. [Google Scholar] [CrossRef]
- Fitzwalter, B.E.; Towers, C.G.; Sullivan, K.D.; Andrysik, Z.; Hoh, M.; Ludwig, M.; O’Prey, J.; Ryan, K.M.; Espinosa, J.M.; Morgan, M.J.; et al. Autophagy Inhibition Mediates Apoptosis Sensitization in Cancer Therapy by Relieving FOXO3a Turnover. Dev. Cell 2018, 44, 555–565.e3. [Google Scholar] [CrossRef] [Green Version]
- Slotta, C.; Schlüter, T.; Ruiz-Perera, L.M.; Kadhim, H.M.; Tertel, T.; Henkel, E.; Hubner, W.; Greiner, J.F.W.; Huser, T.; Kaltschmidt, B.; et al. CRISPR/Cas9-mediated knockout of c-REL in HeLa cells results in profound defects of the cell cycle. PLoS ONE 2017, 12, e0182373. [Google Scholar] [CrossRef] [Green Version]
- Slotta, C.; Storm, J.; Pfisterer, N.; Henkel, E.; Kleinwächter, S.; Pieper, M.; Ruiz-Perera, L.M.; Greiner, J.F.; Kaltschmidt, B.; Kaltschmidt, C. IKK1/2 protect human cells from TNF-mediated RIPK1-dependent apoptosis in an NF-κB-independent manner. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1025–1033. [Google Scholar] [CrossRef]
- Al-Sammarraie, N.; Ray, S.K. Applications of CRISPR-Cas9 technology to genome editing in glioblastoma multiforme. Cells 2021, 10, 2342. [Google Scholar] [CrossRef]
- Tanaka, K.; Kandori, S.; Sakka, S.; Nitta, S.; Tanuma, K.; Shiga, M.; Nagumo, Y.; Negoro, H.; Kojima, T.; Mathis, B.J.; et al. ELOVL2 promotes cancer progression by inhibiting cell apoptosis in renal cell carcinoma. Oncol. Rep. 2021, 47, 23. [Google Scholar] [CrossRef]
- Lohmüller, M.; Roeck, B.F.; Szabo, T.G.; Schapfl, M.A.; Pegka, F.; Herzog, S.; Villunger, A.; Schuler, F. The SKP2-p27 axis defines susceptibility to cell death upon CHK1 inhibition. Mol. Oncol. 2022, 16, 2771–2787. [Google Scholar] [CrossRef]
- Azadbakht, N.; Doosti, A.; Jami, M.-S. CRISPR/Cas9-mediated LINC00511 knockout strategies, increased apoptosis of breast cancer cells via suppressing antiapoptotic genes. Biol. Proced. Online 2022, 24, 8. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.F.A.; Azlan, A.; Lo, K.W.; Azzam, G.; Mohana-Kumaran, N. Dual inhibition of anti-apoptotic proteins BCL-XL and MCL-1 enhances cytotoxicity of Nasopharyngeal carcinoma cells. Discov. Oncol. 2022, 13, 9. [Google Scholar] [CrossRef]
- Thummuri, D.; Khan, S.; Underwood, P.W.; Zhang, P.; Wiegand, J.; Zhang, X.; Budamagunta, V.; Sobh, A.; Tagmount, A.; Loguinov, A.; et al. Overcoming Gemcitabine Resistance in Pancreatic Cancer Using the BCL-XL–Specific Degrader DT2216. Mol. Cancer Ther. 2021, 21, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Nechiporuk, T.; Kurtz, S.E.; Nikolova, O.; Liu, T.; Jones, C.L.; D’Alessandro, A.; Culp-Hill, R.; D’Almeida, A.; Joshi, S.K.; Rosenberg, M.; et al. The TP53 apoptotic network is a primary mediator of resistance to BCL2 inhibition in AML cells. Cancer Discov. 2019, 9, 910–925. [Google Scholar] [CrossRef]
- Yan, X.; Chen, D.; Wang, Y.; Guo, Y.; Tong, C.; Wei, J.; Zhang, Y.; Wu, Z.; Han, W. Identification of NOXA as a pivotal regulator of resistance to CAR T-cell therapy in B-cell malignancies. Signal Transduct. Target. Ther. 2022, 7, 98. [Google Scholar] [CrossRef] [PubMed]
- Kowald, A.; Passos, J.F.; Kirkwood, T.B.L. On the evolution of cellular senescence. Aging Cell 2020, 19, e13270. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: Context-dependent effects of SASP in cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef]
- Wang, L.; de Oliveira, R.L.; Wang, C.; Neto, J.M.F.; Mainardi, S.; Evers, B.; Lieftink, C.; Morris, B.; Jochems, F.; Willemsen, L.; et al. High-Throughput Functional Genetic and Compound Screens Identify Targets for Senescence Induction in Cancer. Cell Rep. 2017, 21, 773–783. [Google Scholar] [CrossRef] [Green Version]
- Schepers, A.; Jochems, F.; Lieftink, C.; Wang, L.; Pogacar, Z.; de Oliveira, R.L.; De Conti, G.; Beijersbergen, R.L.; Bernards, R. Identification of autophagy-related genes as targets for senescence induction using a customizable CRISPR-based suicide switch screen. Mol. Cancer Res. 2021, 19, 1613–1621. [Google Scholar] [CrossRef]
- Gaspar, N.; Hawkins, D.S.; Dirksen, U.; Lewis, I.J.; Ferrari, S.; Le Deley, M.-C.; Kovar, H.; Grimer, R.; Whelan, J.; Claude, L.; et al. Ewing sarcoma: Current management and future approaches through collaboration. J. Clin. Oncol. 2015, 33, 3036–3046. [Google Scholar] [CrossRef] [Green Version]
- Cervera, S.T.; Rodríguez-Martín, C.; Fernández-Tabanera, E.; de Mera, R.M.-F.; Morin, M.; Fernández-Peñalver, S.; Iranzo-Martínez, M.; Amhih-Cardenas, J.; García-García, L.; González-González, L.; et al. Therapeutic potential of ewsr1–fli1 inactivation by crispr/cas9 in ewing sarcoma. Cancers 2021, 13, 3783. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Dering, J.; Conklin, D.; Kalous, O.; Cohen, D.J.; Desai, A.J.; Ginther, C.; Atefi, M.; Chen, I.; Fowst, C.; et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009, 11, R77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpintero-Fernández, P.; Borghesan, M.; Eleftheriadou, O.; Pan-Castillo, B.; Fafián-Labora, J.A.; Mitchell, T.P.; Yuste, A.; Ogrunc, M.; Nightingale, T.D.; Mayan, M.; et al. Genome wide CRISPR/Cas9 screen identifies the coagulation factor IX (F9) as a regulator of senescence. Cell Death Dis. 2022, 13, 163. [Google Scholar] [CrossRef]
- Burgess, M.R.; Hwang, E.; Mroue, R.; Bielski, C.M.; Wandler, A.M.; Huang, B.J.; Firestone, A.J.; Young, A.; Lacap, J.A.; Crocker, L.; et al. KRAS Allelic Imbalance Enhances Fitness and Modulates MAP Kinase Dependence in Cancer. Cell 2017, 168, 817–829.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remacha, L.; Currás-Freixes, M.; Torres-Ruiz, R.; Schiavi, F.; Torres-Pérez, R.; Calsina, B.; Letón, R.; Comino-Méndez, I.; Roldán-Romero, J.M.; Montero-Conde, C.; et al. Gain-of-function mutations in DNMT3A in patients with paraganglioma. Genet. Med. 2018, 20, 1644–1651. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.-H.; Wu, C.-C.; Huang, K.-Y.; Leu, Y.-L.; Yang, S.-C.; Chen, C.-L.; Chen, C.-Y. Integrated Omics Analysis of Non-Small-Cell Lung Cancer Cells Harboring the EGFR C797S Mutation Reveals the Potential of AXL as a Novel Therapeutic Target in TKI-Resistant Lung Cancer. Cancers 2020, 13, 111. [Google Scholar] [CrossRef]
- Li, S.; Garay, J.P.; Tubbs, C.A.; Franco, H.L. CRISPR-based knock-in mutagenesis of the pioneer transcription factor FOXA1: Optimization of strategies for multi-allelic proteins in cancer cells. FEBS Open Bio 2021, 11, 1537–1551. [Google Scholar] [CrossRef]
- Sánchez-Vázquez, R.; Martínez, P.; Blasco, M.A. AKT-dependent signaling of extracellular cues through telomeres impact on tumorigenesis. PLoS Genet. 2021, 17, e1009410. [Google Scholar] [CrossRef]
- Gonzalez, T.L.; Hancock, M.; Sun, S.; Gersch, C.L.; Larios, J.M.; David, W.; Hu, J.; Hayes, D.F.; Wang, S.; Rae, J.M. Targeted degradation of activating estrogen receptor α ligand-binding domain mutations in human breast cancer. Breast Cancer Res. Treat. 2020, 180, 611–622. [Google Scholar] [CrossRef]
- Bahreini, A.; Li, Z.; Wang, P.; Levine, K.M.; Tasdemir, N.; Cao, L.; Weir, H.M.; Puhalla, S.L.; Davidson, N.E.; Stern, A.M.; et al. Mutation site and context dependent effects of ESR1 mutation in genome-edited breast cancer cell models. Breast Cancer Res. 2017, 19, 60. [Google Scholar] [CrossRef]
- Yedier-Bayram, O.; Gokbayrak, B.; Kayabolen, A.; Aksu, A.C.; Cavga, A.D.; Cingöz, A.; Kala, E.Y.; Karabiyik, G.; Günsay, R.; Esin, B.; et al. EPIKOL, a chromatin-focused CRISPR/Cas9-based screening platform, to identify cancer-specific epigenetic vulnerabilities. Cell Death Dis. 2022, 13, 710. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lu, X.; Huang, J.; He, H.; Chen, L.; Liu, Y.; Wang, H.; Xu, Y.; Xing, S.; Ruan, X.; et al. Epigenome screening highlights that JMJD6 confers an epigenetic vulnerability and mediates sunitinib sensitivity in renal cell carcinoma. Clin. Transl. Med. 2021, 11, e328. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chen, C.-W. Epigenetic and Transcriptional Signaling in Ewing Sarcoma—Disease Etiology and Therapeutic Opportunities. Biomedicines 2022, 10, 1325. [Google Scholar] [CrossRef]
- Yuan, S.; Natesan, R.; Sanchez-Rivera, F.J.; Li, J.; Bhanu, N.V.; Yamazoe, T.; Lin, J.H.; Merrell, A.J.; Sela, Y.; Thomas, S.K.; et al. Global regulation of the histone mark H3K36ME2 underlies epithelial plasticity and metastatic progression. Cancer Discov. 2020, 10, 854–871. [Google Scholar] [CrossRef] [Green Version]
- Anastas, J.N.; Zee, B.; Kalin, J.; Kim, M.; Guo, R.; Alexandrescu, S.; Blanco, M.A.; Giera, S.; Gillespie, S.M.; Das, J.; et al. Re-programing Chromatin with a Bifunctional LSD1/HDAC Inhibitor Induces Therapeutic Differentiation in DIPG. Cancer Cell 2019, 36, 528–544.e10. [Google Scholar] [CrossRef]
- Wang, J.; Zhu, X.; Dang, L.; Jiang, H.; Xie, Y.; Li, X.; Guo, J.; Wang, Y.; Peng, Z.; Wang, M.; et al. Epigenomic reprogramming via HRP2-MINA dictates response to proteasome inhibitors in multiple myeloma with t(4;14) translocation. J. Clin. Investig. 2022, 132, e149526. [Google Scholar] [CrossRef] [PubMed]
- Raoof, S.; Mulford, I.J.; Frisco-Cabanos, H.; Nangia, V.; Timonina, D.; Labrot, E.; Hafeez, N.; Bilton, S.J.; Drier, Y.; Ji, F.; et al. Targeting FGFR overcomes EMT-mediated resistance in EGFR mutant non-small cell lung cancer. Oncogene 2019, 38, 6399–6413. [Google Scholar] [CrossRef]
- Kamdar, S.; Isserlin, R.; Van Der Kwast, T.; Zlotta, A.R.; Bader, G.D.; Fleshner, N.E.; Bapat, B. Exploring targets of TET2-mediated methylation reprogramming as potential discriminators of prostate cancer progression. Clin. Epigenet. 2019, 11, 54. [Google Scholar] [CrossRef] [Green Version]
- Benitz, S.; Straub, T.; Mahajan, U.M.; Mutter, J.; Czemmel, S.; Unruh, T.; Wingerath, B.; Deubler, S.; Fahr, L.; Cheng, T.; et al. Ring1b-dependent epigenetic remodelling is an essential prerequisite for pancreatic carcinogenesis. Gut 2019, 68, 2007–2018. [Google Scholar] [CrossRef]
- Khazaei, S.; De Jay, N.; Deshmukh, S.; Hendrikse, L.D.; Jawhar, W.; Chen, C.C.; Mikael, L.G.; Faury, D.; Marchione, D.M.; Lanoix, J.; et al. H3.3 g34w promotes growth and impedes differentiation of osteoblast-like mesenchymal progenitors in giant cell tumor of bone. Cancer Discov. 2020, 10, 1968–1987. [Google Scholar] [CrossRef]
- Law, C.-T.; Wei, L.; Tsang, F.H.; Chan, C.Y.; Xu, I.M.; Lai, R.K.; Ho, D.W.H.; Lee, J.M.; Wong, C.C.L.; Ng, I.O.; et al. HELLS Regulates Chromatin Remodeling and Epigenetic Silencing of Multiple Tumor Suppressor Genes in Human Hepatocellular Carcinoma. Hepatology 2013, 69, 2013–2030. [Google Scholar] [CrossRef] [PubMed]
- Kretzmann, J.A.; Irving, K.L.; Smith, N.M.; Evans, C.W. Modulating gene expression in breast cancer via DNA secondary structure and the CRISPR toolbox. NAR Cancer 2021, 3, zcab048. [Google Scholar] [CrossRef]
- Hodge, D.Q.; Cui, J.; Gamble, M.J.; Guo, W. Histone Variant MacroH2A1 Plays an Isoform-Specific Role in Suppressing Epithelial-Mesenchymal Transition. Sci. Rep. 2018, 8, 841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, X.; Zhang, C.; Xu, Z.; Wang, S.; Li, X.; Stringer-Reasor, E.; Bae, S.; Zeng, L.; Zhao, D.; Liu, R.; et al. Dual CRISPR interference and activation for targeted reactivation of X-linked endogenous FOXP3 in human breast cancer cells. Mol. Cancer 2022, 21, 38. [Google Scholar] [CrossRef] [PubMed]
- Michl, J.; Wang, Y.; Monterisi, S.; Blaszczak, W.; Beveridge, R.; Bridges, E.M.; Koth, J.; Bodmer, W.F.; Swietach, P. CRISPR-Cas9 screen identifies oxidative phosphorylation as essential for cancer cell survival at low extracellular pH. Cell Rep. 2022, 38, 110493. [Google Scholar] [CrossRef]
- Mennuni, M.; Filograna, R.; Felser, A.; Bonekamp, N.A.; Giavalisco, P.; Lytovchenko, O.; Larsson, N. Metabolic resistance to the inhibition of mitochondrial transcription revealed by CRISPR-Cas9 screen. EMBO Rep. 2021, 23, e53054. [Google Scholar] [CrossRef]
- Li, H.; Song, J.; He, Y.; Liu, Y.; Liu, Z.; Sun, W.; Hu, W.; Lei, Q.; Hu, X.; Chen, Z.; et al. CRISPR/Cas9 Screens Reveal that Hexokinase 2 Enhances Cancer Stemness and Tumorigenicity by Activating the ACSL4-Fatty Acid β-Oxidation Pathway. Adv. Sci. 2022, 9, e2105126. [Google Scholar] [CrossRef]
- Tabebi, M.; Dutta, R.K.; Skoglund, C.; Söderkvist, P.; Gimm, O. Loss of SDHB Induces a Metabolic Switch in the hPheo1 Cell Line toward Enhanced OXPHOS. Int. J. Mol. Sci. 2022, 23, 560. [Google Scholar] [CrossRef]
- Torborg, S.R.; Li, Z.; Chan, J.E.; Tammela, T. Cellular and molecular mechanisms of plasticity in cancer. Trends Cancer 2022, 8, 735–746. [Google Scholar] [CrossRef]
- Yasunaga, K.; Ito, T.; Miki, M.; Ueda, K.; Fujiyama, T.; Tachibana, Y.; Fujimori, N.; Kawabe, K.; Ogawa, Y. Using CRISPR/Cas9 to Knock out Amylase in Acinar Cells Decreases Pancreatitis-Induced Autophagy. Biomed. Res. Int. 2018, 2018, 8719397. [Google Scholar] [CrossRef]
- Sanna, A.; Phung, B.; Mitra, S.; Lauss, M.; Choi, J.; Zhang, T.; Njauw, C.-N.J.; Cordero, E.; Harbst, K.; Rosengren, F.; et al. DNA promoter hypermethylation of melanocyte lineage genes determines melanoma phenotype. JCI Insight 2022, 7, e156577. [Google Scholar] [CrossRef] [PubMed]
- Phelps, M.P.; Bailey, J.N.; Vleeshouwer-Neumann, T.; Chen, E.Y. CRISPR screen identifies the NCOR/HDAC3 complex as a major suppressor of differentiation in rhabdomyosarcoma. Proc. Natl. Acad. Sci. USA 2016, 113, 15090–15095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopanja, D.; Chand, V.; O’Brien, E.M.; Mukhopadhyay, N.K.; Zappia, M.P.; Islam, A.B.; Frolov, M.V.; Merrill, B.J.; Raychaudhuri, P. Transcriptional Repression by FoxM1 Suppresses Tumor Differentiation and Promotes Metastasis of Breast Cancer. Cancer Res. 2022, 82, 2458–2471. [Google Scholar] [CrossRef] [PubMed]
- Cumin, C.; Huang, Y.-L.; Rossdam, C.; Ruoff, F.; Céspedes, S.P.; Liang, C.-Y.; Lombardo, F.C.; Coelho, R.; Rimmer, N.; Konantz, M.; et al. Glycosphingolipids are mediators of cancer plasticity through independent signaling pathways. Cell Rep. 2022, 40, 111181. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2018, 20, 69–84. [Google Scholar] [CrossRef]
- Gautron, A.; Bachelot, L.; Aubry, M.; Leclerc, D.; Quéméner, A.M.; Corre, S.; Rambow, F.; Paris, A.; Tardif, N.; Leclair, H.M.; et al. CRISPR screens identify tumor-promoting genes conferring melanoma cell plasticity and resistance. EMBO Mol. Med. 2021, 13, e13466. [Google Scholar] [CrossRef]
- Serresi, M.; Kertalli, S.; Li, L.; Schmitt, M.J.; Dramaretska, Y.; Wierikx, J.; Hulsman, D.; Gargiulo, G. Functional antagonism of chromatin modulators regulates epithelial-mesenchymal transition. Sci. Adv. 2021, 7, eabd7974. [Google Scholar] [CrossRef]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Li, Y.; Liu, H.; Zhang, J.; Wang, J.; Xia, J.; Zhang, Y.; Yu, X.; Ma, J.; Huang, M.; et al. Genome-wide CRISPR/Cas9 library screen identifies PCMT1 as a critical driver of ovarian cancer metastasis. J. Exp. Clin. Cancer Res. 2022, 41, 24. [Google Scholar] [CrossRef]
- Wang, D.; Naydenov, N.G.; Dozmorov, M.G.; Koblinski, J.; Ivanov, A.I. Anillin regulates breast cancer cell migration, growth, and metastasis by non-canonical mechanisms involving control of cell stemness and differentiation. Breast Cancer Res. 2020, 22, 3. [Google Scholar] [CrossRef]
- Neckmann, U.; Wolowczyk, C.; Hall, M.; Almaas, E.; Ren, J.; Zhao, S.; Johannessen, B.; Skotheim, R.I.; Bjørkøy, G.; Dijke, P.T.; et al. GREM1 is associated with metastasis and predicts poor prognosis in ER-negative breast cancer patients. Cell Commun. Signal. 2019, 17, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindberg, M.E.; Stodden, G.R.; King, M.L.; Ii, J.A.M.; Mann, J.L.; DeMayo, F.; Lydon, J.P.; Hayashi, K. Loss of Cdh1 and Pten accelerates cellular invasiveness and angiogenesis in the mouse uterus1. Biol. Reprod. 2013, 89, 8. [Google Scholar] [CrossRef] [Green Version]
- Bajrami, I.; Marlow, R.; van de Ven, M.; Brough, R.; Pemberton, H.N.; Frankum, J.; Song, F.; Rafiq, R.; Konde, A.; Krastev, D.B.; et al. E-Cadherin/ROS1 inhibitor synthetic lethality in breast cancer. Cancer Discov. 2018, 8, 498–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Mulhim, F.; Alqosaibi, A.I.; Al-Muhnna, A.; Farid, K.; Abdel-Ghany, S.; Rizk, H.; Prince, A.-B.; Isichei, A.; Sabit, H. CRISPR/Cas9-mediated activation of CDH1 suppresses metastasis of breast cancer in rats. Electron. J. Biotechnol. 2021, 53, 54–60. [Google Scholar] [CrossRef]
- Wang, K.; Xing, Z.-H.; Jiang, Q.-W.; Yang, Y.; Huang, J.-R.; Yuan, M.-L.; Wei, M.-N.; Li, Y.; Wang, S.-T.; Liu, K.; et al. Targeting uPAR by CRISPR/Cas9 System Attenuates Cancer Malignancy and Multidrug Resistance. Front. Oncol. 2019, 9, 80. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Watanabe, S.; Hibiya, S.; Katsukura, N.; Kitagawa, S.; Sato, A.; Okamoto, R.; Watanabe, M.; Tsuchiya, K. Influence of chronic inflammation on the malignant phenotypes and the plasticity of colorectal cancer cells. Biochem. Biophys. Rep. 2021, 26, 101031. [Google Scholar] [CrossRef]
- Means, A.L.; Freeman, T.J.; Zhu, J.; Woodbury, L.G.; Marincola-Smith, P.; Wu, C.; Meyer, A.R.; Weaver, C.J.; Padmanabhan, C.; An, H.; et al. Epithelial Smad4 Deletion Up-Regulates Inflammation and Promotes Inflammation-Associated Cancer. Cell Mol. Gastroenterol. Hepatol. 2018, 6, 257–276. [Google Scholar] [CrossRef]
- Shi, J.; Yang, F.; Zhou, N.; Jiang, Y.; Zhao, Y.; Zhu, J.; Prelaj, A.; Malhotra, J.; Normanno, N.; Danese, E.; et al. Isochorismatase domain-containing protein 1 (ISOC1) participates in DNA damage repair and inflammation-related pathways to promote lung cancer development. Transl. Lung Cancer Res. 2021, 10, 1444–1456. [Google Scholar] [CrossRef] [PubMed]
- Mengoni, M.; Braun, A.; Gaffal, E.; Tüting, T. The aryl hydrocarbon receptor promotes inflammation-induced dedifferentiation and systemic metastatic spread of melanoma cells. Int. J. Cancer 2020, 147, 2902–2913. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Marchesi, F.; Garlanda, C. Macrophages as tools and targets in cancer therapy. Nat. Rev. Drug Discov. 2022, 21, 799–820. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Liu, Y.; Liu, L.; Chen, M.; Wang, X.; Yang, J.; Gong, Y.; Ding, B.-S.; Wei, Y.; Wei, X. Tumor cells induce LAMP2a expression in tumor-associated macrophage for cancer progression. EBioMedicine 2019, 40, 118–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vajdic, C.M.; van Leeuwen, M.T. Cancer incidence and risk factors after solid organ transplantation. Int. J. Cancer 2009, 125, 1747–1754. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.W.L.; Swann, J.B.; Koebel, C.M.; Schreiber, R.D.; Smyth, M.J. Immune-mediated dormancy: An equilibrium with cancer. J. Leukoc. Biol. 2008, 84, 988–993. [Google Scholar] [CrossRef] [Green Version]
- Pagès, F.; Galon, J.; Dieu-Nosjean, M.-C.; Tartour, E.; Sautès-Fridman, C.; Fridman, W.-H. Immune infiltration in human tumors: A prognostic factor that should not be ignored. Oncogene 2009, 29, 1093–1102. [Google Scholar] [CrossRef] [Green Version]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Hu, B.; Shao, J.; Shen, B.; Du, J.; Du, Y.; Zhou, J.; Yu, L.; Zhang, L.; Chen, F.; et al. CRISPR-Cas9 mediated efficient PD-1 disruption on human primary T cells from cancer patients. Sci. Rep. 2016, 6, 20070. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.; Yang, N.; He, J.; Gong, W.; Lai, Z.; Xie, L.; Tao, L.; Xu, C.; Wang, H.; Zhang, G.; et al. Generation of cancer-specific cytotoxic PD-1—T cells using liposome-encapsulated CRISPR/cas system with dendritic/tumor fusion cells. J. Biomed. Nanotechnol. 2019, 15, 593–601. [Google Scholar] [CrossRef]
- Xu, Y.; Chen, C.; Guo, Y.; Hu, S.; Sun, Z. Effect of CRISPR/Cas9-Edited PD-1/PD-L1 on Tumor Immunity and Immunotherapy. Front. Immunol. 2022, 13, 848327. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mohseni, M.; Grauel, A.; Diez, J.E.; Guan, W.; Liang, S.; Choi, J.E.; Pu, M.; Chen, D.; Laszewski, T.; et al. SHP2 blockade enhances anti-tumor immunity via tumor cell intrinsic and extrinsic mechanisms. Sci. Rep. 2021, 11, 1399. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, Z.; Shen, M.; Wang, Y.; Wang, L.; Li, J.; Yang, W.; Li, J.; Li, H.; Wang, X.; et al. Programmable Unlocking Nano-Matryoshka-CRISPR Precisely Reverses Immunosuppression to Unleash Cascade Amplified Adaptive Immune Response. Adv. Sci. 2021, 8, 2100292. [Google Scholar] [CrossRef] [PubMed]
- Freen-van Heeren, J.J. Using CRISPR to enhance T cell effector function for therapeutic applications. Cytokine X 2021, 3, 100049. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Chow, R.D.; Bai, Z.; Zhu, L.; Errami, Y.; Dai, X.; Dong, M.B.; Ye, L.; Zhang, X.; Renauer, P.A.; et al. Multiplexed activation of endogenous genes by CRISPRa elicits potent antitumor immunity. Nat. Immunol. 2019, 20, 1494–1505. [Google Scholar] [CrossRef]
- Volpert, O.V.; Dameron, K.M.; Bouck, N. Sequential development of an angiogenic phenotype by human® broblasts progressing to tumorigenicity. Oncogene 1997, 14, 1495–1502. [Google Scholar] [CrossRef] [Green Version]
- Ronca, R.; Benkheil, M.; Mitola, S.; Struyf, S.; Liekens, S. Tumor angiogenesis revisited: Regulators and clinical implications. Med. Res. Rev. 2017, 37, 1231–1274. [Google Scholar] [CrossRef] [Green Version]
- Hariprabu, K.N.G.; Sathya, M.; Vimalraj, S. CRISPR/Cas9 in cancer therapy: A review with a special focus on tumor angiogenesis. Int. J. Biol. Macromol. 2021, 192, 913–930. [Google Scholar] [CrossRef]
- Zhu, W.; Liu, C.; Lu, T.; Zhang, Y.; Zhang, S.; Chen, Q.; Deng, N. Knockout of EGFL6 by CRISPR/Cas9 Mediated Inhibition of Tumor Angiogenesis in Ovarian Cancer. Front. Oncol. 2020, 10, 1451. [Google Scholar] [CrossRef]
- Chen, S.; Xue, Y.; Wu, X.; Le, C.; Bhutkar, A.; Bell, E.L.; Zhang, F.; Langer, R.; Sharp, P.A. Global microRNA depletion suppresses tumor angiogenesis. Genes Dev. 2014, 28, 1054–1067. [Google Scholar] [CrossRef]
- Tsai, M.-L.; Lee, C.-H.; Huang, L.-C.; Chen, Y.-H.; Liu, W.-N.; Lin, C.-Y.; Hsu, K.-W.; Lee, A.-W.; Lin, C.-L. CRISPR-mediated knockout of VEGFR2/KDR inhibits cell growth in a squamous thyroid cancer cell line. FEBS Open Bio 2022, 12, 993–1005. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Izard, J.; Walsh, E.; Batich, K.; Chongsathidkiet, P.; Clarke, G.; Sela, D.A.; Muller, A.J.; Mullin, J.M.; Albert, K.; et al. The host microbiome regulates and maintains human health: A primer and perspective for non-microbiologists. Cancer Res. 2017, 77, 1783–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sears, C.L.; Garrett, W.S. Microbes, microbiota, and colon cancer. Cell Host Microbe 2014, 15, 317–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dzutsev, A.; Badger, J.H.; Perez-Chanona, E.; Roy, S.; Salcedo, R.; Smith, C.K.; Trinchieri, G. Microbes and Cancer. Annu. Rev. Immunol. 2017, 35, 199–228. [Google Scholar] [CrossRef] [PubMed]
- Fessler, J.; Matson, V.; Gajewski, T.F. Exploring the emerging role of the microbiome in cancer immunotherapy. J. Immuno Ther. Cancer 2019, 7, 108. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Model | District | Technique | Target | Suggested Mechanism (If Applicable) | Ref. |
|---|---|---|---|---|---|
| MCF-7 | Breast | KO | DCAF13 | Accumulation of PERP * | [12] |
| MOLM-13 | AML | CRISPR screening | KAT2A | Inhibition of leukemogenic transcriptional programs, induction to differentiation leading to apoptosis | [13] |
| OCI-AML2 | |||||
| OCI-AML3 | |||||
| MDA-MB-231 | Breast | Inducible KO | RLIP | Downregulation of surviving and Bcl-2; upregulation of Bim | [14] |
| MCF-7 | |||||
| NCI-H460 | NSCLC | CRISPR screening | MDM2 | Removal of p53-inhibiting factor | [15] |
| A549 | |||||
| OSRC | RCC | dCas13b-dependent methylation | ZNF677 | - | [16] |
| CAK12 | |||||
| MDA-MB-231 | Breast | Cas9 knockdown | B2AR | Disruption of the B2AR-MOR interaction resulted in less aggressive phenotype | [17] |
| MDA-MB-468 | MOR | ||||
| MDA-MB-231 | Breast | Kinome-wide CRISPR screening | PLK1 | - | [18] |
| MDA-MB-468 | |||||
| T47D | Breast | KO | HO-1 | - | [19] |
| HCT116 | CRC | KO | OLA1 | Downregulation of CA9 and HIF-1α | [20] |
| Lovo | |||||
| Kyse-30 | Esophagus | CRISPR/dCas9 | ZNF154 | Targeted demethylation of ZNF154 promoter induced ZNF154 expression and inhibited proliferation | [21] |
| Kyse-140 | |||||
| MCF-7 | Breast | CRISPR screening | ARID1A | - | [22] |
| FaDu | HNSCC | KO | SEC62 | - | [23] |
| PDX366 | PDA | CRISPR screening | ISL2 | - | [24] |
| HCT116 | CRC | KO | ATF2 | Inhibition of the cancer driver TROP2 | [25] |
| HT29 | |||||
| OCM1 | UM | CRISPR screening | GPS2 | Upregulation of oncogenic MAPK and PI3K-Akt pathways and downregulation of Slit/Robo pathway | [26] |
| MCF-7 | Breast | KO | Linc-RoR (lncRNA) | Increase in the protein stability of DUSP7 decreasing ERK phosphorylation | [27] |
| AGS | GC | CRISPR screening | METTL1 | [28] | |
| Huh7 | HCC | KO | SMPDL3A | Suppression of tumor proliferation and promotion of apoptosis through ERH | [29] |
| HepG2 | |||||
| Neuro2a | Neuroblastoma | KO | PLAUR | p38 activation and decreased p53-mediated chemosensitivity | [30] |
| GIC | Glioblastoma | KO | CD95 | Acquired resistance to CD95L-induced apoptosis | [31] |
| Model | District | Target | Mutation | Effect | Ref. |
|---|---|---|---|---|---|
| HCT116 | CRC | KRAS | G13D | Increasing zygosity of the mutant increased sensitivity to MAPK inhibitors | [75] |
| HeLa * | - | DNMT3A | K299I | The mutation altered the methylation pattern of the genome | [76] |
| H1975 | NSCLC | EGFR | C797S | The mutation recapitulated the resistance to third generation TKis and showed upregulation of AXL | [77] |
| MCF-7 | Breast | FOXA1 | K295A | Permanent acetylation mimic of FOXA1 in breast cancer | [78] |
| HEK293T * | - | TRF1 | T273A/T358A | Inhibition of PI3K/Akt pathway | [79] |
| MCF-7 | Breast | ESR1 | D538G/Y537S | Increased sensitivity to ERD-148, new generation PROTAC | [80] |
| MCF-7 | Breast | ESR1 | D538G/Y537S | The mutations recapitulated the ligand independent ERα transcriptional activity, ligand-independent growth and endocrine resistance | [81] |
| T47D |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capoferri, D.; Filiberti, S.; Faletti, J.; Tavani, C.; Ronca, R. Ten Years of CRISPRing Cancers In Vitro. Cancers 2022, 14, 5746. https://doi.org/10.3390/cancers14235746
Capoferri D, Filiberti S, Faletti J, Tavani C, Ronca R. Ten Years of CRISPRing Cancers In Vitro. Cancers. 2022; 14(23):5746. https://doi.org/10.3390/cancers14235746
Chicago/Turabian StyleCapoferri, Davide, Serena Filiberti, Jessica Faletti, Camilla Tavani, and Roberto Ronca. 2022. "Ten Years of CRISPRing Cancers In Vitro" Cancers 14, no. 23: 5746. https://doi.org/10.3390/cancers14235746