
Epigenetic Mechanisms Underlying Melanoma Resistance to Immune and Targeted Therapies

Abstract
:Simple Summary
Abstract
1. Introduction
2. Role of the Epigenome in Therapeutic Resistance
2.1. Interferon-Mediated Viral Mimicry Facilitated by ERV De-Repression Enhances Response to Immunotherapy
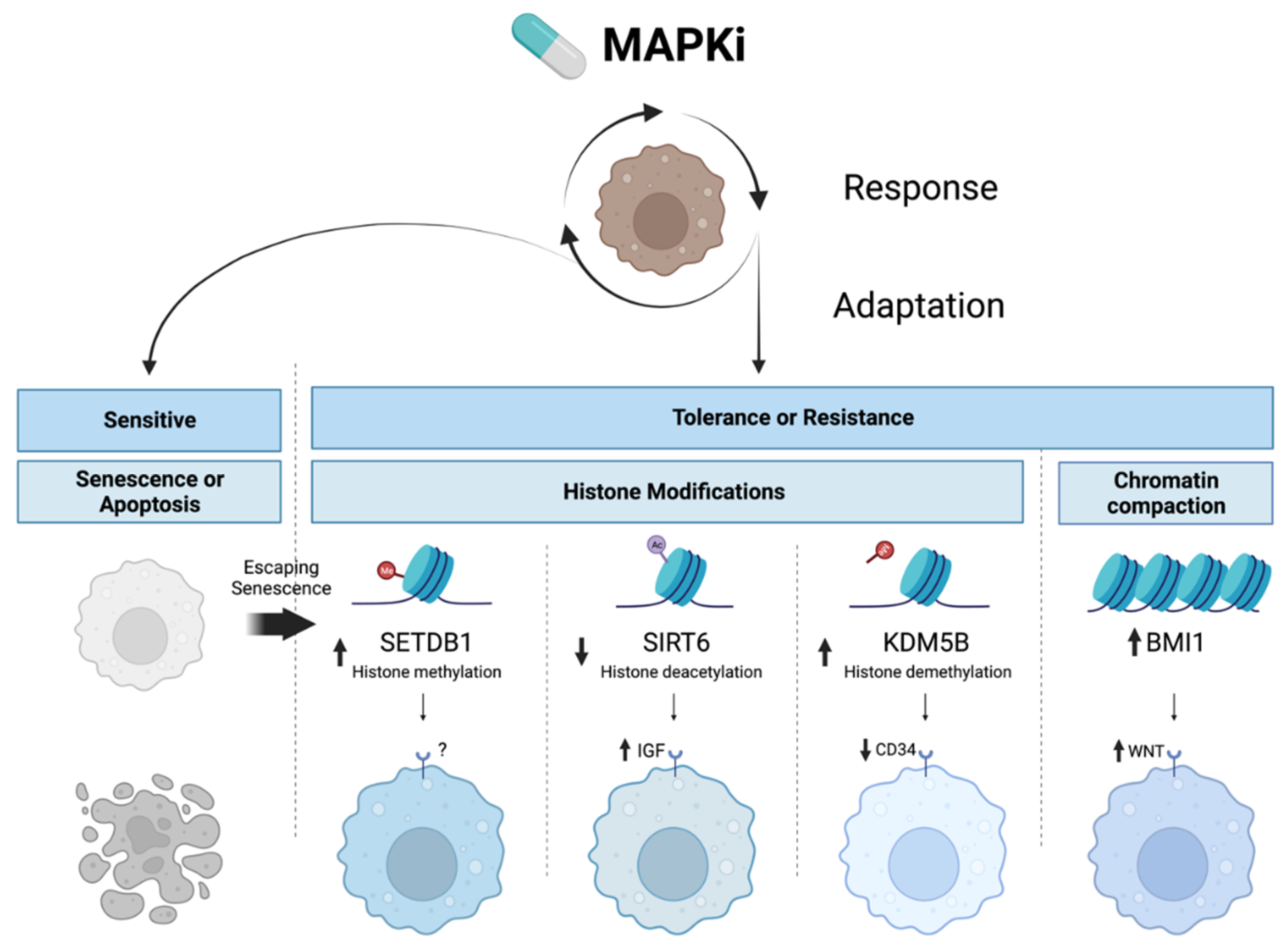
2.2. Targeted Therapy Resistance via Epigenetic Upregulation of Survival Pathways Parallel to MAPK Signaling
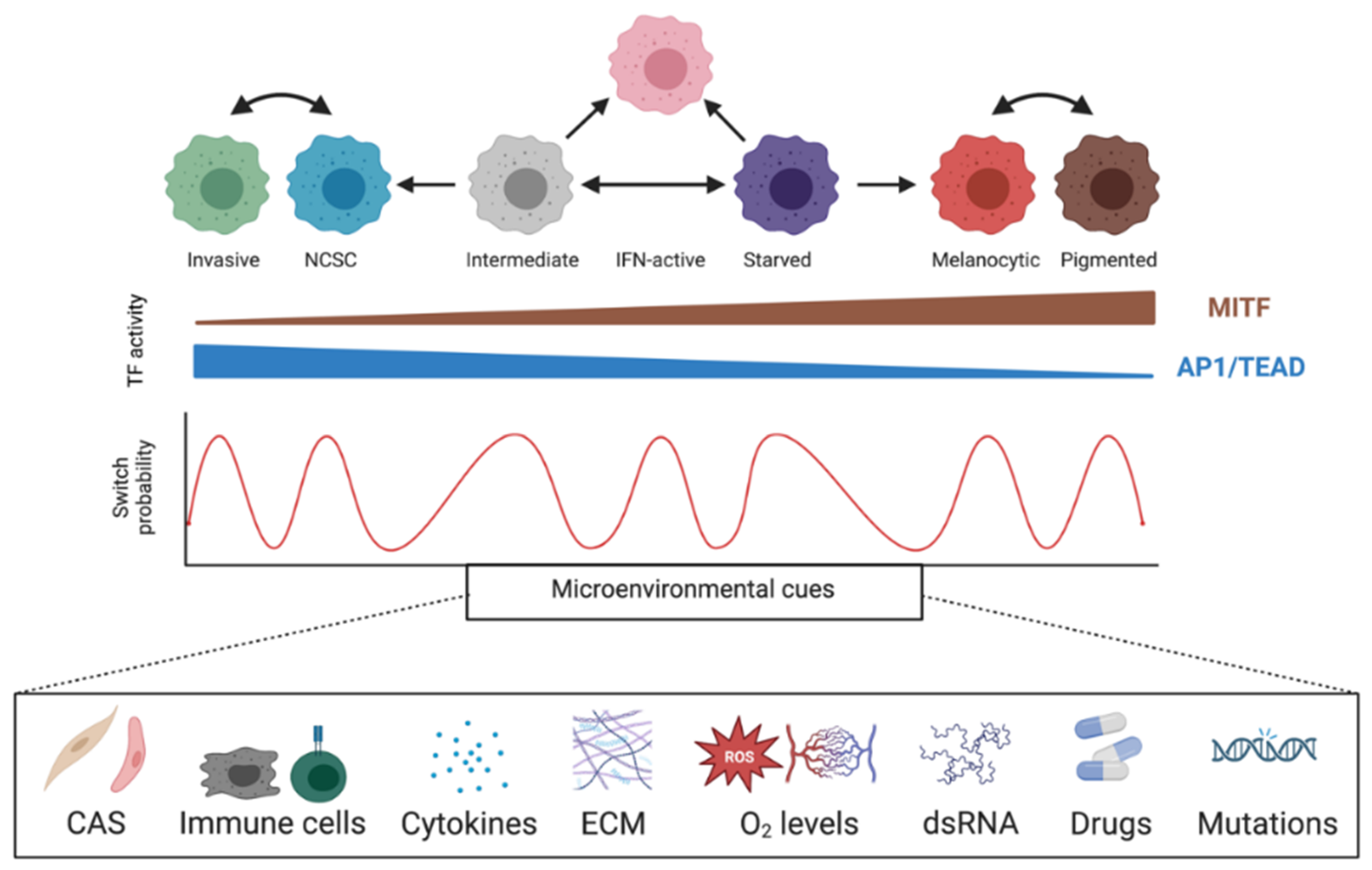
2.3. Phenotype Switching and Targeted Therapy Resistance

{kind=link}
{kind=link}
{kind=link}
| Gene | Function | Mechanism |
|---|---|---|
| SETDB1 [190] | Histone methyltransferase | SETDB1 inhibition is cytotoxic to BRAFi-resistant melanoma cells, and inhibition synergizes with BRAFi and MEKi. |
| SIRT6 [153] | Histone deacetylase | SIRT6 haploinsufficiency results in H3K56 acetylation at the IGFBP2 locus, activating PI3K/AKT/mTOR, and facilitating melanoma resistance to MAPKi. IGF-1R inhibition restores sensitivity to MAPKi. Complete SIRT6 loss activates DNA damage response due to global chromosomal instability and increases MAPKi sensitivity. |
| BMI1 [149] | Polycomb ring finger oncogene | BMI1 expression shifts melanoma to a metastatic state by inducing an invasive gene signature through WNT5A, ROR2, EGFR, and PDGFR, without a decrease in proliferation. WNT signaling maintains MITF expression, preventing proliferation defects typical of the invasive state, while concurrent EGFR and PDGFR upregulation maintains BRAFi resistance. BMI1 inhibition restored BRAFi sensitivity through WNT5A. |
| KDM5B [189] | Histone demethylase | KDM5B upregulation following BRAFi treatment decreases H3K4me3 levels and triggers the conversion of melanoma cells from drug-sensitive CD34+ to drug-resistant CD34- cell states. |
2.4. Non-Coding RNAs Involved in MAPKi Therapy Resistance
3. Concluding Remarks
4. Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- National Cancer Institute. Melanoma of the Skin-Cancer Stat Facts. Available online: https://seer.cancer.gov/statfacts/html/melan.html (accessed on 7 July 2022).
- Arnold, M.; De Vries, E.; Whiteman, D.C.; Jemal, A.; Bray, F.; Parkin, D.M.; Soerjomataram, I. Global burden of cutaneous melanoma attributable to ultraviolet radiation in 2012. Int. J. Cancer 2018, 143, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Köhler, C.; Nittner, D.; Rambow, F.; Radaelli, E.; Stanchi, F.; Vandamme, N.; Baggiolini, A.; Sommer, L.; Berx, G.; Oord, J.J.V.D.; et al. Mouse Cutaneous Melanoma Induced by Mutant BRaf Arises from Expansion and Dedifferentiation of Mature Pigmented Melanocytes. Cell Stem Cell 2017, 21, 679–693.e6. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.; Donahue, L.R.; Choi, E.; Scumpia, P.O.; Lowry, W.E.; Grenier, J.K.; Zhu, J.; White, A.C. Melanocyte Stem Cell Activation and Translocation Initiate Cutaneous Melanoma in Response to UV Exposure. Cell Stem Cell 2017, 21, 665–678.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Q.; Lee, W.; Mohri, Y.; Takeo, M.; Lim, C.H.; Xu, X.; Myung, P.; Atit, R.P.; Taketo, M.M.; Moubarak, R.S.; et al. A novel mouse model demonstrates that oncogenic melanocyte stem cells engender melanoma resembling human disease. Nat. Commun. 2019, 10, 5023. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Lo, S.N.; Scolyer, R.A.; Thompson, J.F. Long-Term Survival of Patients with Thin (T1) Cutaneous Melanomas: A Breslow Thickness Cut Point of 0.8 mm Separates Higher-Risk and Lower-Risk Tumors. Ann. Surg. Oncol. 2018, 25, 894–902. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Kirkwood, J.M.; Grob, J.-J.; Simeone, E.; Grimaldi, A.M.; Maio, M.; Palmieri, G.; Testori, A.; Marincola, F.M.; Mozzillo, N. The role of BRAF V600 mutation in melanoma. J. Transl. Med. 2012, 10, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alqathama, A. BRAF in malignant melanoma progression and metastasis: Potentials and challenges. Am. J. Cancer Res. 2020, 10, 1103–1114. [Google Scholar] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion Sileni, V.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Vuoristo, M.-S.; Hahka-Kemppinen, M.; Parvinen, L.-M.; Pyrhönen, S.; Seppä, H.; Korpela, M.; Kellokumpu-Lehtinen, P. Randomized trial of dacarbazine versus bleomycin, vincristine, lomustine and dacarbazine (BOLD) chemotherapy combined with natural or recombinant interferon-alpha in patients with advanced melanoma. Melanoma Res. 2005, 15, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Griffin, M.; Scotto, D.; Josephs, D.H.; Mele, S.; Crescioli, S.; Bax, H.J.; Pellizzari, G.; Wynne, M.D.; Nakamura, M.; Hoffmann, R.M.; et al. BRAF inhibitors: Resistance and the promise of combination treatments for melanoma. Oncotarget 2017, 8, 78174–78192. [Google Scholar] [CrossRef] [Green Version]
- Patel, H.; Yacoub, N.; Mishra, R.; White, A.; Yuan, L.; Alanazi, S.; Garrett, J.T. Current Advances in the Treatment of BRAF-Mutant Melanoma. Cancers 2020, 12, 482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef] [Green Version]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.-K.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, H.; Mishra, R.; Yacoub, N.; Alanazi, S.; Kilroy, M.K.; Garrett, J.T. IGF1R/IR Mediates Resistance to BRAF and MEK Inhibitors in BRAF-Mutant Melanoma. Cancers 2021, 13, 5863. [Google Scholar] [CrossRef]
- Taniguchi, H.; Yamada, T.; Wang, R.; Tanimura, K.; Adachi, Y.; Nishiyama, A.; Tanimoto, A.; Takeuchi, S.; Araujo, L.H.; Boroni, M.; et al. AXL confers intrinsic resistance to osimertinib and advances the emergence of tolerant cells. Nat. Commun. 2019, 10, 259. [Google Scholar] [CrossRef] [PubMed]
- Maertens, O.; Johnson, B.; Hollstein, P.; Frederick, D.T.; Cooper, Z.A.; Messiaen, L.; Bronson, R.T.; McMahon, M.; Granter, S.; Flaherty, K.; et al. Elucidating distinct roles for NF1 in melanomagenesis. Cancer Discov. 2013, 3, 338–349. [Google Scholar] [CrossRef] [Green Version]
- Whittaker, S.R.; Theurillat, J.-P.; Van Allen, E.; Wagle, N.; Hsiao, J.; Cowley, G.S.; Schadendorf, D.; Root, D.E.; Garraway, L.A. A genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer Discov. 2013, 3, 350–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagle, N.; Emery, C.; Berger, M.F.; Davis, M.J.; Sawyer, A.; Pochanard, P.; Kehoe, S.M.; Johannessen, C.M.; MacConaill, L.E.; Hahn, W.C.; et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J. Clin. Oncol. 2011, 29, 3085–3096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, G.V.; Fung, C.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Hyman, J.; Shahheydari, H.; Tembe, V.; Thompson, J.F.; Saw, R.P.; et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat. Commun. 2014, 5, 5694. [Google Scholar] [CrossRef] [Green Version]
- Johannessen, C.M.; Boehm, J.S.; Kim, S.Y.; Thomas, S.R.; Wardwell, L.; Johnson, L.A.; Emery, C.M.; Stransky, N.; Cogdill, A.P.; Barretina, J.; et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 2010, 468, 968–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, F.; Persaud, Y.; A Pratilas, C.; Taylor, B.S.; Janakiraman, M.; She, Q.-B.; Gallardo, H.; Liu, C.; Merghoub, T.; Hefter, B.; et al. Concurrent loss of the PTEN and RB1 tumor suppressors attenuates RAF dependence in melanomas harboring (V600E)BRAF. Oncogene 2012, 31, 446–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villanueva, J.; Vultur, A.; Lee, J.T.; Somasundaram, R.; Fukunaga-Kalabis, M.; Cipolla, A.K.; Wubbenhorst, B.; Xu, X.; Gimotty, P.A.; Kee, D.; et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010, 18, 683–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corcoran, R.B.; Dias-Santagata, D.; Bergethon, K.; Iafrate, A.J.; Settleman, J.; Engelman, J.A. BRAF gene amplification can promote acquired resistance to MEK inhibitors in cancer cells harboring the BRAF V600E mutation. Sci. Signal. 2010, 3, ra84. [Google Scholar] [CrossRef] [Green Version]
- Poulikakos, P.I.; Persaud, Y.; Janakiraman, M.; Kong, X.; Ng, C.; Moriceau, G.; Shi, H.; Atefi, M.; Titz, B.; Gabay, M.T.; et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011, 480, 387–390. [Google Scholar] [CrossRef] [Green Version]
- Pachella, L.A.; Madsen, L.T.; Dains, J.E. The Toxicity and Benefit of Various Dosing Strategies for Interleukin-2 in Metastatic Melanoma and Renal Cell Carcinoma. J. Adv. Pract. Oncol. 2015, 6, 212–221. [Google Scholar]
- Schuchter, L.M. Adjuvant interferon therapy for melanoma: High-dose, low-dose, no dose, which dose? J. Clin. Oncol. 2004, 22, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Sznol, M.; McDermott, D.F.; Kluger, H.M.; Carvajal, R.D.; Sharfman, W.H.; Brahmer, J.R.; Lawrence, D.P.; Atkins, M.B.; Powderly, J.D.; et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J. Clin. Oncol. 2014, 32, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.-A.; Reed, K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [Green Version]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Long-Term Outcomes With Nivolumab Plus Ipilimumab or Nivolumab Alone Versus Ipilimumab in Patients With Advanced Melanoma. J. Clin. Oncol. 2022, 40, 127–137. [Google Scholar] [CrossRef]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [Green Version]
- Keilholz, U.; Mehnert, J.M.; Bauer, S.; Bourgeois, H.; Patel, M.R.; Gravenor, D.; Nemunaitis, J.J.; Taylor, M.H.; Wyrwicz, L.; Lee, K.-W.; et al. Avelumab in patients with previously treated metastatic melanoma: Phase 1b results from the JAVELIN Solid Tumor trial. J. Immunother. Cancer 2019, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Gutiérrez, E.C.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N. Engl. J. Med. 2022, 386, 24–34. [Google Scholar] [CrossRef]
- Kalbasi, A.; Ribas, A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 2020, 20, 25–39. [Google Scholar] [CrossRef] [PubMed]
- van Rooij, N.; Van Buuren, M.M.; Philips, D.; Velds, A.; Toebes, M.; Heemskerk, B.; Van Dijk, L.J.; Behjati, S.; Hilkmann, H.; El Atmioui, D.; et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J. Clin. Oncol. 2013, 31, e439–e442. [Google Scholar] [CrossRef]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.-C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Sha, D.; Jin, Z.; Budczies, J.; Kluck, K.; Stenzinger, A.; Sinicrope, F.A. Tumor Mutational Burden as a Predictive Biomarker in Solid Tumors. Cancer Discov. 2020, 10, 1808–1825. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [Green Version]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Wang, P.; Chen, Y.; Wang, C. Beyond Tumor Mutation Burden: Tumor Neoantigen Burden as a Biomarker for Immunotherapy and Other Types of Therapy. Front. Oncol. 2021, 11, 672677. [Google Scholar] [CrossRef]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017, 19, 1189–1201. [Google Scholar] [CrossRef] [Green Version]
- Cui, C.; Xu, C.; Yang, W.; Chi, Z.; Sheng, X.; Si, L.; Xie, Y.; Yu, J.; Wang, S.; Yu, R.; et al. Ratio of the interferon-γ signature to the immunosuppression signature predicts anti-PD-1 therapy response in melanoma. NPJ Genom. Med. 2021, 6, 7. [Google Scholar] [CrossRef]
- Kirkwood, J.M.; Iannotti, N.; Cho, D.; O’Day, S.; Gibney, G.; Hodi, F.S.; Munster, P.; Hoyle, P.; Owens, S.; Smith, M.; et al. Abstract CT176: Effect of JAK/STAT or PI3Kδ plus PD-1 inhibition on the tumor microenvironment: Biomarker results from a phase Ib study in patients with advanced solid tumors. Cancer Res. 2018, 78, CT176. [Google Scholar] [CrossRef]
- Propper, D.J.; Chao, D.; Braybrooke, J.P.; Bahl, P.; Thavasu, P.; Balkwill, F.; Turley, H.; Dobbs, N.; Gatter, K.; Talbot, D.C.; et al. Low-dose IFN-gamma induces tumor MHC expression in metastatic malignant melanoma. Clin. Cancer Res. 2003, 9, 84–92. [Google Scholar] [PubMed]
- Liu, D.; Schilling, B.; Liu, D.; Sucker, A.; Livingstone, E.; Jerby-Arnon, L.; Zimmer, L.; Gutzmer, R.; Satzger, I.; Loquai, C.; et al. Author Correction: Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat. Med. 2020, 26, 1147. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Gao, Y.; Zhang, P.; Chu, Q. Acquired Resistance to Immune Checkpoint Blockades: The Underlying Mechanisms and Potential Strategies. Front. Immunol. 2021, 12, 693609. [Google Scholar] [CrossRef]
- Kim, Y.J.; Sheu, K.M.; Tsoi, J.; Abril-Rodriguez, G.; Medina, E.; Grasso, C.S.; Torrejon, D.Y.; Champhekar, A.S.; Litchfield, K.; Swanton, C.; et al. Melanoma dedifferentiation induced by IFN-gamma epigenetic remodeling in response to anti-PD-1 therapy. J. Clin. Invest. 2021, 131, e145859. [Google Scholar] [CrossRef] [PubMed]
- Shields, B.D.; Koss, B.; Taylor, E.M.; Storey, A.J.; West, K.L.; Byrum, S.D.; Mackintosh, S.G.; Edmondson, R.; Mahmoud, F.; Shalin, S.C.; et al. Loss of E-Cadherin Inhibits CD103 Antitumor Activity and Reduces Checkpoint Blockade Responsiveness in Melanoma. Cancer Res. 2019, 79, 1113–1123. [Google Scholar] [CrossRef] [Green Version]
- Trujillo, J.A.; Luke, J.J.; Zha, Y.; Segal, J.P.; Ritterhouse, L.L.; Spranger, S.; Matijevich, K.; Gajewski, T.F. Secondary resistance to immunotherapy associated with beta-catenin pathway activation or PTEN loss in metastatic melanoma. J. Immunother. Cancer 2019, 7, 295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grasso, C.S.; Tsoi, J.; Onyshchenko, M.; Abril-Rodriguez, G.; Ross-Macdonald, P.; Wind-Rotolo, M.; Champhekar, A.; Medina, E.; Torrejon, D.Y.; Shin, D.S.; et al. Conserved Interferon-gamma Signaling Drives Clinical Response to Immune Checkpoint Blockade Therapy in Melanoma. Cancer Cell 2020, 38, 500–515.e3. [Google Scholar] [CrossRef]
- Prendergast, L.; McClurg, U.L.; Hristova, R.; Berlinguer-Palmini, R.; Greener, S.; Veitch, K.; Hernandez, I.; Pasero, P.; Rico, D.; Higgins, J.M.G.; et al. Resolution of R-loops by INO80 promotes DNA replication and maintains cancer cell proliferation and viability. Nat. Commun. 2020, 11, 4534. [Google Scholar] [CrossRef]
- Zhou, B.; Wang, L.; Zhang, S.; Bennett, B.D.; He, F.; Zhang, Y.; Xiong, C.; Han, L.; Diao, L.; Li, P.; et al. INO80 governs superenhancer-mediated oncogenic transcription and tumor growth in melanoma. Genes Dev. 2016, 30, 1440–1453. [Google Scholar] [CrossRef] [Green Version]
- Dar, A.A.; Majid, S.; Bezrookove, V.; Phan, B.; Ursu, S.; Nosrati, M.; De Semir, D.; Sagebiel, R.W.; Miller, J.R.; Debs, R.; et al. BPTF transduces MITF-driven prosurvival signals in melanoma cells. Proc. Natl. Acad. Sci. USA 2016, 113, 6254–6258. [Google Scholar] [CrossRef] [Green Version]
- Eckey, M.; Kuphal, S.; Straub, T.; Rümmele, P.; Kremmer, E.; Bosserhoff, A.; Becker, P.B. Nucleosome remodeler SNF2L suppresses cell proliferation and migration and attenuates Wnt signaling. Mol. Cell. Biol. 2012, 32, 2359–2371. [Google Scholar] [CrossRef] [PubMed]
- Dreier, M.R.; de la Serna, I.L. SWI/SNF Chromatin Remodeling Enzymes in Melanoma. Epigenomes 2022, 6, 10. [Google Scholar] [CrossRef]
- Laurette, P.; Strub, T.; Koludrovic, D.; Keime, C.; Le Gras, S.; Seberg, H.; Van Otterloo, E.; Imrichova, H.; Siddaway, R.; Aerts, S.; et al. Transcription factor MITF and remodeller BRG1 define chromatin organisation at regulatory elements in melanoma mcells. Elife 2015, 4, e06857. [Google Scholar] [CrossRef] [PubMed]
- Carcamo, S.; Nguyen, C.B.; Grossi, E.; Filipescu, D.; Alpsoy, A.; Dhiman, A.; Sun, D.; Narang, S.; Imig, J.; Martin, T.C.; et al. Altered BAF occupancy and transcription factor dynamics in PBAF-deficient melanoma. Cell Rep. 2022, 39, 110637. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Zucconi, B.E.; Wu, M.; Nocco, S.E.; Meyers, D.J.; McGee, J.S.; Venkatesh, S.; Cohen, D.L.; Gonzalez, E.C.; Ryu, B.; et al. MITF Expression Predicts Therapeutic Vulnerability to p300 Inhibition in Human Melanoma. Cancer Res. 2019, 79, 2649–2661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woan, K.V.; Lienlaf, M.; Perez-Villaroel, P.; Lee, C.; Cheng, F.; Knox, T.; Woods, D.M.; Barrios, K.; Powers, J.; Sahakian, E.; et al. Targeting histone deacetylase 6 mediates a dual anti-melanoma effect: Enhanced antitumor immunity and impaired cell proliferation. Mol. Oncol. 2015, 9, 1447–1457. [Google Scholar] [CrossRef] [Green Version]
- Ceol, C.J.; Houvras, Y.; Jane-Valbuena, J.; Bilodeau, S.; Orlando, D.A.; Battisti, V.; Fritsch, L.; Lin, W.M.; Hollmann, T.J.; Ferré, F.; et al. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature 2011, 471, 513–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moubarak, R.S.; de Pablos-Aragoneses, A.; Ortiz-Barahona, V.; Gong, Y.; Gowen, M.; Dolgalev, I.; Shadaloey, S.A.A.; Argibay, D.; Karz, A.; Von Itter, R.; et al. The histone demethylase PHF8 regulates TGFβ signaling and promotes melanoma metastasis. Sci. Adv. 2022, 8, eabi7127. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Goldberg, M.S.; Cumberland, L.K.; Ratnakumar, K.; Segura, M.F.; Emanuel, P.O.; Menendez, S.; Vardabasso, C.; LeRoy, G.; Vidal, C.I.; et al. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature 2010, 468, 1105–1109. [Google Scholar] [CrossRef] [Green Version]
- Vardabasso, C.; Gaspar-Maia, A.; Hasson, D.; Pünzeler, S.; Valle-Garcia, D.; Straub, T.; Keilhauer, E.C.; Strub, T.; Dong, J.; Panda, T.; et al. Histone Variant H2A.Z.2 Mediates Proliferation and Drug Sensitivity of Malignant Melanoma. Mol. Cell 2015, 59, 75–88. [Google Scholar] [CrossRef] [Green Version]
- Duarte, L.F.; Young, A.; Wang, Z.; Wu, H.-A.; Panda, T.; Kou, Y.; Kapoor, A.; Hasson, D.; Mills, N.R.; Ma’Ayan, A.; et al. Histone H3.3 and its proteolytically processed form drive a cellular senescence programme. Nat. Commun. 2014, 5, 5210. [Google Scholar] [CrossRef] [PubMed]
- Micevic, G.; Theodosakis, N.; Bosenberg, M. Aberrant DNA methylation in melanoma: Biomarker and therapeutic opportunities. Clin. Epigenet. 2017, 9, 34. [Google Scholar] [CrossRef] [Green Version]
- Koroknai, V.; Szász, I.; Hernandez-Vargas, H.; Fernandez-Jimenez, N.; Cuenin, C.; Herceg, Z.; Vízkeleti, L.; Ádány, R.; Ecsedi, S.; Balázs, M. DNA hypermethylation is associated with invasive phenotype of malignant melanoma. Exp. Dermatol. 2020, 29, 39–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonvin, E.; Radaelli, E.; Bizet, M.; Luciani, F.; Calonne, E.; Putmans, P.; Nittner, D.; Singh, N.K.; Santagostino, S.F.; Petit, V.; et al. TET2-Dependent Hydroxymethylome Plasticity Reduces Melanoma Initiation and Progression. Cancer Res. 2019, 79, 482–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Zhou, Z.; Ma, L.; Li, C.; Lin, Y.; Yu, T.; Wei, J.-F.; Zhu, L.; Yao, G. Effects of RNA methylation N6-methyladenosine regulators on malignant progression and prognosis of melanoma. Cancer Cell Int. 2021, 21, 453. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.-Y.; Wang, T.; Su, X.; Guo, S. Identification of the m6A RNA Methylation Regulators WTAP as a Novel Prognostic Biomarker and Genomic Alterations in Cutaneous Melanoma. Front. Mol. Biosci. 2021, 8, 665222. [Google Scholar] [CrossRef]
- Meng, J.; Huang, X.; Qiu, Y.; Yu, M.; Lu, J.; Yao, J. Characterization of m6A-Related Genes Landscape in Skin Cutaneous Melanoma to Aid Immunotherapy and Assess Prognosis. Int. J. Gen. Med. 2021, 14, 5345–5361. [Google Scholar] [CrossRef]
- Yu, X.; Zheng, H.; Tse, G.; Chan, M.T.; Wu, W.K. Long non-coding RNAs in melanoma. Cell Prolif. 2018, 51, e12457. [Google Scholar] [CrossRef] [Green Version]
- Leucci, E.; Vendramin, R.; Spinazzi, M.; Laurette, P.; Fiers, M.; Wouters, J.; Radaelli, E.; Eyckerman, S.; Leonelli, C.; Vanderheyden, K.; et al. Melanoma addiction to the long non-coding RNA SAMMSON. Nature 2016, 531, 518–522. [Google Scholar] [CrossRef]
- Vendramin, R.; Verheyden, Y.; Ishikawa, H.; Goedert, L.; Nicolas, E.; Saraf, K.; Armaos, A.; Ponti, R.D.; Izumikawa, K.; Mestdagh, P.; et al. SAMMSON fosters cancer cell fitness by concertedly enhancing mitochondrial and cytosolic translation. Nat. Struct. Mol. Biol. 2018, 25, 1035–1046. [Google Scholar] [CrossRef]
- Gambi, G.; Mengus, G.; Davidson, G.; Demesmaeker, E.; Cuomo, A.; Bonaldi, T.; Katopodi, V.; Malouf, G.G.; Leucci, E.; Davidson, I. The lncRNA LENOX interacts with RAP2C to regulate metabolism and promote resistance to MAPK inhibition in melanoma. Cancer Res. 2022. [Google Scholar] [CrossRef] [PubMed]
- Varrone, F.; Caputo, E. The miRNAs Role in Melanoma and in Its Resistance to Therapy. Int. J. Mol. Sci. 2020, 21, 878. [Google Scholar] [CrossRef] [Green Version]
- Couts, K.L.; Anderson, E.M.; Gross, M.M.; Sullivan, K.; Ahn, N.G. Oncogenic B-Raf signaling in melanoma cells controls a network of microRNAs with combinatorial functions. Oncogene 2013, 32, 1959–1970. [Google Scholar] [CrossRef] [PubMed]
- Choe, M.H.; Yoon, Y.; Kim, J.; Hwang, S.-G.; Han, Y.-H.; Kim, J.-S. miR-550a-3-5p acts as a tumor suppressor and reverses BRAF inhibitor resistance through the direct targeting of YAP. Cell Death Dis. 2018, 9, 640. [Google Scholar] [CrossRef]
- Hallajzadeh, J.; Amirani, E.; Mirzaei, H.; Shafabakhsh, R.; Mirhashemi, S.M.; Sharifi, M.; Yousefi, B.; A Mansournia, M.; Asemi, Z. Circular RNAs: New genetic tools in melanoma. Biomark. Med. 2020, 14, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Qian, P.; Linbo, L.; Xiaomei, Z.; Hui, P. Circ_0002770, acting as a competitive endogenous RNA, promotes proliferation and invasion by targeting miR-331-3p in melanoma. Cell Death Dis. 2020, 11, 264. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Cui, W.; Yang, C.; Du, L.P. Circular RNA ZNF609 drives tumor progression by regulating the miR-138-5p/SIRT7 axis in melanoma. Aging 2021, 13, 19822–19834. [Google Scholar] [CrossRef]
- Fukumoto, T.; Lin, J.; Fatkhutdinov, N.; Liu, P.; Somasundaram, R.; Herlyn, M.; Zhang, R.; Nishigori, C. ARID2 Deficiency Correlates with the Response to Immune Checkpoint Blockade in Melanoma. J. Investig. Dermatol. 2021, 141, 1564–1572.e4. [Google Scholar] [CrossRef]
- Filipski, K.; Scherer, M.; Zeiner, K.N.; Bucher, A.; Kleemann, J.; Jurmeister, P.; Hartung, T.I.; Meissner, M.; Plate, K.H.; Fenton, T.R.; et al. DNA methylation-based prediction of response to immune checkpoint inhibition in metastatic melanoma. J. Immunother. Cancer 2021, 9, e002226. [Google Scholar] [CrossRef]
- Yang, S.; Wei, J.; Cui, Y.-H.; Park, G.; Shah, P.; Deng, Y.; Aplin, A.E.; Lu, Z.; Hwang, S.; He, C.; et al. m6A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat. Commun. 2019, 10, 2782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, F.; Li, H.; Li, Y.; Liu, Y.; Li, X.; Dang, N.; Chu, Q.; Yan, J.; Fang, Z.; Wu, H.; et al. Identification of m6A Regulator-Associated Methylation Modification Clusters and Immune Profiles in Melanoma. Front. Cell Dev. Biol. 2021, 9, 761134. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Kryczek, I.; Nam, J.; Li, X.; Li, S.; Li, J.; Wei, S.; Grove, S.; Vatan, L.; Zhou, J.; et al. LIMIT is an immunogenic lncRNA in cancer immunity and immunotherapy. Nat. Cell Biol. 2021, 23, 526–537. [Google Scholar] [CrossRef]
- Wei, C.-Y.; Zhu, M.-X.; Lu, N.-H.; Liu, J.-Q.; Yang, Y.-W.; Zhang, Y.; Shi, Y.-D.; Feng, Z.-H.; Li, J.-X.; Qi, F.-Z.; et al. Circular RNA circ_0020710 drives tumor progression and immune evasion by regulating the miR-370-3p/CXCL12 axis in melanoma. Mol. Cancer 2020, 19, 84. [Google Scholar] [CrossRef]
- Mastroianni, J.; Stickel, N.; Andrlova, H.; Hanke, K.; Melchinger, W.; Duquesne, S.; Schmidt, D.; Falk, M.; Andrieux, G.; Pfeifer, D.; et al. miR-146a Controls Immune Response in the Melanoma Microenvironment. Cancer Res. 2019, 79, 183–195. [Google Scholar] [CrossRef] [Green Version]
- Huber, V.; Vallacchi, V.; Fleming, V.; Hu, X.; Cova, A.; Dugo, M.; Shahaj, E.; Sulsenti, R.; Vergani, E.; Filipazzi, P.; et al. Tumor-derived microRNAs induce myeloid suppressor cells and predict immunotherapy resistance in melanoma. J. Clin. Investig. 2018, 128, 5505–5516. [Google Scholar] [CrossRef] [Green Version]
- Echevarría-Vargas, I.M.; Reyes-Uribe, P.I.; Guterres, A.; Yin, X.; Kossenkov, A.V.; Liu, Q.; Zhang, G.; Krepler, C.; Cheng, C.; Wei, Z.; et al. Co-targeting BET and MEK as salvage therapy for MAPK and checkpoint inhibitor-resistant melanoma. EMBO Mol. Med. 2018, 10, e8446. [Google Scholar] [CrossRef]
- Grigore, F.; Yang, H.; Hanson, N.D.; VanBrocklin, M.W.; Sarver, A.L.; Robinson, J.P. BRAF inhibition in melanoma is associated with the dysregulation of histone methylation and histone methyltransferases. Neoplasia 2020, 22, 376–389. [Google Scholar] [CrossRef]
- Zakharia, Y.; Monga, V.; Swami, U.; Bossler, A.D.; Freesmeier, M.; Frees, M.; Khan, M.; Frydenlund, N.; Srikantha, R.; Vanneste, M.; et al. Targeting epigenetics for treatment of BRAF mutated metastatic melanoma with decitabine in combination with vemurafenib: A phase lb study. Oncotarget 2017, 8, 89182–89193. [Google Scholar] [CrossRef] [Green Version]
- Dar, A.A.; Nosrati, M.; Bezrookove, V.; de Semir, D.; Majid, S.; Thummala, S.; Sun, V.; Tong, S.; Leong, S.P.L.; Minor, D.; et al. The role of BPTF in melanoma progression and in response to BRAF-targeted therapy. J. Natl. Cancer Inst. 2015, 107, djv034. [Google Scholar] [CrossRef] [Green Version]
- Vergani, E.; Di Guardo, L.; Dugo, M.; Rigoletto, S.; Tragni, G.; Ruggeri, R.; Perrone, F.; Tamborini, E.; Gloghini, A.; Arienti, F.; et al. Overcoming melanoma resistance to vemurafenib by targeting CCL2-induced miR-34a, miR-100 and miR-125b. Oncotarget 2016, 7, 4428–4441. [Google Scholar] [CrossRef] [Green Version]
- Kolenda, T.; Rutkowski, P.; Michalak, M.; Kozak, K.; Guglas, K.; Ryś, M.; Galus, Ł.; Woźniak, S.; Ługowska, I.; Gos, A.; et al. Plasma lncRNA expression profile as a prognostic tool in BRAF-mutant metastatic melanoma patients treated with BRAF inhibitor. Oncotarget 2019, 10, 3879–3893. [Google Scholar] [CrossRef] [PubMed]
- Koetz-Ploch, L.; Hanniford, D.; Dolgalev, I.; Sokolova, E.; Zhong, J.; Díaz-Martínez, M.; Bernstein, E.; Darvishian, F.; Flaherty, K.T.; Chapman, P.B.; et al. MicroRNA-125a promotes resistance to BRAF inhibitors through suppression of the intrinsic apoptotic pathway. Pigment Cell Melanoma Res. 2017, 30, 328–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizos, H.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Fung, C.; Hyman, J.; Haydu, L.E.; Mijatov, B.; Becker, T.M.; Boyd, S.C.; et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: Spectrum and clinical impact. Clin. Cancer Res. 2014, 20, 1965–1977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madore, J.; Strbenac, D.; Vilain, R.; Menzies, A.M.; Yang, J.Y.H.; Thompson, J.F.; Long, G.V.; Mann, G.J.; Scolyer, R.A.; Wilmott, J.S. PD-L1 Negative Status is Associated with Lower Mutation Burden, Differential Expression of Immune-Related Genes, and Worse Survival in Stage III Melanoma. Clin. Cancer Res. 2016, 22, 3915–3923. [Google Scholar] [CrossRef] [Green Version]
- Al Emran, A.; Chatterjee, A.; Rodger, E.J.; Tiffen, J.C.; Gallagher, S.J.; Eccles, M.R.; Hersey, P. Targeting DNA Methylation and EZH2 Activity to Overcome Melanoma Resistance to Immunotherapy. Trends Immunol 2019, 40, 328–344. [Google Scholar] [CrossRef] [Green Version]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef] [Green Version]
- Pandey, S.; Djibo, R.; Darracq, A.; Calendo, G.; Zhang, H.; Henry, R.A.; Andrews, A.J.; Baylin, S.B.; Madzo, J.; Najmanovich, R.; et al. Selective CDK9 Inhibition by Natural Compound Toyocamycin in Cancer Cells. Cancers 2022, 14, 3340. [Google Scholar] [CrossRef]
- Zhang, H.; Pandey, S.; Travers, M.; Sun, H.; Morton, G.; Madzo, J.; Chung, W.; Khowsathit, J.; Perez-Leal, O.; Barrero, C.A.; et al. Targeting CDK9 Reactivates Epigenetically Silenced Genes in Cancer. Cell 2018, 175, 1244–1258.e26. [Google Scholar] [CrossRef] [Green Version]
- Menon, D.R.; Das, S.; Krepler, C.; Vultur, A.; Rinner, B.; Schauer, S.; Kashofer, K.; Wagner, K.; Zhang, G.; Rad, E.B.; et al. A stress-induced early innate response causes multidrug tolerance in melanoma. Oncogene 2015, 34, 4545. [Google Scholar] [CrossRef] [Green Version]
- Rambow, F.; Rogiers, A.; Marin-Bejar, O.; Aibar, S.; Femel, J.; Dewaele, M.; Karras, P.; Brown, D.; Chang, Y.H.; Debiec-Rychter, M.; et al. Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 2018, 174, 843–855.e19. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef]
- Su, Y.; Wei, W.; Robert, L.; Xue, M.; Tsoi, J.; Garcia-Diaz, A.; Moreno, B.H.; Kim, J.; Ng, R.H.; Lee, J.W.; et al. Single-cell analysis resolves the cell state transition and signaling dynamics associated with melanoma drug-induced resistance. Proc. Natl. Acad. Sci. USA 2017, 114, 13679–13684. [Google Scholar] [CrossRef] [Green Version]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [Green Version]
- Hurst, T.P.; Magiorkinis, G. Epigenetic Control of Human Endogenous Retrovirus Expression: Focus on Regulation of Long-Terminal Repeats (LTRs). Viruses 2017, 9, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, I.; Kurata, S.-I. Association of endogenous retroviruses and long terminal repeats with human disorders. Front. Oncol. 2013, 3, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tristem, M. Identification and characterization of novel human endogenous retrovirus families by phylogenetic screening of the human genome mapping project database. J. Virol. 2000, 74, 3715–3730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Löwer, R.; Tönjes, R.R.; Korbmacher, C.; Kurth, R.; Löwer, J. Identification of a Rev-related protein by analysis of spliced transcripts of the human endogenous retroviruses HTDV/HERV-K. J. Virol. 1995, 69, 141–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armbruester, V.; Sauter, M.; Krautkraemer, E.; Meese, E.; Kleiman, A.; Best, B.; Roemer, K.; Mueller-Lantzsch, N. A novel gene from the human endogenous retrovirus K expressed in transforMed. cells. Clin. Cancer Res. 2002, 8, 1800–1807. [Google Scholar]
- Contreras-Galindo, R.; Kaplan, M.H.; Dube, D.; Gonzalez-Hernandez, M.J.; Chan, S.; Meng, F.; Dai, M.; Omenn, G.S.; Gitlin, S.D.; Markovitz, D.M. Human Endogenous Retrovirus Type K (HERV-K) Particles Package and Transmit HERV-K-Related Sequences. J. Virol. 2015, 89, 7187–7201. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Kaye, S.; Gore, M.E.; McClure, M.O.; Bunker, C.B. The role of human endogenous retroviruses in melanoma. Br. J. Dermatol. 2009, 161, 1225–1231. [Google Scholar] [CrossRef]
- Singh, M.; Cai, H.; Bunse, M.; Feschotte, C.; Izsvak, Z. Human Endogenous Retrovirus K Rec forms a Regulatory Loop with MITF that Opposes the Progression of Melanoma to an Invasive Stage. Viruses 2020, 12, 1303. [Google Scholar] [CrossRef] [PubMed]
- Greenig, M. HERVs, immunity, and autoimmunity: Understanding the connection. PeerJ 2019, 7, e6711. [Google Scholar] [CrossRef] [PubMed]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science 2016, 351, 1083–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buscher, K.; Trefzer, U.; Hofmann, M.; Sterry, W.; Kurth, R.; Denner, J. Expression of human endogenous retrovirus K in melanomas and melanoma cell lines. Cancer Res. 2005, 65, 4172–4180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.; Kaye, S.; Francis, N.; Peston, D.; Gore, M.; McClure, M.; Bunker, C. Human endogenous retrovirus K (HERV-K) rec mRNA is expressed in primary melanoma but not in benign naevi or normal skin. Pigment Cell Melanoma Res. 2013, 26, 426–428. [Google Scholar] [CrossRef] [PubMed]
- Schiavetti, F.; Thonnard, J.; Colau, D.; Boon, T.; Coulie, P.G. A human endogenous retroviral sequence encoding an antigen recognized on melanoma by cytolytic T lymphocytes. Cancer Res. 2002, 62, 5510–5516. [Google Scholar]
- Krishnamurthy, J.; Rabinovich, B.A.; Mi, T.; Switzer, K.C.; Olivares, S.; Maiti, S.N.; Plummer, J.B.; Singh, H.; Kumaresan, P.R.; Huls, H.M.; et al. Genetic Engineering of T Cells to Target HERV-K, an Ancient Retrovirus on Melanoma. Clin. Cancer Res. 2015, 21, 3241–3251. [Google Scholar] [CrossRef] [Green Version]
- Bannert, N.; Hofmann, H.; Block, A.; Hohn, O. HERVs New Role in Cancer: From Accused Perpetrators to Cheerful Protectors. Front. Microbiol. 2018, 9, 178. [Google Scholar] [CrossRef]
- Chen, R.; Ishak, C.A.; De Carvalho, D.D. Endogenous Retroelements and the Viral Mimicry Response in Cancer Therapy and Cellular Homeostasis. Cancer Discov. 2021, 11, 2707–2725. [Google Scholar] [CrossRef]
- Chiaro, J.; Kasanen, H.H.; Whalley, T.; Capasso, C.; Grönholm, M.; Feola, S.; Peltonen, K.; Hamdan, F.; Hernberg, M.; Mäkelä, S.; et al. Viral Molecular Mimicry Influences the Antitumor Immune Response in Murine and Human Melanoma. Cancer Immunol. Res. 2021, 9, 981–993. [Google Scholar] [CrossRef]
- Yang, Y.; Bedford, M.T. Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 2013, 13, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Richard, S. Cellular pathways influenced by protein arginine methylation: Implications for cancer. Mol. Cell 2021, 81, 4357–4368. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Liu, X.; Cai, X.; Ouyang, G.; Fan, S.; Wang, J.; Xiao, W. Zebrafish prmt7 negatively regulates antiviral responses by suppressing the retinoic acid-inducible gene-I-like receptor signaling. FASEB J. 2020, 34, 988–1000. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Li, X.; Cai, X.; Zha, H.; Zhou, Z.; Sun, X.; Rong, F.; Tang, J.; Zhu, C.; Liu, X.; et al. Arginine monomethylation by PRMT7 controls MAVS-mediated antiviral innate immunity. Mol. Cell 2021, 81, 3171–3186.e8. [Google Scholar] [CrossRef]
- Srour, N.; Villarreal, O.D.; Hardikar, S.; Yu, Z.; Preston, S.; Miller, W.H.; Szewczyk, M.M.; Barsyte-Lovejoy, D.; Xu, H.; Chen, T.; et al. PRMT7 ablation stimulates anti-tumor immunity and sensitizes melanoma to immune checkpoint blockade. Cell Rep. 2022, 38, 110582. [Google Scholar] [CrossRef]
- Hwang, J.W.; Cho, Y.; Bae, G.U.; Kim, S.N.; Kim, Y.K. Protein arginine methyltransferases: Promising targets for cancer therapy. Exp. Mol. Med. 2021, 53, 788–808. [Google Scholar] [CrossRef]
- Szewczyk, M.M.; Ishikawa, Y.; Organ, S.; Sakai, N.; Li, F.; Halabelian, L.; Ackloo, S.; Couzens, A.L.; Eram, M.; Dilworth, D.; et al. Pharmacological inhibition of PRMT7 links arginine monomethylation to the cellular stress response. Nat. Commun. 2020, 11, 2396. [Google Scholar] [CrossRef]
- Sheng, W.; LaFleur, M.W.; Nguyen, T.H.; Chen, S.; Chakravarthy, A.; Conway, J.R.; Li, Y.; Chen, H.; Yang, H.; Hsu, P.-H.; et al. LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell 2018, 174, 549–563.e19. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Liao, G.; Yu, B. LSD1/KDM1A inhibitors in clinical trials: Advances and prospects. J. Hematol. Oncol. 2019, 12, 129. [Google Scholar] [CrossRef] [Green Version]
- Griffin, G.K.; Wu, J.; Iracheta-Vellve, A.; Patti, J.C.; Hsu, J.; Davis, T.; Dele-Oni, D.; Du, P.P.; Halawi, A.G.; Ishizuka, J.J.; et al. Epigenetic silencing by SETDB1 suppresses tumour intrinsic immunogenicity. Nature 2021, 595, 309–314. [Google Scholar] [CrossRef]
- Dranoff, G.; Jaffee, E.; Lazenby, A.; Golumbek, P.; Levitsky, H.; Brose, K.; Jackson, V.; Hamada, H.; Pardoll, D.; Mulligan, R.C.; et al. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc. Natl. Acad. Sci. USA 1993, 90, 3539–3543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.-M.; Cai, W.L.; Liu, X.; Thakral, D.; Luo, J.; Chan, L.H.; McGeary, M.K.; Song, E.; Blenman, K.R.M.; Micevic, G.; et al. KDM5B promotes immune evasion by recruiting SETDB1 to silence retroelements. Nature 2021, 598, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Khodadadi-Jamayran, A.; Dolgalev, I.; Cho, H.; Badri, S.; Chiriboga, L.A.; Zeck, B.; Gregorio, M.L.D.R.; Dowling, C.M.; Labbe, K.; et al. Targeting the Atf7ip-Setdb1 Complex Augments Antitumor Immunity by Boosting Tumor Immunogenicity. Cancer Immunol. Res. 2021, 9, 1298–1315. [Google Scholar] [CrossRef] [PubMed]
- Lazaro-Camp, V.J.; Salari, K.; Meng, X.; Yang, S. SETDB1 in cancer: Overexpression and its therapeutic implications. Am. J. Cancer Res. 2021, 11, 1803–1827. [Google Scholar]
- Federico, A.; Steinfass, T.; Larribère, L.; Novak, D.; Morís, F.; Núñez, L.-E.; Umansky, V.; Utikal, J. Mithramycin A and Mithralog EC-8042 Inhibit SETDB1 Expression and Its Oncogenic Activity in Malignant Melanoma. Mol. Ther. Oncolytics 2020, 18, 83–99. [Google Scholar] [CrossRef]
- Jose, A.; Shenoy, G.G.; Rodrigues, G.S.; Kumar, N.A.N.; Munisamy, M.; Thomas, L.; Kolesar, J.; Rai, G.; Rao, P.P.N.; Rao, M. Histone Demethylase KDM5B as a Therapeutic Target for Cancer Therapy. Cancers 2020, 12, 2121. [Google Scholar] [CrossRef]
- Zheng, Y.-C.; Chang, J.; Wang, L.-C.; Ren, H.-M.; Pang, J.-R.; Liu, H.-M. Lysine demethylase 5B (KDM5B): A potential anti-cancer drug target. Eur. J. Med. Chem. 2019, 161, 131–140. [Google Scholar] [CrossRef]
- Ferretti, R.; Bhutkar, A.; McNamara, M.C.; Lees, J.A. BMI1 induces an invasive signature in melanoma that promotes metastasis and chemoresistance. Genes Dev. 2016, 30, 18–33. [Google Scholar] [CrossRef] [Green Version]
- Badodi, S.; Pomella, N.; Lim, Y.M.; Brandner, S.; Morrison, G.; Pollard, S.M.; Zhang, X.; Zabet, N.R.; Marino, S. Combination of BMI1 and MAPK/ERK inhibitors is effective in medulloblastoma. Neuro-Oncology 2022, 24, 1273–1285. [Google Scholar] [CrossRef]
- Eberle-Singh, J.A.; Sagalovskiy, I.; Maurer, H.C.; Sastra, S.A.; Palermo, C.F.; Decker, A.R.; Kim, M.J.; Sheedy, J.; Mollin, A.; Cao, L.; et al. Effective Delivery of a Microtubule Polymerization Inhibitor Synergizes with Standard Regimens in Models of Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2019, 25, 5548–5560. [Google Scholar] [CrossRef]
- Jernigan, F.; Branstrom, A.; Baird, J.D.; Cao, L.; Dali, M.; Furia, B.; Kim, M.J.; O’Keefe, K.; Kong, R.; Laskin, O.L.; et al. Preclinical and Early Clinical Development of PTC596, a Novel Small-Molecule Tubulin-Binding Agent. Mol. Cancer Ther. 2021, 20, 1846–1857. [Google Scholar] [CrossRef] [PubMed]
- Strub, T.; Ghiraldini, F.G.; Carcamo, S.; Li, M.; Wroblewska, A.; Singh, R.; Goldberg, M.S.; Hasson, D.; Wang, Z.; Gallagher, S.; et al. SIRT6 haploinsufficiency induces BRAF(V600E) melanoma cell resistance to MAPK inhibitors via IGF signalling. Nat. Commun. 2018, 9, 3440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelsen, T.S.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [Green Version]
- Mattei, J.; Ballhausen, A.; Bassett, R.; Shephard, M.; Chattopadhyay, C.; Hudgens, C.; Tetzlaff, M.; Woodman, S.; Sato, T.; Patel, S.P. A phase II study of the insulin-like growth factor type I receptor inhibitor IMC-A12 in patients with metastatic uveal melanoma. Melanoma Res. 2020, 30, 574–579. [Google Scholar] [CrossRef]
- Smith, T.J.; Kahaly, G.J.; Ezra, D.G.; Fleming, J.C.; Dailey, R.A.; Tang, R.A.; Harris, G.J.; Antonelli, A.; Salvi, M.; Goldberg, R.A.; et al. Teprotumumab for Thyroid-Associated Ophthalmopathy. N. Engl. J. Med. 2017, 376, 1748–1761. [Google Scholar] [CrossRef]
- Fiorentino, F.; Mai, A.; Rotili, D. Emerging Therapeutic Potential of SIRT6 Modulators. J. Med. Chem. 2021, 64, 9732–9758. [Google Scholar] [CrossRef]
- Palamaris, K.; Moutafi, M.; Gakiopoulou, H.; Theocharis, S. Histone Deacetylase (HDAC) Inhibitors: A Promising Weapon to Tackle Therapy Resistance in Melanoma. Int. J. Mol. Sci. 2022, 23, 3660. [Google Scholar] [CrossRef]
- Yamada, T.; Hasegawa, S.; Inoue, Y.; Date, Y.; Yamamoto, N.; Mizutani, H.; Nakata, S.; Matsunaga, K.; Akamatsu, H. Wnt/beta-catenin and kit signaling sequentially regulate melanocyte stem cell differentiation in UVB-induced epidermal pigmentation. J. Investig. Dermatol. 2013, 133, 2753–2762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larribere, L.; Hasegawa, S.; Inoue, Y.; Date, Y.; Yamamoto, N.; Mizutani, H.; Nakata, S.; Matsunaga, K.; Akamatsu, H. PI3K mediates protection against TRAIL-induced apoptosis in primary human melanocytes. Cell Death Differ. 2004, 11, 1084–1091. [Google Scholar] [CrossRef] [Green Version]
- Edmondson, S.R.; Russo, V.C.; McFarlane, A.C.; Wraight, C.J.; Werther, G.A. Interactions between growth hormone, insulin-like growth factor I, and basic fibroblast growth factor in melanocyte growth. J. Clin. Endocrinol. Metab. 1999, 84, 1638–1644. [Google Scholar] [CrossRef]
- Hoek, K.S.; Goding, C.R. Cancer stem cells versus phenotype-switching in melanoma. Pigment Cell Melanoma Res. 2010, 23, 746–759. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, A.; Imrichova, H.; Atak, Z.K.; Dewaele, M.; Rambow, F.; Hulselmans, G.; Christiaens, V.; Svetlichnyy, D.; Luciani, F.; Van den Mooter, L.; et al. Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat. Commun. 2015, 6, 6683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoek, K.S.; Eichhoff, O.M.; Schlegel, N.C.; Döbbeling, U.; Kobert, N.; Schaerer, L.; Hemmi, S.; Dummer, R. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. 2008, 68, 650–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsoi, J.; Robert, L.; Paraiso, K.; Galvan, C.; Sheu, K.M.; Lay, J.; Wong, D.J.; Atefi, M.; Shirazi, R.; Wang, X.; et al. Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 2018, 33, 890–904.e5. [Google Scholar] [CrossRef] [PubMed]
- Wouters, J.; Kalender-Atak, Z.; Minnoye, L.; Spanier, K.I.; De Waegeneer, M.; González-Blas, C.B.; Mauduit, D.; Davie, K.; Hulselmans, G.; Najem, A.; et al. Robust gene expression programs underlie recurrent cell states and phenotype switching in melanoma. Nat. Cell Biol. 2020, 22, 986–998. [Google Scholar] [CrossRef]
- Rambow, F.; Marine, J.-C.; Goding, C.R. Melanoma plasticity and phenotypic diversity: Therapeutic barriers and opportunities. Genes Dev. 2019, 33, 1295–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertolotto, C.; Lesueur, F.; Giuliano, S.; Strub, T.; De Lichy, M.; Bille, K.; Dessen, P.; D’Hayer, B.; Mohamdi, H.; Remenieras, A.; et al. A SUMOylation-defective MITF germline mutation predisposes to melanoma and renal carcinoma. Nature 2011, 480, 94–98. [Google Scholar] [CrossRef]
- Ngeow, K.C.; Friedrichsen, H.J.; Li, L.; Zeng, Z.; Andrews, S.; Volpon, L.; Brunsdon, H.; Berridge, G.; Picaud, S.; Fischer, R.; et al. BRAF/MAPK and GSK3 signaling converges to control MITF nuclear export. Proc. Natl. Acad. Sci. USA 2018, 115, E8668–E8677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schepsky, A.; Bruser, K.; Gunnarsson, G.J.; Goodall, J.; Hallsson, J.H.; Goding, C.R.; Steingrimsson, E.; Hecht, A. The microphthalmia-associated transcription factor Mitf interacts with beta-catenin to determine target gene expression. Mol. Cell Biol. 2006, 26, 8914–8927. [Google Scholar] [CrossRef] [Green Version]
- de la Serna, I.L.; Ohkawa, Y.; Higashi, C.; Dutta, C.; Osias, J.; Kommajosyula, N.; Tachibana, T.; Imbalzano, A.N. The microphthalmia-associated transcription factor requires SWI/SNF enzymes to activate melanocyte-specific genes. J. Biol. Chem. 2006, 281, 20233–20241. [Google Scholar] [CrossRef] [Green Version]
- Bemis, L.T.; Chen, R.; Amato, C.M.; Classen, E.H.; Robinson, S.E.; Coffey, D.G.; Erickson, P.F.; Shellman, Y.G.; Robinson, W.A. MicroRNA-137 targets microphthalmia-associated transcription factor in melanoma cell lines. Cancer Res. 2008, 68, 1362–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arts, N.; Cané, S.; Hennequart, M.; Lamy, J.; Bommer, G.; Van Den Eynde, B.; De Plaen, E. microRNA-155, induced by interleukin-1ss, represses the expression of microphthalmia-associated transcription factor (MITF-M) in melanoma cells. PLoS ONE 2015, 10, e0122517. [Google Scholar] [CrossRef] [Green Version]
- Qian, H.; Yang, C.; Yang, Y. MicroRNA-26a inhibits the growth and invasiveness of malignant melanoma and directly targets on MITF gene. Cell Death Discov. 2017, 3, 17028. [Google Scholar] [CrossRef] [PubMed]
- Haflidadóttir, B.S.; Bergsteinsdóttir, K.; Praetorius, C.; Steingrímsson, E. miR-148 regulates Mitf in melanoma cells. PLoS ONE 2010, 5, e11574. [Google Scholar] [CrossRef] [PubMed]
- Louphrasitthiphol, P.; Siddaway, R.; Loffreda, A.; Pogenberg, V.; Friedrichsen, H.; Schepsky, A.; Zeng, Z.; Lu, M.; Strub, T.; Freter, R.; et al. Tuning Transcription Factor Availability through Acetylation-Mediated Genomic Redistribution. Mol. Cell 2020, 79, 472–487.e10. [Google Scholar] [CrossRef] [PubMed]
- Falletta, P.; Sanchez-Del-Campo, L.; Chauhan, J.; Effern, M.; Kenyon, A.; Kershaw, C.J.; Siddaway, R.; Lisle, R.; Freter, R.; Daniels, M.J.; et al. Translation reprogramming is an evolutionarily conserved driver of phenotypic plasticity and therapeutic resistance in melanoma. Genes Dev. 2017, 31, 18–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, J.; Smith, M.; Zudaire, I.; Wellbrock, C.; Arozarena, I. Glucose availability controls ATF4-mediated MITF suppression to drive melanoma cell growth. Oncotarget 2017, 8, 32946–32959. [Google Scholar] [CrossRef] [Green Version]
- Hartman, M.L.; Talar, B.; Noman, M.Z.; Gajos-Michniewicz, A.; Chouaib, S.; Czyz, M. Gene expression profiling identifies microphthalmia-associated transcription factor (MITF) and Dickkopf-1 (DKK1) as regulators of microenvironment-driven alterations in melanoma phenotype. PLoS ONE 2014, 9, e95157. [Google Scholar] [CrossRef] [Green Version]
- Landsberg, J.; Kohlmeyer, J.; Renn, M.; Bald, T.; Rogava, M.; Cron, M.; Fatho, M.; Lennerz, V.; Wölfel, T.; Hölzel, M.; et al. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature 2012, 490, 412–416. [Google Scholar] [CrossRef]
- Riesenberg, S.; Groetchen, A.; Siddaway, R.; Bald, T.; Reinhardt, J.; Smorra, D.; Kohlmeyer, J.; Renn, M.; Phung, B.; Aymans, P.; et al. MITF and c-Jun antagonism interconnects melanoma dedifferentiation with pro-inflammatory cytokine responsiveness and myeloid cell recruitment. Nat. Commun. 2015, 6, 8755. [Google Scholar] [CrossRef] [Green Version]
- Golan, T.; Messer, A.R.; Amitai-Lange, A.; Melamed, Z.; Ohana, R.; Bell, R.E.; Kapitansky, O.; Lerman, G.; Greenberger, S.; Khaled, M.; et al. Interactions of Melanoma Cells with Distal Keratinocytes Trigger Metastasis via Notch Signaling Inhibition of MITF. Mol. Cell 2015, 59, 664–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheli, Y.; Giuliano, S.; Fenouille, N.; Allegra, M.; Hofman, V.; Hofman, P.; Bahadoran, P.; Lacour, J.-P.; Tartare-Deckert, S.; Bertolotto, C.; et al. Hypoxia and MITF control metastatic behaviour in mouse and human melanoma cells. Oncogene 2012, 31, 2461–2470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feige, E.; Yokoyama, S.; Levy, C.; Khaled, M.; Igras, V.; Lin, R.J.; Lee, S.; Widlund, H.R.; Granter, S.R.; Kung, A.L.; et al. Hypoxia-induced transcriptional repression of the melanoma-associated oncogene MITF. Proc. Natl. Acad. Sci. USA 2011, 108, E924–E933. [Google Scholar] [CrossRef] [Green Version]
- Miskolczi, Z.; Smith, M.; Rowling, E.J.; Ferguson, J.; Barriuso, J.; Wellbrock, C. Collagen abundance controls melanoma phenotypes through lineage-specific microenvironment sensing. Oncogene 2018, 37, 3166–3182. [Google Scholar] [CrossRef]
- Reinhardt, J.; Landsberg, J.; Schmid-Burgk, J.L.; Ramis, B.B.; Bald, T.; Glodde, N.; Lopez-Ramos, D.; Young, A.; Ngiow, S.F.; Nettersheim, D.; et al. MAPK Signaling and Inflammation Link Melanoma Phenotype Switching to Induction of CD73 during Immunotherapy. Cancer Res. 2017, 77, 4697–4709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berico, P.; Cigrang, M.; Davidson, G.; Braun, C.; Sandoz, J.; Legras, S.; Vokshi, B.H.; Slovic, N.; Peyresaubes, F.; Robles, C.M.G.; et al. CDK7 and MITF repress a transcription program involved in survival and drug tolerance in melanoma. EMBO Rep. 2021, 22, e51683. [Google Scholar] [CrossRef] [PubMed]
- García-Jiménez, C.; Goding, C.R. Starvation and Pseudo-Starvation as Drivers of Cancer Metastasis through Translation Reprogramming. Cell Metab. 2019, 29, 254–267. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhang, S.-M.; McGeary, M.K.; Krykbaeva, I.; Lai, L.; Jansen, D.J.; Kales, S.C.; Simeonov, A.; Hall, M.D.; Kelly, D.P.; et al. KDM5B Promotes Drug Resistance by Regulating Melanoma-Propagating Cell Subpopulations. Mol. Cancer Ther. 2019, 18, 706–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orouji, E.; Federico, A.; Larribère, L.; Novak, D.; Lipka, D.B.; Assenov, Y.; Sachindra, S.; Hüser, L.; Granados, K.; Gebhardt, C.; et al. Histone methyltransferase SETDB1 contributes to melanoma tumorigenesis and serves as a new potential therapeutic target. Int. J. Cancer 2019, 145, 3462–3477. [Google Scholar] [CrossRef]
- Borsotti, P.; Ghilardi, C.; Ostano, P.; Silini, A.; Dossi, R.; Pinessi, D.; Foglieni, C.; Scatolini, M.; Lacal, P.M.; Ferrari, R.; et al. Thrombospondin-1 is part of a Slug-independent motility and metastatic program in cutaneous melanoma, in association with VEGFR-1 and FGF-2. Pigment Cell Melanoma Res. 2015, 28, 73–81. [Google Scholar] [CrossRef]
- Jayachandran, A.; Anaka, M.; Prithviraj, P.; Hudson, C.; McKeown, S.J.; Lo, P.-H.; Vella, L.J.; Goding, C.R.; Cebon, J.; Behren, A. Thrombospondin 1 promotes an aggressive phenotype through epithelial-to-mesenchymal transition in human melanoma. Oncotarget 2014, 5, 5782–5797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeanne, A.; Boulagnon-Rombi, C.; Devy, J.; Théret, L.; Fichel, C.; Bouland, N.; Diebold, M.-D.; Martiny, L.; Schneider, C.; Dedieu, S. Matricellular TSP-1 as a target of interest for impeding melanoma spreading: Towards a therapeutic use for TAX2 peptide. Clin. Exp. Metastasis 2016, 33, 637–649. [Google Scholar] [CrossRef]
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-coding RNA networks in cancer. Nat. Rev. Cancer 2018, 18, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Ramilowski, J.A.; Yip, C.W.; Agrawal, S.; Chang, J.-C.; Ciani, Y.; Kulakovskiy, I.V.; Mendez, M.; Ooi, J.L.C.; Ouyang, J.F.; Parkinson, N.; et al. Functional annotation of human long noncoding RNAs via molecular phenotyping. Genome Res. 2020, 30, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef] [Green Version]
- Iyer, M.K.; Niknafs, Y.S.; Malik, R.; Singhal, U.; Sahu, A.; Hosono, Y.; Barrette, T.R.; Prensner, J.R.; Evans, J.R.; Zhao, S.; et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015, 47, 199–208. [Google Scholar] [CrossRef]
- Schmitt, A.M.; Chang, H.Y. Long Noncoding RNAs in Cancer Pathways. Cancer Cell 2016, 29, 452–463. [Google Scholar] [CrossRef] [Green Version]
- Melixetian, M.; Bossi, D.; Mihailovich, M.; Punzi, S.; Barozzi, I.; Marocchi, F.; Cuomo, A.; Bonaldi, T.; Testa, G.; Marine, J.; et al. Long non-coding RNA TINCR suppresses metastatic melanoma dissemination by preventing ATF4 translation. EMBO Rep. 2021, 22, e50852. [Google Scholar] [CrossRef]
- Sun, X.; Li, J.; Sun, Y.; Zhang, Y.; Dong, L.; Shen, C.; Yang, L.; Yang, M.; Li, Y.; Shen, G.; et al. miR-7 reverses the resistance to BRAFi in melanoma by targeting EGFR/IGF-1R/CRAF and inhibiting the MAPK and PI3K/AKT signaling pathways. Oncotarget 2016, 7, 53558–53570. [Google Scholar] [CrossRef] [Green Version]
- Vitiello, M.; D’Aurizio, R.; Poliseno, L. Biological role of miR-204 and miR-211 in melanoma. Oncoscience 2018, 5, 248–251. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Martinez, M.; Benito-Jardon, L.; Teixido, J. New insights in melanoma resistance to BRAF inhibitors: A role for microRNAs. Oncotarget 2018, 9, 35374–35375. [Google Scholar] [CrossRef] [PubMed]
- Ennen, M.; Keime, C.; Gambi, G.; Kieny, A.; Coassolo, S.; Thibault-Carpentier, C.; Margerin-Schaller, F.; Davidson, G.; Vagne, C.; Lipsker, D.; et al. MITF-High and MITF-Low Cells and a Novel Subpopulation Expressing Genes of Both Cell States Contribute to Intra- and Intertumoral Heterogeneity of Primary Melanoma. Clin. Cancer Res. 2017, 23, 7097–7107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Titov, D.V.; Gilman, B.; He, Q.-L.; Bhat, S.; Low, W.-K.; Dang, Y.; Smeaton, M.; Demain, A.L.; Miller, P.S.; Kugel, J.F.; et al. XPB, a subunit of TFIIH, is a target of the natural product triptolide. Nat. Chem. Biol. 2011, 7, 182–188. [Google Scholar] [CrossRef] [Green Version]
- Nuñez, G.S.; Robles, C.M.G.; Giraudon, C.; Martínez-Leal, J.F.; Compe, E.; Coin, F.; Aviles, P.; Galmarini, C.M.; Egly, J.-M. Lurbinectedin Specifically Triggers the Degradation of Phosphorylated RNA Polymerase II and the Formation of DNA Breaks in Cancer Cells. Mol. Cancer Ther. 2016, 15, 2399–2412. [Google Scholar] [CrossRef] [PubMed]
- Skorupan, N.; I Ahmad, M.; Steinberg, S.M.; Trepel, J.B.; Cridebring, D.; Han, H.; Von Hoff, D.D.; Alewine, C. A phase II trial of the super-enhancer inhibitor MinnelideTM in advanced refractory adenosquamous carcinoma of the pancreas. Future Oncol. 2022, 18, 2475–2481. [Google Scholar] [CrossRef] [PubMed]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef] [Green Version]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Parolia, A.; Qiao, Y.; Bawa, P.; Eyunni, S.; Mannan, R.; Carson, S.E.; Chang, Y.; Wang, X.; Zhang, Y.; et al. Targeting SWI/SNF ATPases in enhancer-addicted prostate cancer. Nature 2022, 601, 434–439. [Google Scholar] [CrossRef]
- Kress, T.R.; Sabò, A.; Amati, B. MYC: Connecting selective transcriptional control to global RNA production. Nat. Rev. Cancer 2015, 15, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Jain, A.D.; Truica, M.I.; Izquierdo-Ferrer, J.; Anker, J.F.; Lysy, B.; Sagar, V.; Luan, Y.; Chalmers, Z.R.; Unno, K.; et al. Small-Molecule MYC Inhibitors Suppress Tumor Growth and Enhance Immunotherapy. Cancer Cell 2019, 36, 483–497.e15. [Google Scholar] [CrossRef]
- Quemener, A.M.; Bachelot, L.; Forestier, A.; Donnou-Fournet, E.; Gilot, D.; Galibert, M.-D. The powerful world of antisense oligonucleotides: From bench to bedside. Wiley Interdiscip. Rev. RNA 2020, 11, e1594. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T.; Wang, S.; Vickers, T.A.; Shen, W.; Liang, X.-H. Cellular uptake and trafficking of antisense oligonucleotides. Nat. Biotechnol. 2017, 35, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Dowdy, S.F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 2017, 35, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Geary, R.S.; Norris, D.; Yu, R.; Bennett, C.F. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 46–51. [Google Scholar] [CrossRef]
- Ämmälä, C.; Drury, W.J.; Knerr, L.; Ahlstedt, I.; Stillemark-Billton, P.; Wennberg-Huldt, C.; Andersson, E.-M.; Valeur, E.; Jansson-Löfmark, R.; Janzén, D.; et al. Targeted delivery of antisense oligonucleotides to pancreatic beta-cells. Sci. Adv. 2018, 4, eaat3386. [Google Scholar] [CrossRef] [Green Version]
- Mazur, C.; Powers, B.; Zasadny, K.; Sullivan, J.M.; Dimant, H.; Kamme, F.; Hesterman, J.; Matson, J.; Oestergaard, M.; Seaman, M.; et al. Brain pharmacology of intrathecal antisense oligonucleotides revealed through multimodal imaging. JCI Insight 2019, 4, e129240. [Google Scholar] [CrossRef]

| Gene | Function | Mechanism |
|---|---|---|
| PRMT7 [136] | Protein arginine methyl transferase (PRMT) | PRMT7 inhibition represses DNMTs regulating ERV expression. ERVs cause dsRNA stress, detected by increased MDA5 and RIG-I due to H3K4me3 and H4R3me2 hypomethylation of their promoters. Shifting the cell into viral mimicry, the resultant IFN expression enhances antigen presentation and improves PD-1 & CTLA-4 checkpoint blockade via increased CD8+ T cell and reduced MDSC infiltration. |
| LSD1 [139] | Histone demethylase | LSD1 ablation increases ERV transcription and reduces core RISC complex proteins, generating cellular dsRNA stress detected by TLR3 and MDA5, inducing viral mimicry, activating IFN signaling, and increasing MHC-I and PD-L1 expression. Enhanced immunogenicity improves CD8+ T cell infiltration and boosts response to PD-1 checkpoint blockade. |
| SETDB1 [141] | Histone methyltransferase | SETDB1 loss de-represses TEs encoding viral antigens and immunostimulatory genes. Intact viral ORFs in induced TEs facilitate generation of MHC-I peptides and trigger CD8+ T cell responses. In B16F10-GVAX melanoma, SETDB1 knockout sensitized cells to PD-1 checkpoint blockade. Rare bi-directional TE transcription prevented the generation of dsRNA and interferon signaling and thwarted viral mimicry. |
| KDM5B and SETDB1 [143] | Histone demethylase and histone methyltransferase | KDM5B recruits and regulates SETDB1 in a proteosome-dependent manner. KDM5B loss increases bi-directional ERV transcription, accumulating dsRNA stress. MDA5 and cGAS depletion ablate IFN-I activation, MHC-I signaling, and induction of ISGs following KDM5B knockout. Increased CD8+ T cell activity following KDM5B knockout sensitized treatment-resistant YUMM1.7 melanoma cells to anti-PD-1 in vivo treatment. |
| ATF7IP and SETDB1 [144] | SETDB1 adaptor and histone methyltransferase | ATF7IP interacts with SETDB1 to repress ERV expression. ATF7IP deficiency reduces H3K9me3 deposition by SETDB1 and inhibits spliceosome activity, significantly increasing mRNA intron retention. Interferon signaling through IRF7 and IRF9 increase ERV antigen presentation, enhancing immunogenicity and facilitating clearance via elevated T cell infiltration. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rubanov, A.; Berico, P.; Hernando, E. Epigenetic Mechanisms Underlying Melanoma Resistance to Immune and Targeted Therapies. Cancers 2022, 14, 5858. https://doi.org/10.3390/cancers14235858
Rubanov A, Berico P, Hernando E. Epigenetic Mechanisms Underlying Melanoma Resistance to Immune and Targeted Therapies. Cancers. 2022; 14(23):5858. https://doi.org/10.3390/cancers14235858
Chicago/Turabian StyleRubanov, Andrey, Pietro Berico, and Eva Hernando. 2022. "Epigenetic Mechanisms Underlying Melanoma Resistance to Immune and Targeted Therapies" Cancers 14, no. 23: 5858. https://doi.org/10.3390/cancers14235858