
Targeting FTO Suppresses Pancreatic Carcinogenesis via Regulating Stem Cell Maintenance and EMT Pathway


Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Stable Knockdown of FTO in Pancreatic Cancer Cells and Overexpression in HPDE Cells
2.3. Cell Viability
2.4. Anchorage-Dependent and Anchorage-Independent Colony Formation Assay
2.5. Apoptosis and Cell Cycle Analysis
2.6. Sphere Formation Assay
2.7. Migration and Invasion Assay
2.8. RNA Isolation, cDNA Synthesis, and qPCR
2.9. Western Blot Analysis
2.10. In Vivo Tumor Xenograft Study
2.11. Statistical Analysis
3. Results
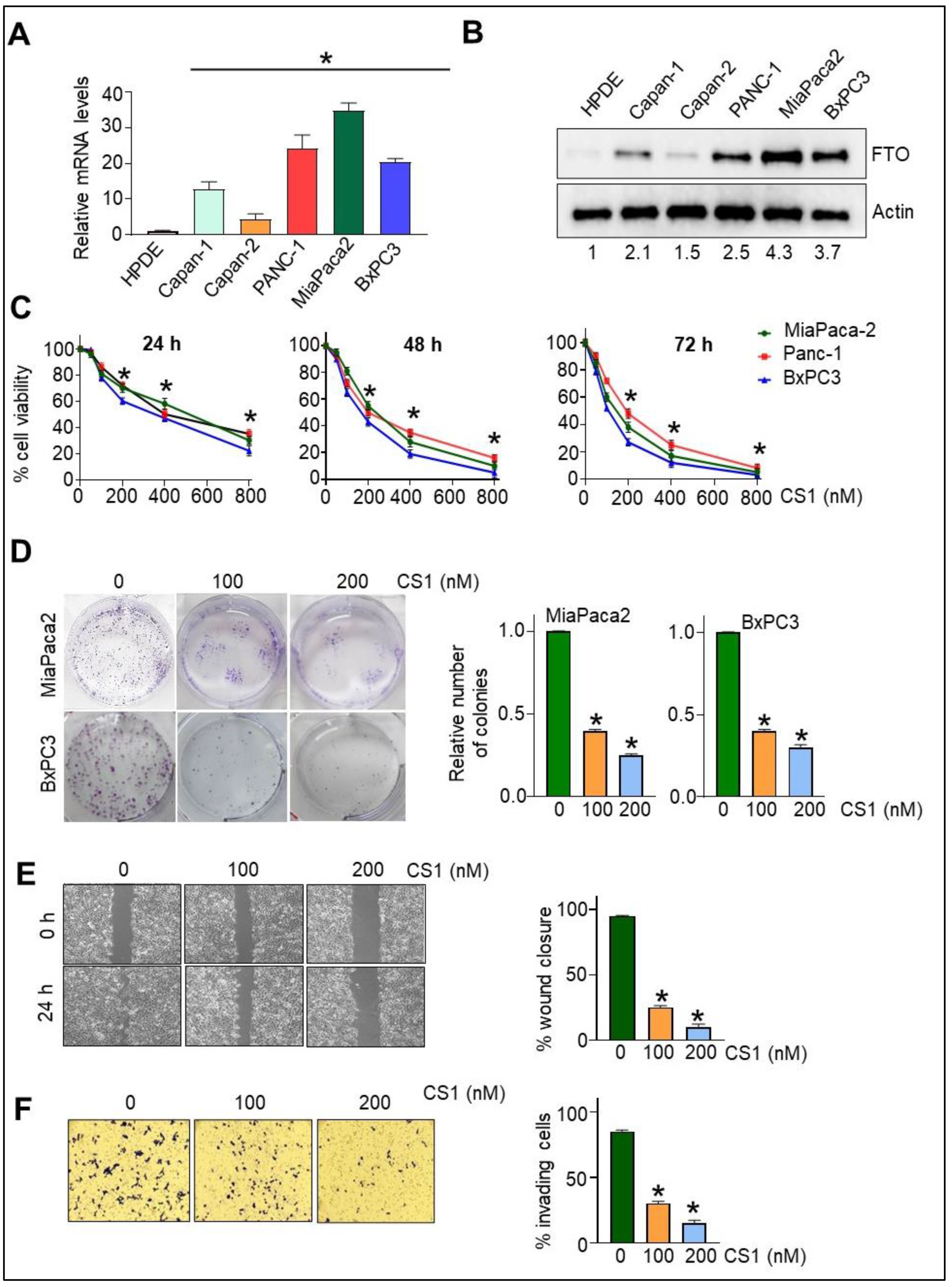
3.1. FTO Is Overexpressed in Pancreatic Cancer Cells
3.2. FTO Inhibition Affects Cell Viability, Migration, and Invasiveness of Pancreatic Cancer Cells
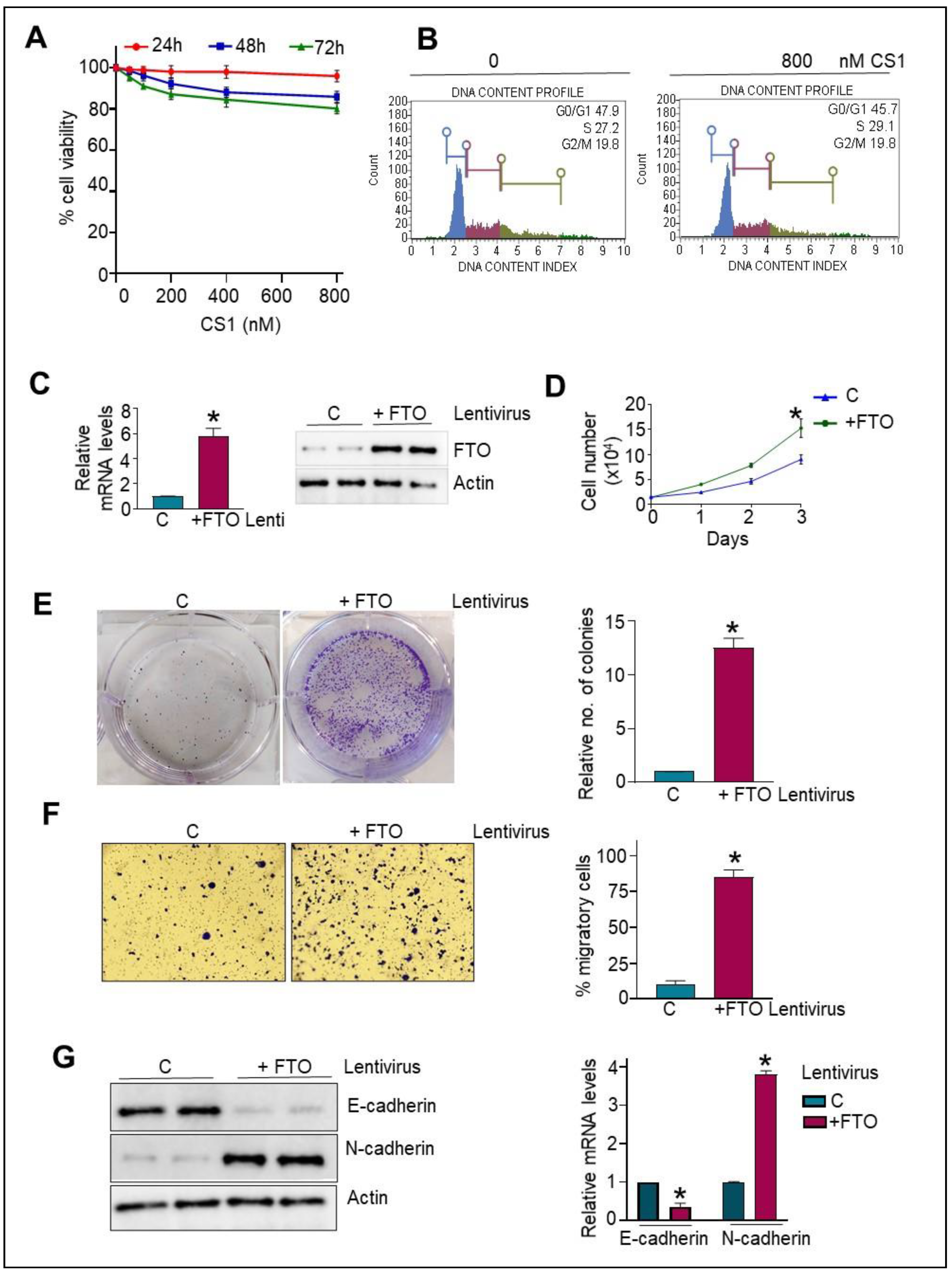
3.3. Normal HPDE Cells Remain Unaltered following CS1 Treatment
3.4. FTO Overexpression in Normal HPDE Cells Promotes Proliferative and Migratory Phenotype via Modulating EMT Traits
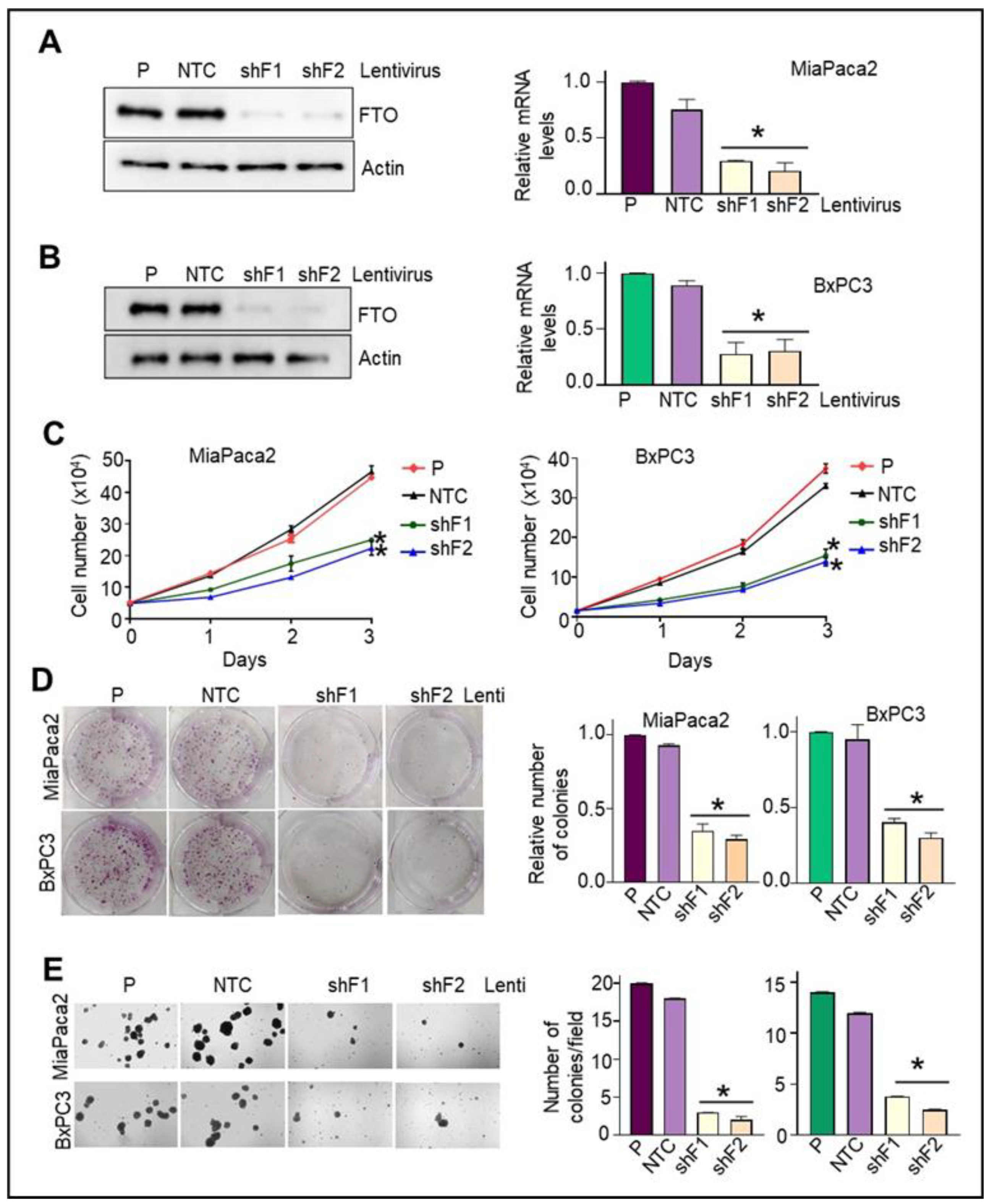
3.5. Stable FTO Depletion in Pancreatic Cancer Cells Impairs Their Cell Growth and Proliferative Capabilities
3.6. Loss of FTO Alters the Cell Cycle Profile and Induces Apoptosis in Pancreatic Cancer Cells
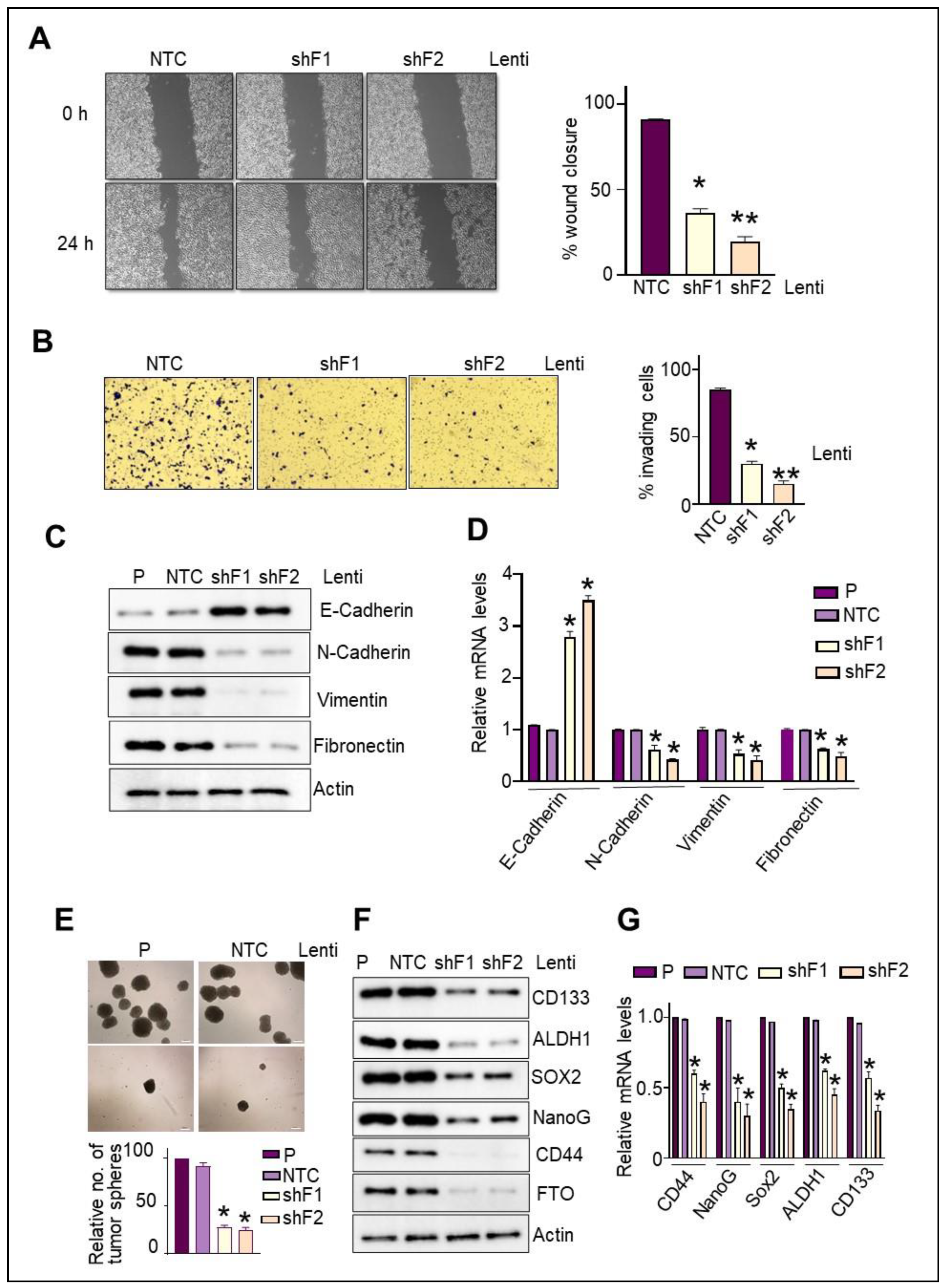
3.7. FTO Is Required for the Migratory and Invasive Aptitude of Pancreatic Cancer Cells
3.8. FTO Is Required for the Maintenance of Stemness Traits and Self-Renewal Potential of Pancreatic CSCs
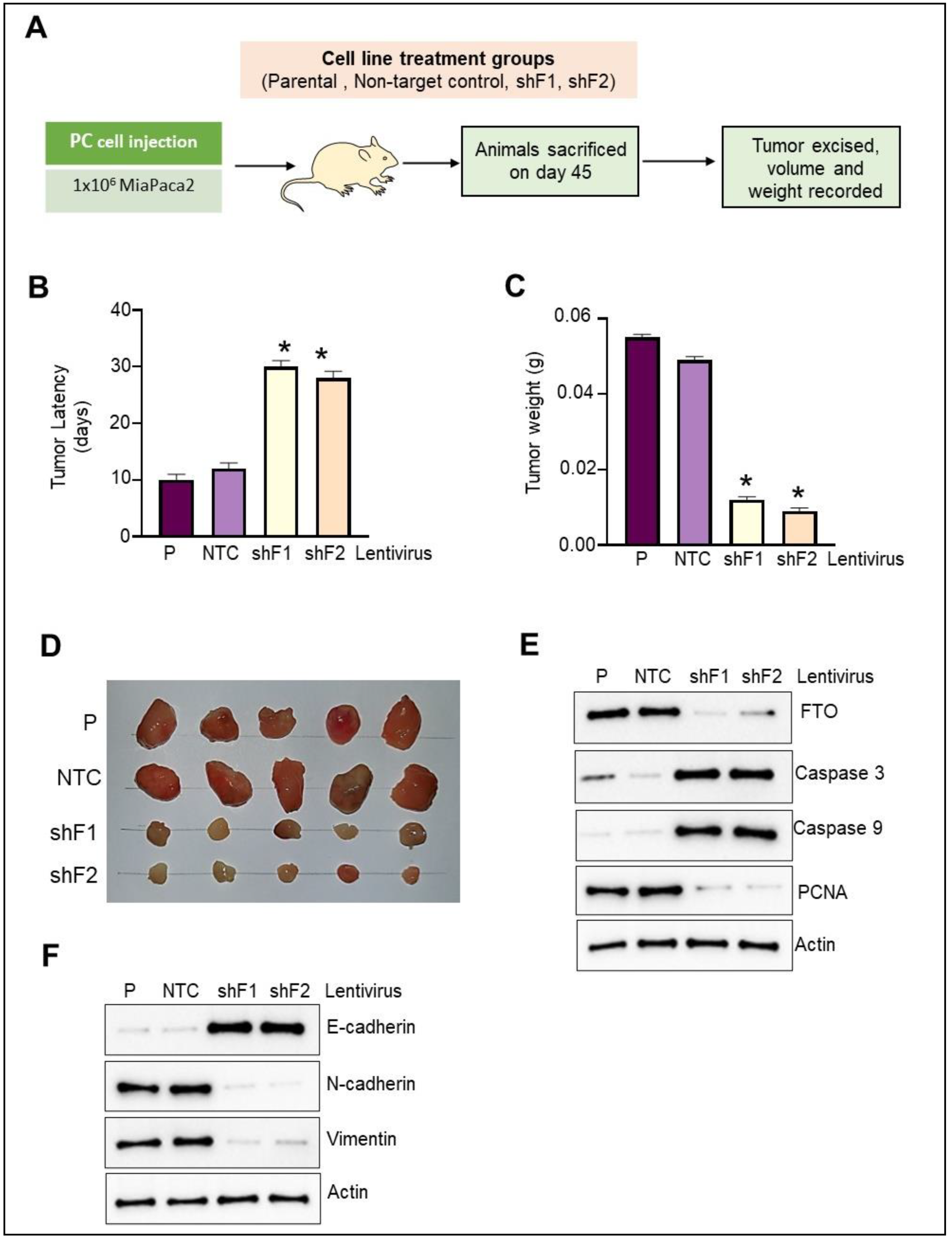
3.9. FTO Depletion Inhibits Pancreatic Cancer Cell Xenograft Growth
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- da Costa, W.L., Jr.; Oluyomi, A.O.; Thrift, A.P. Trends in the Incidence of Pancreatic Adenocarcinoma in All 50 United States Examined Through an Age-Period-Cohort Analysis. JNCI Cancer Spectr. 2020, 4, pkaa033. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Y.; Ahmad, A.; Banerjee, S.; Azmi, A.S.; Kong, D.; Sarkar, F.H. Pancreatic cancer: Understanding and overcoming chemoresistance. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Luo, G.; Gong, Y.; Xu, H.; Qian, Y.; Deng, S.; Huang, Q.; Yang, C.; Cheng, H.; Jin, K.; et al. Prognostic Value of the C-Reactive Protein/Lymphocyte Ratio in Pancreatic Cancer. Ann. Surg. Oncol. 2020, 27, 4017–4025. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Nanta, R.; Sharma, J.; Gunewardena, S.; Singh, K.P.; Shankar, S.; Srivastava, R.K. PI3K/AKT/mTOR and sonic hedgehog pathways cooperate together to inhibit human pancreatic cancer stem cell characteristics and tumor growth. Oncotarget 2015, 6, 32039–32060. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Xu, X.Y. PI3K/Akt/mTOR signaling pathway in cancer stem cells: From basic research to clinical application. Am. J. Cancer Res. 2015, 5, 1602–1609. [Google Scholar] [PubMed]
- Floor, S.; van Staveren, W.C.; Larsimont, D.; Dumont, J.E.; Maenhaut, C. Cancer cells in epithelial-to-mesenchymal transition and tumor-propagating-cancer stem cells: Distinct, overlapping or same populations. Oncogene 2011, 30, 4609–4621. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [Green Version]
- Lonardo, E.; Hermann, P.C.; Heeschen, C. Pancreatic cancer stem cells—Update and future perspectives. Mol. Oncol. 2010, 4, 431–442. [Google Scholar] [CrossRef] [Green Version]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef]
- Chen, X.Y.; Zhang, J.; Zhu, J.S. The role of m(6)A RNA methylation in human cancer. Mol. Cancer 2019, 18, 103. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Zhou, K.I.; Parisien, M.; Dai, Q.; Diatchenko, L.; Pan, T. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic. Acids Res. 2017, 45, 6051–6063. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Lv, J.; Yu, H.; Han, J.; Yang, X.; Feng, D.; Wu, Q.; Yuan, B.; Lu, Q.; Yang, H. Mechanism of RNA modification N6-methyladenosine in human cancer. Mol. Cancer 2020, 19, 104. [Google Scholar] [CrossRef]
- Wei, J.; Liu, F.; Lu, Z.; Fei, Q.; Ai, Y.; He, P.C.; Shi, H.; Cui, X.; Su, R.; Klungland, A.; et al. Differential m(6)A, m(6)Am, and m(1)A Demethylation Mediated by FTO in the Cell Nucleus and Cytoplasm. Mol. Cell 2018, 71, 973–985.e975. [Google Scholar] [CrossRef] [Green Version]
- Han, S.H.; Choe, J. Diverse molecular functions of m(6)A mRNA modification in cancer. Exp. Mol. Med. 2020, 52, 738–749. [Google Scholar] [CrossRef]
- Zhang, J.; Cheng, X.; Wang, J.; Huang, Y.; Yuan, J.; Guo, D. Gene signature and prognostic merit of M6a regulators in colorectal cancer. Exp. Biol. Med. 2020, 245, 1344–1354. [Google Scholar] [CrossRef]
- Kandimalla, R.; Gao, F.; Li, Y.; Huang, H.; Ke, J.; Deng, X.; Zhao, L.; Zhou, S.; Goel, A.; Wang, X. RNAMethyPro: A biologically conserved signature of N6-methyladenosine regulators for predicting survival at pan-cancer level. NPJ Precis Oncol. 2019, 3, 13. [Google Scholar] [CrossRef] [Green Version]
- Yue, B.; Song, C.; Yang, L.; Cui, R.; Cheng, X.; Zhang, Z.; Zhao, G. METTL3-mediated N6-methyladenosine modification is critical for epithelial-mesenchymal transition and metastasis of gastric cancer. Mol. Cancer 2019, 18, 142. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Zhao, B.S.; Zhou, A.; Lin, K.; Zheng, S.; Lu, Z.; Chen, Y.; Sulman, E.P.; Xie, K.; Bogler, O.; et al. m(6)A Demethylase ALKBH5 Maintains Tumorigenicity of Glioblastoma Stem-like Cells by Sustaining FOXM1 Expression and Cell Proliferation Program. Cancer Cell 2017, 31, 591–606.e596. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.Z.; Yang, F.; Zhou, C.C.; Liu, F.; Yuan, J.H.; Wang, F.; Wang, T.T.; Xu, Q.G.; Zhou, W.P.; Sun, S.H. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N(6) -methyladenosine-dependent primary MicroRNA processing. Hepatology 2017, 65, 529–543. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, M.; Ge, S.; Huang, W.; Lin, X.; Gao, J.; Gong, J.; Shen, L. Reduced m6A modification predicts malignant phenotypes and augmented Wnt/PI3K-Akt signaling in gastric cancer. Cancer Med. 2019, 8, 4766–4781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwok, C.T.; Marshall, A.D.; Rasko, J.E.; Wong, J.J. Genetic alterations of m(6)A regulators predict poorer survival in acute myeloid leukemia. J. Hematol. Oncol. 2017, 10, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Weng, H.; Su, R.; Weng, X.; Zuo, Z.; Li, C.; Huang, H.; Nachtergaele, S.; Dong, L.; Hu, C.; et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N(6)-Methyladenosine RNA Demethylase. Cancer Cell 2017, 31, 127–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, A.; Dang, Y.; Chen, G.; Mo, Z. Overexpression of the fat mass and obesity associated gene (FTO) in breast cancer and its clinical implications. Int. J. Clin. Exp. Pathol. 2015, 8, 13405–13410. [Google Scholar] [PubMed]
- Sigurdson, A.J.; Brenner, A.V.; Roach, J.A.; Goudeva, L.; Muller, J.A.; Nerlich, K.; Reiners, C.; Schwab, R.; Pfeiffer, L.; Waldenberger, M.; et al. Selected single-nucleotide polymorphisms in FOXE1, SERPINA5, FTO, EVPL, TICAM1 and SCARB1 are associated with papillary and follicular thyroid cancer risk: Replication study in a German population. Carcinogenesis 2016, 37, 677–684. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Shao, W.; Jiang, Y.; Wang, X.; Liu, Y.; Liu, X. FTO expression is associated with the occurrence of gastric cancer and prognosis. Oncol. Rep. 2017, 38, 2285–2292. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Shen, J.; Gao, L.; Feng, Y. Estrogen promotes fat mass and obesity-associated protein nuclear localization and enhances endometrial cancer cell proliferation via the mTOR signaling pathway. Oncol Rep. 2016, 35, 2391–2397. [Google Scholar] [CrossRef] [Green Version]
- Cui, Q.; Shi, H.; Ye, P.; Li, L.; Qu, Q.; Sun, G.; Sun, G.; Lu, Z.; Huang, Y.; Yang, C.G.; et al. m(6)A RNA Methylation Regulates the Self-Renewal and Tumorigenesis of Glioblastoma Stem Cells. Cell Rep. 2017, 18, 2622–2634. [Google Scholar] [CrossRef]
- Lin, Y.; Ueda, J.; Yagyu, K.; Ishii, H.; Ueno, M.; Egawa, N.; Nakao, H.; Mori, M.; Matsuo, K.; Kikuchi, S. Association between variations in the fat mass and obesity-associated gene and pancreatic cancer risk: A case-control study in Japan. BMC Cancer 2013, 13, 337. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, K.; Toden, S.; Ravindranathan, P.; Han, H.; Goel, A. Curcumin sensitizes pancreatic cancer cells to gemcitabine by attenuating PRC2 subunit EZH2, and the lncRNA PVT1 expression. Carcinogenesis 2017, 38, 1036–1046. [Google Scholar] [CrossRef]
- Garg, R.; Blando, J.M.; Perez, C.J.; Lal, P.; Feldman, M.D.; Smyth, E.M.; Ricciotti, E.; Grosser, T.; Benavides, F.; Kazanietz, M.G. COX-2 mediates pro-tumorigenic effects of PKCepsilon in prostate cancer. Oncogene 2018, 37, 4735–4749. [Google Scholar] [CrossRef] [PubMed]
- Shimura, T.; Sharma, P.; Sharma, G.G.; Banwait, J.K.; Goel, A. Enhanced anti-cancer activity of andrographis with oligomeric proanthocyanidins through activation of metabolic and ferroptosis pathways in colorectal cancer. Sci. Rep. 2021, 11, 7548. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, C.; Goel, A. Andrographis overcomes 5-fluorouracil-associated chemoresistance through inhibition of DKK1 in colorectal cancer. Carcinogenesis 2021, 42, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Su, R.; Dong, L.; Li, Y.; Gao, M.; Han, L.; Wunderlich, M.; Deng, X.; Li, H.; Huang, Y.; Gao, L.; et al. Targeting FTO Suppresses Cancer Stem Cell Maintenance and Immune Evasion. Cancer Cell 2020, 38, 79–96.e11. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Dominissini, D.; Rechavi, G.; He, C. Gene expression regulation mediated through reversible m(6)A RNA methylation. Nat. Rev. Genet. 2014, 15, 293–306. [Google Scholar] [CrossRef]
- Zhao, B.S.; Roundtree, I.A.; He, C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 2017, 18, 31–42. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Li, H.; Wu, A.; Peng, Y.; Shu, G.; Yin, G. Functions of N6-methyladenosine and its role in cancer. Mol. Cancer 2019, 18, 176. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Cheng, L.; He, S.; Li, W.; Zhou, C.; Zhou, B.; Liu, J.; Xu, J.; Yu, X.; Zhu, H. Multiomics integrative analysis for gene signatures and prognostic values of m(6)A regulators in pancreatic adenocarcinoma: A retrospective study in The Cancer Genome Atlas project. Aging 2020, 12, 20587–20610. [Google Scholar] [CrossRef]
- Meng, Z.; Yuan, Q.; Zhao, J.; Wang, B.; Li, S.; Offringa, R.; Jin, X.; Wu, H. The m(6)A-Related mRNA Signature Predicts the Prognosis of Pancreatic Cancer Patients. Mol. Ther. Oncolytics. 2020, 17, 460–470. [Google Scholar] [CrossRef]
- Zhou, S.; Bai, Z.L.; Xia, D.; Zhao, Z.J.; Zhao, R.; Wang, Y.Y.; Zhe, H. FTO regulates the chemo-radiotherapy resistance of cervical squamous cell carcinoma (CSCC) by targeting beta-catenin through mRNA demethylation. Mol. Carcinog. 2018, 57, 590–597. [Google Scholar] [CrossRef]
- Tang, X.; Liu, S.; Chen, D.; Zhao, Z.; Zhou, J. The role of the fat mass and obesity-associated protein in the proliferation of pancreatic cancer cells. Oncol. Lett. 2019, 17, 2473–2478. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Wu, D.; Ning, J.; Liu, W.; Zhang, D. Changes of N6-methyladenosine modulators promote breast cancer progression. BMC Cancer 2019, 19, 326. [Google Scholar] [CrossRef] [Green Version]
- Ruan, D.-Y.; Li, T.; Wang, Y.; Meng, Q.; Li, Y.; Yu, K.; Wang, M.; Lin, J.; Luo, L.; Wang, D.-S.; et al. FTO downregulation mediated by hypoxia facilitates colorectal cancer metastasis. Oncogene 2021, 40, 5168–5181. [Google Scholar] [CrossRef]
- Yang, X.; Shao, F.; Guo, D.; Wang, W.; Wang, J.; Zhu, R.; Gao, Y.; He, J.; Lu, Z. WNT/beta-catenin-suppressed FTO expression increases m(6)A of c-Myc mRNA to promote tumor cell glycolysis and tumorigenesis. Cell Death Dis. 2021, 12, 462. [Google Scholar] [CrossRef]
- Yi, W.; Yu, Y.; Li, Y.; Yang, J.; Gao, S.; Xu, L. The tumor-suppressive effects of alpha-ketoglutarate-dependent dioxygenase FTO via N6-methyladenosine RNA methylation on bladder cancer patients. Bioengineered 2021, 12, 5323–5333. [Google Scholar] [CrossRef]
- Wen, L.; Pan, X.; Yu, Y.; Yang, B. Down-regulation of FTO promotes proliferation and migration, and protects bladder cancer cells from cisplatin-induced cytotoxicity. BMC Urology 2020, 20, 39. [Google Scholar] [CrossRef]
- Huang, Y.; Su, R.; Sheng, Y.; Dong, L.; Dong, Z.; Xu, H.; Ni, T.; Zhang, Z.S.; Zhang, T.; Li, C.; et al. Small-Molecule Targeting of Oncogenic FTO Demethylase in Acute Myeloid Leukemia. Cancer Cell 2019, 35, 677–691.e610. [Google Scholar] [CrossRef] [Green Version]
- Hirayama, M.; Wei, F.Y.; Chujo, T.; Oki, S.; Yakita, M.; Kobayashi, D.; Araki, N.; Takahashi, N.; Yoshida, R.; Nakayama, H.; et al. FTO Demethylates Cyclin D1 mRNA and Controls Cell-Cycle Progression. Cell Rep. 2020, 31, 107464. [Google Scholar] [CrossRef]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [Green Version]
- Georgakopoulos-Soares, I.; Chartoumpekis, D.V.; Kyriazopoulou, V.; Zaravinos, A. EMT Factors and Metabolic Pathways in Cancer. Front. Oncol. 2020, 10, 499. [Google Scholar] [CrossRef]
- Ribatti, D.; Tamma, R.; Annese, T. Epithelial-Mesenchymal Transition in Cancer: A Historical Overview. Transl. Oncol. 2020, 13, 100773. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Aznar, E.; Wiesmuller, L.; Sainz, B., Jr.; Hermann, P.C. EMT and Stemness-Key Players in Pancreatic Cancer Stem Cells. Cancers 2019, 11, 1136. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garg, R.; Melstrom, L.; Chen, J.; He, C.; Goel, A. Targeting FTO Suppresses Pancreatic Carcinogenesis via Regulating Stem Cell Maintenance and EMT Pathway. Cancers 2022, 14, 5919. https://doi.org/10.3390/cancers14235919
Garg R, Melstrom L, Chen J, He C, Goel A. Targeting FTO Suppresses Pancreatic Carcinogenesis via Regulating Stem Cell Maintenance and EMT Pathway. Cancers. 2022; 14(23):5919. https://doi.org/10.3390/cancers14235919
Chicago/Turabian StyleGarg, Rachana, Laleh Melstrom, Jianjun Chen, Chuan He, and Ajay Goel. 2022. "Targeting FTO Suppresses Pancreatic Carcinogenesis via Regulating Stem Cell Maintenance and EMT Pathway" Cancers 14, no. 23: 5919. https://doi.org/10.3390/cancers14235919
APA StyleGarg, R., Melstrom, L., Chen, J., He, C., & Goel, A. (2022). Targeting FTO Suppresses Pancreatic Carcinogenesis via Regulating Stem Cell Maintenance and EMT Pathway. Cancers, 14(23), 5919. https://doi.org/10.3390/cancers14235919