
PKCβ Facilitates Leukemogenesis in Chronic Lymphocytic Leukaemia by Promoting Constitutive BCR-Mediated Signalling
 , , and
, , and
Abstract
Simple Summary
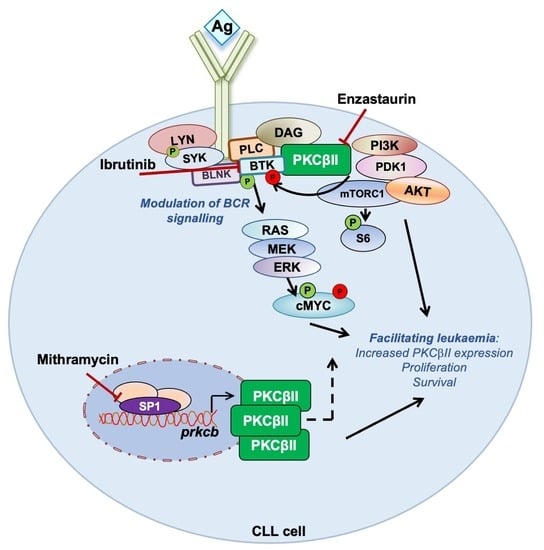
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Primary Cells and Cell Lines
2.2. Surface and Intracellular Staining and Flow Cytometric Analysis
2.3. Cell Stimulation/Drug Treatment
2.4. Cell Counting Beads
2.5. Proliferation/Apoptosis Assays
2.6. Lentiviral Knockdown of prkcb
2.7. qPCR
2.8. Chromatin Immunoprecipitation (ChIP)
2.9. Microarrays
2.10. Western Blots
2.11. Migration Assay
2.12. Statistics
3. Results
3.1. PKCβ Promotes Leukemogenesis in the PKCα-KR CLL-like Mouse Model
3.2. Sp1 Regulates Similar Transcriptional Networks in the PKCα-KR CLL-like Cells and Primary Human CLL Samples
3.3. BCR Signalling Components Are Dysregulated in PKCα-KR CLL-like Cells
3.4. PKCα-KR CLL-like and Primary Human CLL Cells Share Similar Responses to BCR-Targeted Inhibitors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stevenson, F.K.; Forconi, F.; Packham, G. The meaning and relevance of B-cell receptor structure and function in chronic lymphocytic leukemia. Semin. Hematol. 2014, 51, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Hamblin, T.J.; Davis, Z.; Gardiner, A.; Oscier, D.G.; Stevenson, F.K. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999, 94, 1848–1854. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Chiorazzi, N. B cell receptor signaling in chronic lymphocytic leukemia. Trends Immunol. 2013, 34, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Slupsky, J.R. Does B cell receptor signaling in chronic lymphocytic leukemia cells differ from that in other B cell types. Scientifica 2014, 2014, 208928. [Google Scholar] [CrossRef]
- Park, H.; Wahl, M.I.; Afar, D.E.; Turck, C.W.; Rawlings, D.J.; Tam, C.; Scharenberg, A.M.; Kinet, J.P.; Witte, O.N. Regulation of Btk function by a major autophosphorylation site within the SH3 domain. Immunity 1996, 4, 515–525. [Google Scholar] [CrossRef]
- Michie, A.M.; Nakagawa, R. Elucidating the role of protein kinase C in chronic lymphocytic leukaemia. Hematol. Oncol. 2006, 24, 134–138. [Google Scholar] [CrossRef]
- Shinohara, H.; Yasuda, T.; Aiba, Y.; Sanjo, H.; Hamadate, M.; Watarai, H.; Sakurai, H.; Kurosaki, T. PKCbeta regulates BCR-mediated IKK activation by facilitating the interaction between TAK1 and CARMA1. J. Exp. Med. 2005, 202, 1423–1431. [Google Scholar] [CrossRef]
- Abrams, S.T.; Lakum, T.; Lin, K.; Jones, G.M.; Treweeke, A.T.; Farahani, M.; Hughes, M.; Zuzel, M.; Slupsky, J.R. B-cell receptor signaling in chronic lymphocytic leukemia cells is regulated by overexpressed active protein kinase CbetaII. Blood 2007, 109, 1193–1201. [Google Scholar] [CrossRef][Green Version]
- zum Büschenfelde, C.M.; Wagner, M.; Lutzny, G.; Oelsner, M.; Feuerstacke, Y.; Decker, T.; Bogner, C.; Peschel, C.; Ringshausen, I. Recruitment of PKC-betaII to lipid rafts mediates apoptosis-resistance in chronic lymphocytic leukemia expressing ZAP-70. Leukemia 2010, 24, 141–152. [Google Scholar] [CrossRef]
- Holler, C.; Piñón, J.D.; Denk, U.; Heyder, C.; Hofbauer, S.; Greil, R.; Egle, A. PKCbeta is essential for the development of chronic lymphocytic leukemia in the TCL1 transgenic mouse model: Validation of PKCbeta as a therapeutic target in chronic lymphocytic leukemia. Blood 2009, 113, 2791–2794. [Google Scholar] [CrossRef]
- Nakagawa, R.; Soh, J.W.; Michie, A.M. Subversion of PKCα signaling in hematopoietic progenitor cells results in the generation of a B-CLL-like population in vivo. Cancer Res. 2006, 66, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, R.; Vukovic, M.; Tarafdar, A.; Cosimo, E.; Dunn, K.; McCaig, A.M.; Holroyd, A.; McClanahan, F.; Ramsay, A.G.; Gribben, J.G.; et al. Generation of a poor prognostic chronic lymphocytic leukemia-like disease model: PKCα subversion induces an upregulation of PKCβII expression in B lymphocytes. Haematologica 2015, 100, 499–510. [Google Scholar] [CrossRef] [PubMed]
- McCaig, A.M.; Cosimo, E.; Leach, M.T.; Michie, A.M. Dasatinib inhibits B cell receptor signalling in chronic lymphocytic leukaemia but novel combination approaches are required to overcome additional pro-survival microenvironmental signals. Br. J. Haematol. 2011, 153, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Cosimo, E.; McCaig, A.M.; Carter-Brzezinski, L.J.; Wheadon, H.; Leach, M.T.; Le Ster, K.; Berthou, C.; Durieu, E.; Oumata, N.; Galons, H.; et al. Inhibition of NF-κB-mediated signaling by the cyclin-dependent kinase inhibitor CR8 overcomes prosurvival stimuli to induce apoptosis in chronic lymphocytic leukemia cells. Clin. Cancer Res. 2013, 19, 2393–2405. [Google Scholar] [CrossRef] [PubMed]
- Harris, W.J.; Huang, X.; Lynch, J.T.; Spencer, G.J.; Hitchin, J.R.; Li, Y.; Ciceri, F.; Blaser, J.G.; Greystoke, B.F.; Jordan, A.M.; et al. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell. 2012, 17, 473–487. [Google Scholar] [CrossRef]
- Tarafdar, A.; Dobbin, E.; Corrigan, P.; Freeburn, R.; Wheadon, H. Canonical Wnt signaling promotes early hematopoietic progenitor formation and erythroid specification during embryonic stem cell differentiation. PLoS ONE 2013, 8, e81030. [Google Scholar] [CrossRef]
- Al-Sanabra, O.; Duckworth, A.D.; Glenn, M.A.; Brown, B.R.; Angelillo, P.; Lee, K.; Herbert, J.; Falciani, F.; Kalakonda, N.; Slupsky, J.R. Transcriptional mechanism of vascular endothelial growth factor-induced expression of protein kinase CβII in chronic lymphocytic leukaemia cells. Sci. Rep. 2017, 7, 43228. [Google Scholar] [CrossRef]
- Ray, R.; Snyder, R.C.; Thomas, S.; Koller, C.A.; Miller, D.M. Mithramycin blocks protein binding and function of the SV40 early promoter. J. Clin. Investig. 1989, 83, 2003–2007. [Google Scholar] [CrossRef]
- de Rooij, M.F.M.; Kuil, A.; Geest, C.R.; Eldering, E.; Chang, B.Y.; Buggy, J.J.; Pals, S.T.; Spaargaren, M. The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood 2012, 119, 2590–2594. [Google Scholar] [CrossRef]
- Zupo, S.; Isnardi, L.; Megna, M.; Massara, R.; Malavasi, F.; Dono, M.; Cosulich, E.; Ferrarini, M. CD38 expression distinguishes two groups of B-cell chronic lymphocytic leukemias with difference responses to anti-IgM antibodies and propensity to apoptosis. Blood 1996, 88, 1365. [Google Scholar] [CrossRef]
- Lanham, S.; Hamblin, T.; Oscier, D.; Ibbotson, R.; Stevenson, F.; Packham, G. Differential signaling via surface IgM is associated with VH gene mutational status and CD38 expression in chronic lymphocytic leukemia. Blood 2003, 101, 1087. [Google Scholar] [CrossRef] [PubMed]
- Leitges, M.; Schmedt, C.; Guinamard, R.; Davoust, J.; Schaal, S.; Stabel, S.; Tarakhovsky, A. Immunodeficiency in protein kinase cbeta-deficient mice. Science 1996, 273, 788–791. [Google Scholar] [CrossRef] [PubMed]
- Azar, A.A.; Michie, A.M.; Tarafdar, A.; Malik, N.; Menon, G.K.; Till, K.J.; Vlatkovic, N.; Slipsky, J.R. A novel transgenic mouse strain expressing PKCβII demonstrates expansion of B1 and marginal zone B cell populations. Sci. Rep. 2020, 10, 13156. [Google Scholar] [CrossRef]
- Pfeifhofer, C.; Gruber, T.; Letschka, T.; Thuille, N.; Lutz-Nicoladoni, C.; Hermann-Kleiter, N.; Braun, U.; Leitges, M.; Baier, G. Defective Ig2a/2b class switching in PKC alpha-/- mice. J. Immunol. 2006, 176, 6004. [Google Scholar] [CrossRef]
- Gökmen-Polar, Y.; Murray, N.R.; Velasco, M.A.; Gatalica, Z.; Fields, A.P. Elevated protein kinase C betaII is an early promotive event in colon carcinogenesis. Cancer Res. 2001, 61, 1375–1381. [Google Scholar] [PubMed]
- Lutzny, G.; Kocher, T.; Schmidt-Supprian, M.; Rudelius, M.; Klein-Hitpass, L.; Finch, A.J.; Duris, J.; Wagner, M.; Haferlach, C.; Seifert, M.; et al. Protein kinase c-β-dependent activation of NF-κB in stromal cells is indispensable for the survival of chronic lymphocytic leukemia B cells in vivo. Cancer Cell. 2013, 23, 77–92. [Google Scholar] [CrossRef]
- Park, E.; Chen, J.; Moore, A.; Mangolini, M.; Santoro, A.; Boyd, J.R.; Schjerven, H.; Ecker, V.; Buchner, M.; Williamson, J.C.; et al. Stromal cell protein kinase C-β inhibition enhances chemosensitivity in B cell malignancies and overcomes drug resistance. Sci. Transl. Med. 2020, 12, eaax9340. [Google Scholar] [CrossRef]
- Murray, N.R.; Davidson, L.A.; Chapkin, R.S.; Clay Gustafson, W.; Schattenberg, D.G.; Fields, A.P. Overexpression of protein kinase C betaII induces colonic hyperproliferation and increased sensitivity to colon carcinogenesis. J. Cell Biol. 1999, 145, 699–711. [Google Scholar] [CrossRef]
- Nguyen, P.H.; Fedorchenko, O.; Rosen, N.; Koch, M.; Barthel, R.; Winarski, T.; Florin, A.; Wunderlich, F.T.; Reinary, N.; Hallek, M. LYN kinase in the tumor microenvironment is essential for the progression of chronic lymphocytic leukemia. Cancer Cell. 2016, 30, 610–622. [Google Scholar] [CrossRef]
- Wu, W.; Zhu, H.; Fu, Y.; Shen, W.; Miao, K.; Hong, M.; Xiu, W.; Fan, L.; Young, K.H.; Liu, P.; et al. High LEF1 expression predicts adverse prognosis in chronic lymphocytic leukemia and may be targeted by ethacrynic acid. Oncotarget 2016, 7, 21631–21643. [Google Scholar] [CrossRef]
- Chiang, C.L.; Goswami, S.; Frissora, F.W.; Xie, Z.; Yan, P.S.; Bundschuh, R.; Walker, L.A.; Huang, X.; Mani, R.; Mo, X.M.; et al. ROR1-targeted delivery of miR-29b induces cell cycle arrest and therapeutic benefit in vivo in a CLL mouse model. Blood 2019, 134, 432–444. [Google Scholar] [CrossRef]
- Lorenzo-Herrero, S.; Sordo-Bahamonde, C.; Bretones, G.; Payer, Á.R.; González-Rodríguez, A.P.; González-García, E.; Perez-Escuredo, M.; Villa-Alvarez, J.; Nunez, L.E.; Moris, F.; et al. The Mithralog EC-7072 Induces Chronic Lymphocytic Leukemia Cell Death by Targeting Tonic B-Cell Receptor Signaling. Front. Immunol. 2019, 10, 2455. [Google Scholar] [CrossRef] [PubMed]
- Rivas, J.R.; Liu, Y.; Alhakeem, S.S.; Eckenrode, J.M.; Marti, F.; Collard, J.P.; Zhang, Y.; Shaaban, K.A.; Muthusamy, N.; Hilderbrandt, G.C.; et al. Interleukin-10 suppression enhances T-cell antitumor immunity and responses to checkpoint blockade in chronic lymphocytic leukemia. Leukemia, 2021; online ahead of print. [Google Scholar]
- Schleiss, C.; Carapito, R.; Fornecker, L.M.; Muller, L.; Paul, N.; Tahar, O.; Pichot, A.; Tavian, M.; Nicolae, A.; Miguet, L.; et al. Temporal multiomic modeling reveals a B-cell receptor proliferative program in chronic lymphocytic leukemia. Leukemia 2021, 35, 1463–1474. [Google Scholar] [CrossRef]
- Cosimo, E.; Tarafdar, A.; Moles, M.W.; Holroyd, A.K.; Malik, N.; Catherwood, M.A.; Hay, J.; Dunn, K.M.; Macdonald, A.M.; Guichard, S.M.; et al. AKT/mTORC2 inhibition activates FOXO1 function in CLL cells reducing B cell receptor-mediated survival. Clin. Cancer Res. 2019, 25, 1574–1587. [Google Scholar] [CrossRef]
- Wahl, M.I.; Fluckiger, A.C.; Kato, R.M.; Park, H.; Witte, O.N.; Rawlings, D.J. Phosphorylation of two regulatory tyrosine residues in the activation of Bruton’s tyrosine kinase via alternative receptors. Proc. Natl. Acad. Sci. USA 1997, 94, 11526–11533. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.R.; McNulty, A.M.; Hanna, K.R.; Konicek, B.W.; Lynch, R.L.; Bailey, S.N.; Banks, C.; Capen, A.; Goode, R.; Lewis, J.E.; et al. The protein kinase Cbeta-selective inhibitor Enzastaurin (LY317615.HCl), suppresses signalling through the AKT pathway, induces apoptosis, and suppresses growth of human colon cancer and glioblastoma xenografts. Cancer Res. 2005, 65, 7462–7469. [Google Scholar] [CrossRef]
- Kang, S.W.; Wahl, M.I.; Chu, J.; Kitaura, J.; Kawakami, Y.; Kato, R.M.; Tabuchi, R.; Tarakhovsky, A.; Kawakami, T.; Turck, C.W.; et al. PKCbeta modulates antigen receptor signaling via regulation of Btk membrane localization. EMBO J. 2001, 20, 5692–5702. [Google Scholar] [CrossRef] [PubMed]
- Coutré, S.E.; Furman, R.R.; Flinn, I.W.; Burger, J.A.; Blum, K.; Sharman, J.; Jones, J.; Wierda, W.; Zhao, W.; Heerema, N.A.; et al. Extended Treatment with Single-Agent Ibrutinib at the 420 mg Dose Leads to Durable Responses in Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma. Clin. Cancer Res. 2017, 23, 1149–1155. [Google Scholar] [CrossRef]
- Treon, S.P.; Tripsas, C.K.; Meid, K.; Warren, D.; Varma, G.; Green, R.; Argyropoulos, K.V.; Yang, G.; Cao, Y.; Xu, L.; et al. Ibrutinib in previously treated Waldenstrom’s macroglobulinemia. N. Engl. J. Med. 2015, 372, 1430–1440. [Google Scholar] [CrossRef]
- Wang, M.L.; Rule, S.; Martin, P.; Goy, A.; Auer, R.; Kahl, B.S.; Jurczak, W.; Advani, R.H.; Romaguera, J.E.; Williams, M.E.; et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N. Engl. J. Med. 2013, 369, 507–516. [Google Scholar] [CrossRef]
- Crump, M.; Leppä, S.; Fayad, L.; Lee, J.J.; Rocco, A.D.; Ogura, M.; Hagberg, H.; Schnell, F.; Rifkin, R.; Mackensen, A.; et al. Randomized, Double-Blind, Phase III Trial of Enzastaurin Versus Placebo in Patients Achieving Remission After First-Line Therapy for High-Risk Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2016, 34, 2484–2492. [Google Scholar] [CrossRef] [PubMed]
- El-Gamal, D.; Williams, K.; LaFollette, T.D.; Cannon, M.; Blachly, J.S.; Zhong, Y.; Woyach, J.A.; Williams, E.; Awan, F.T.; Jones, J.; et al. PKC-β as a therapeutic target in CLL: PKC inhibitor AEB071 demonstrates preclinical activity in CLL. Blood 2014, 124, 1481–1491. [Google Scholar] [CrossRef] [PubMed]
- Naylor, T.L.; Tang, H.; Ratsch, B.A.; Enns, A.; Loo, A.; Chen, L.; Lenz, P.; Waters, N.J.; Schuler, W.; Donken, B.; et al. Protein kinase C inhibitor sotrastaurin selectively inhibits the growth of CD79 mutant diffuse large B-cell lymphomas. Cancer Res. 2011, 71, 2643–2653. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hay, J.; Tarafdar, A.; Holroyd, A.K.; Moka, H.A.; Dunn, K.M.; Alshayeb, A.; Lloyd, B.H.; Cassels, J.; Malik, N.; Khan, A.F.; et al. PKCβ Facilitates Leukemogenesis in Chronic Lymphocytic Leukaemia by Promoting Constitutive BCR-Mediated Signalling. Cancers 2022, 14, 6006. https://doi.org/10.3390/cancers14236006
Hay J, Tarafdar A, Holroyd AK, Moka HA, Dunn KM, Alshayeb A, Lloyd BH, Cassels J, Malik N, Khan AF, et al. PKCβ Facilitates Leukemogenesis in Chronic Lymphocytic Leukaemia by Promoting Constitutive BCR-Mediated Signalling. Cancers. 2022; 14(23):6006. https://doi.org/10.3390/cancers14236006
Chicago/Turabian StyleHay, Jodie, Anuradha Tarafdar, Ailsa K. Holroyd, Hothri A. Moka, Karen M. Dunn, Alzahra Alshayeb, Bryony H. Lloyd, Jennifer Cassels, Natasha Malik, Ashfia F. Khan, and et al. 2022. "PKCβ Facilitates Leukemogenesis in Chronic Lymphocytic Leukaemia by Promoting Constitutive BCR-Mediated Signalling" Cancers 14, no. 23: 6006. https://doi.org/10.3390/cancers14236006
APA StyleHay, J., Tarafdar, A., Holroyd, A. K., Moka, H. A., Dunn, K. M., Alshayeb, A., Lloyd, B. H., Cassels, J., Malik, N., Khan, A. F., Sou, I., Lees, J., Almuhanna, H. N. B., Kalakonda, N., Slupsky, J. R., & Michie, A. M. (2022). PKCβ Facilitates Leukemogenesis in Chronic Lymphocytic Leukaemia by Promoting Constitutive BCR-Mediated Signalling. Cancers, 14(23), 6006. https://doi.org/10.3390/cancers14236006