
N6-Isopentenyladenosine Impairs Mitochondrial Metabolism through Inhibition of EGFR Translocation on Mitochondria in Glioblastoma Cells
 ,
,
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Cell Cultures and Reagents
2.2. Preparation of Glioblastoma Primary Cell Lines
2.3. Western Blot Analysis
2.4. Immunofluorescence
2.5. Mitochondrial Protein Fractionation
2.6. PUMA Depletion by RNA Interference
2.7. Primary Glioblastoma Characterization
2.8. IDH1 and IDH2 Mutation Status
2.9. MGMT Methylation Assessment
- -
- Methylated MGMT:
- forward primer 5′-TTTCGACGTTCGTAGGTTTTCGC-3′,
- reverse primer 5′-GCACTCTTCCGAAAACGAAACG-3′.
- -
- Unmethylated MGMT:
- forward primer 5′-TTTGTGTTTTGATGTTTGTAGGTTTTTGT-3′,
- reverse primer 5′-AACTCCACACTCTT CCAAAAACAAAACA-3′.
2.10. Seahorse Analysis
2.11. Cell Transfection
2.12. Statistical Analysis
3. Results
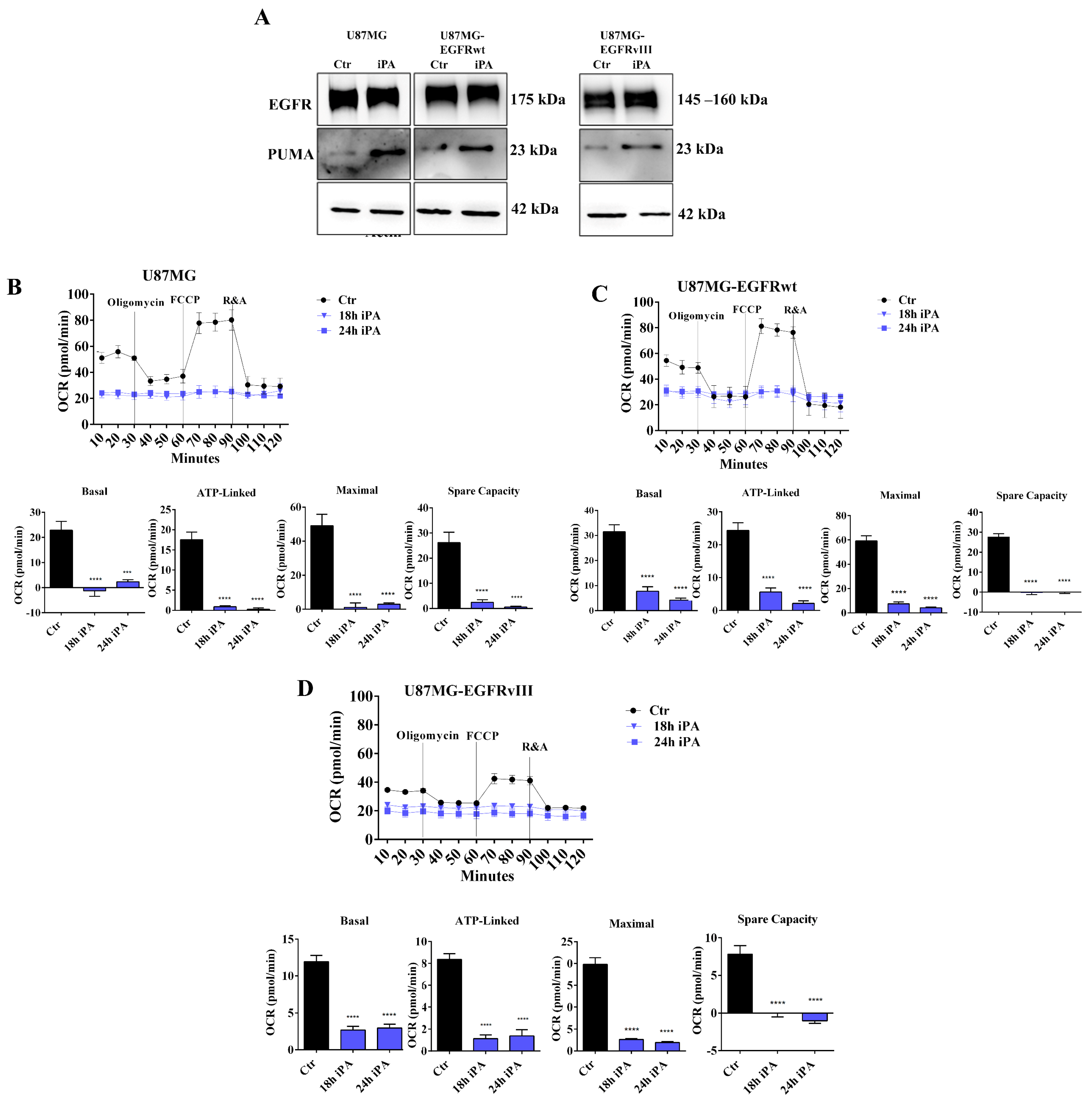
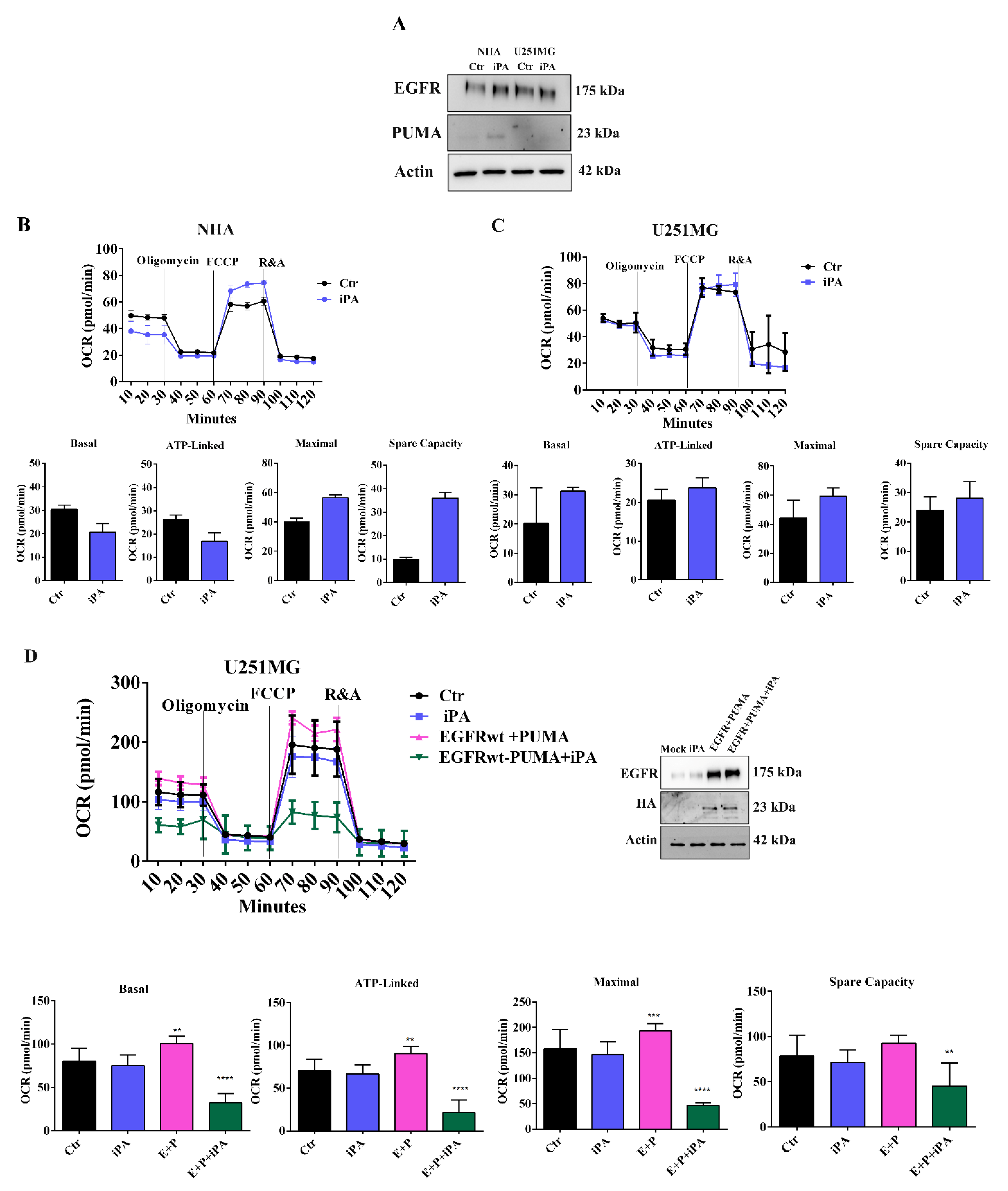
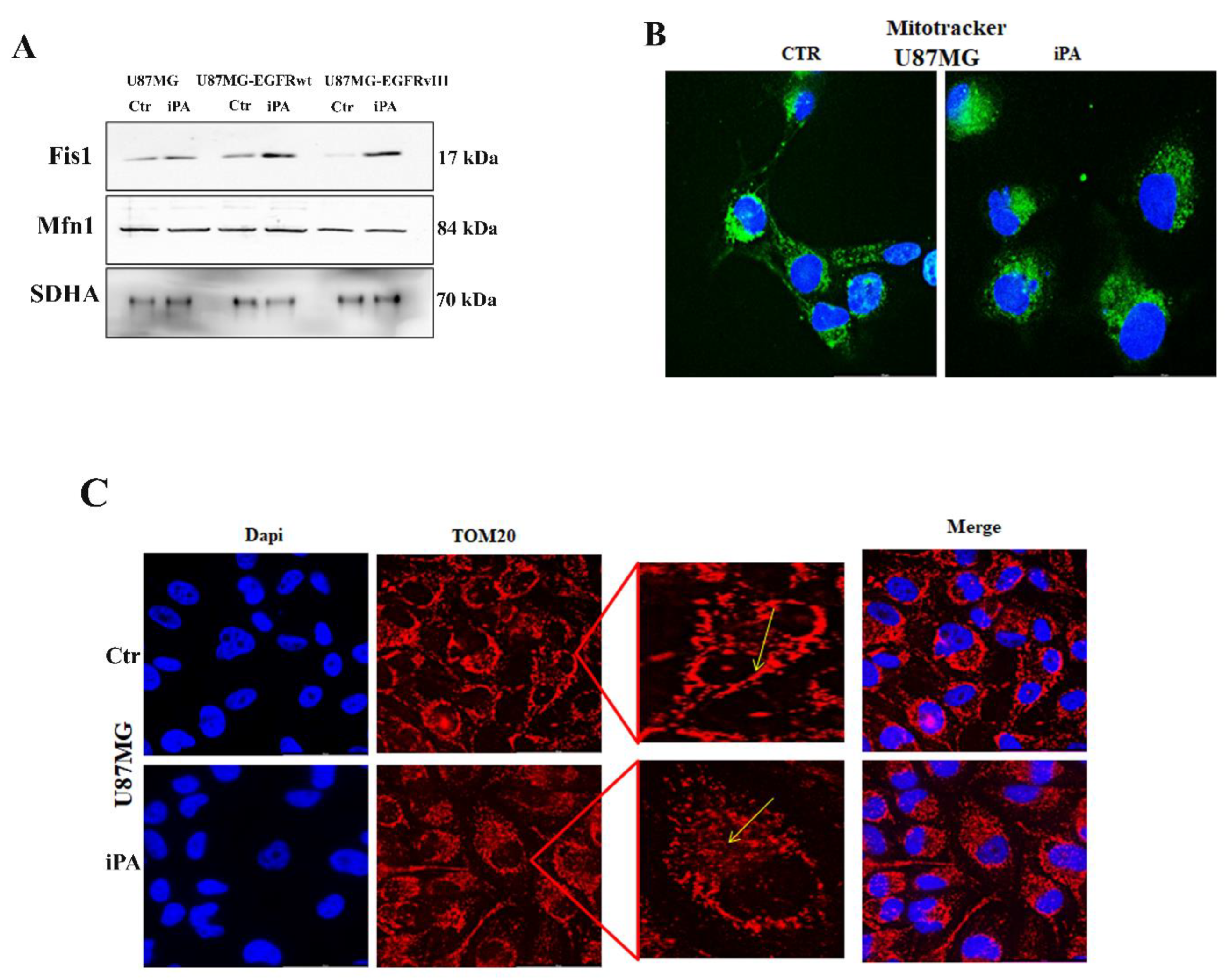
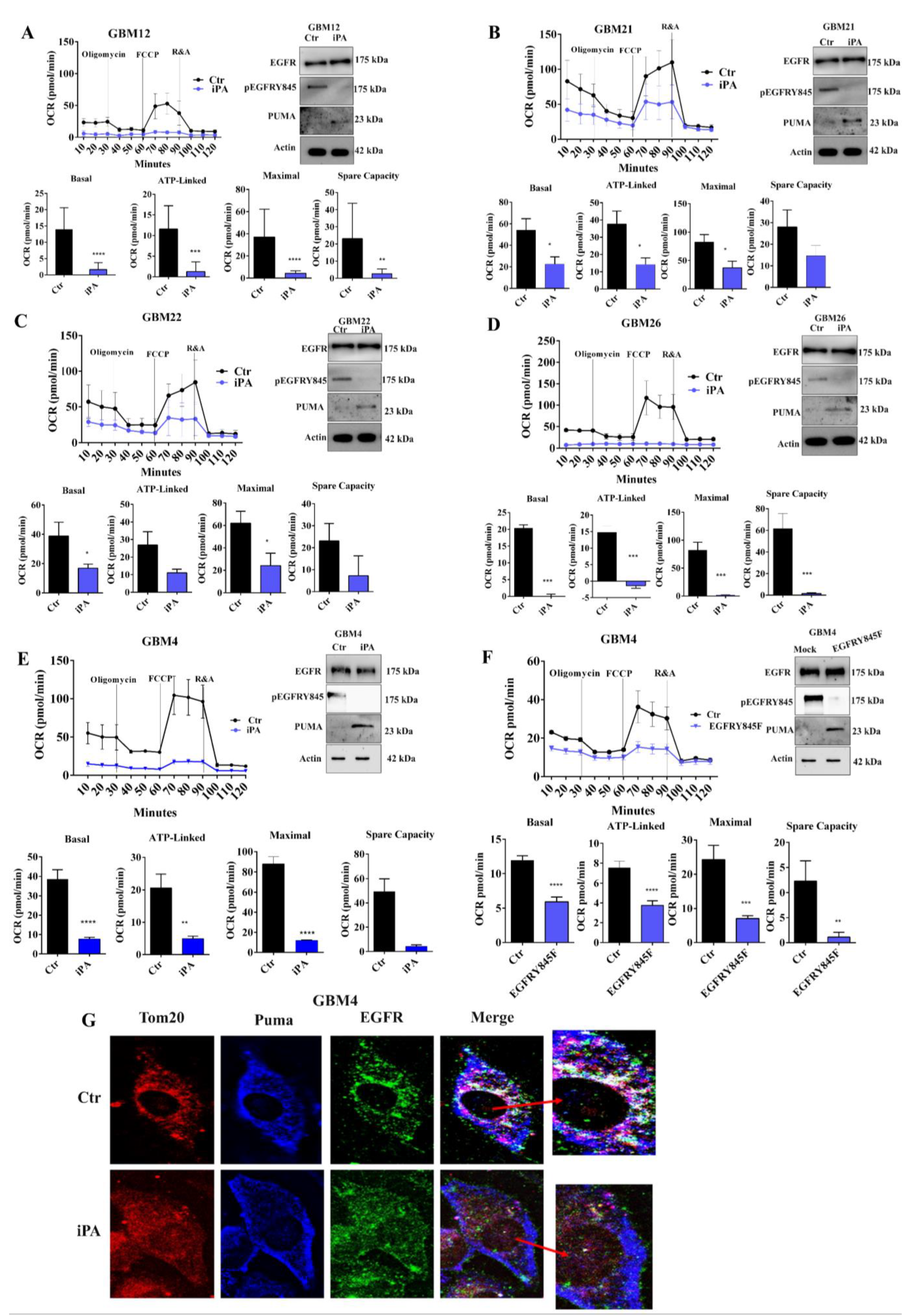
3.1. iPA Impairs the Mitochondrial Metabolism in GBM Cell Lines
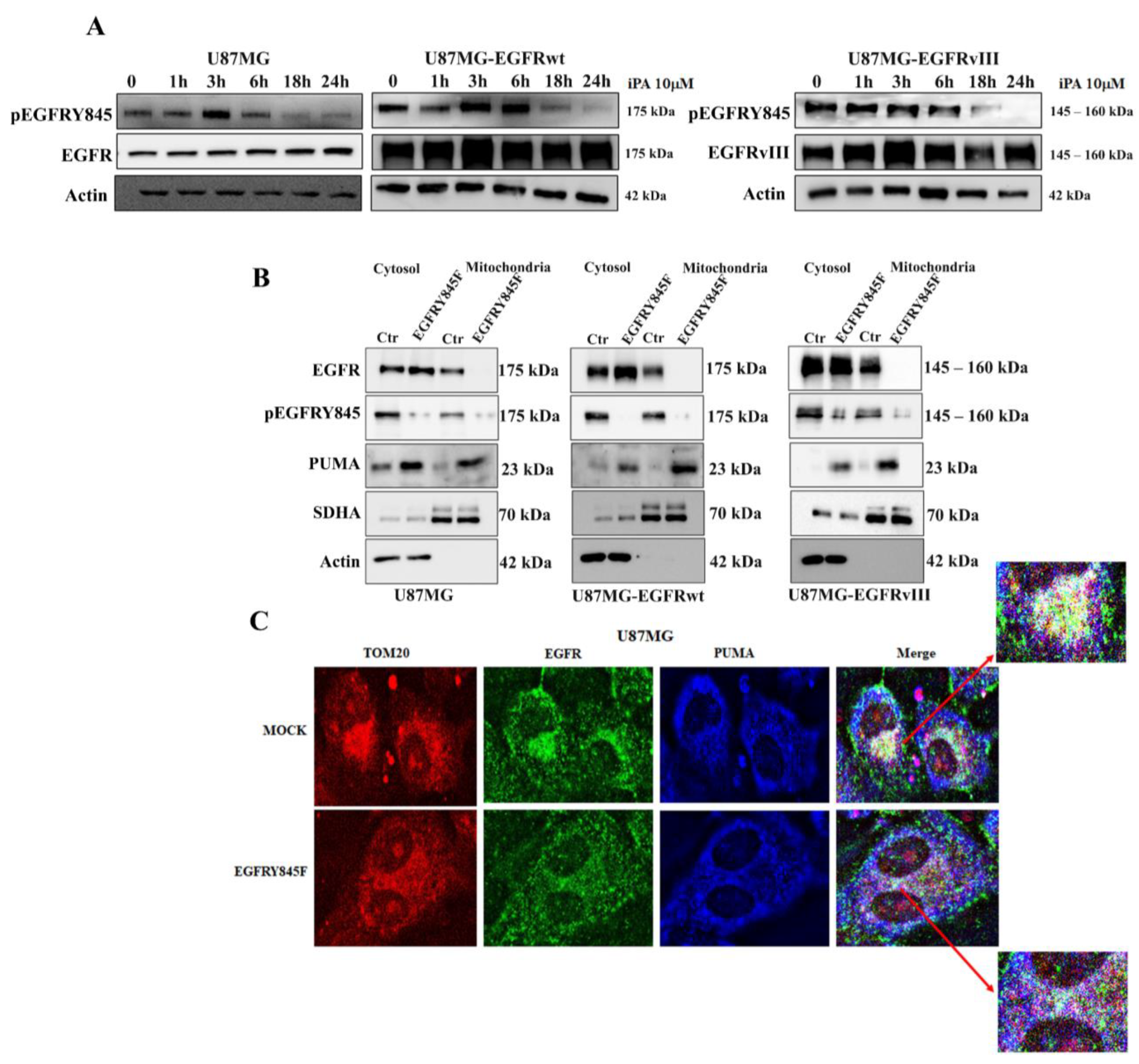
3.2. iPA Inhibits the EGFR/EGFRvIII Translocation on Mitochondria in GBM Cells
3.3. iPA Inhibits the Y845 Phosphorylation of EGFR and EGFRvIII in GBM Cells
3.4. The Y845 Phosphorylation of EGFR Is Important for Mitochondrial Activity
3.5. iPA Selectively Inhibits Mitochondrial Metabolism in GBM Primary Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cloughesy, T.F.; Cavenee, W.K.; Mischel, P.S. Glioblastoma: From molecular pathology to targeted treatment. Annu. Rev. Pathol. 2014, 9, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Karl Belanger, M.D.; Alba, A.; Brandes, M.D.; Christine Marosi, M.D.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [Green Version]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [Green Version]
- Hambardzumyan, D.; Bergers, G. Glioblastoma: Defining Tumor Niches. Trends Cancer 2015, 1, 252–265. [Google Scholar] [CrossRef] [Green Version]
- Ou, A.; Yung, W.K.A.; Majd, N. Molecular Mechanisms of Treatment Resistance in Glioblastoma. Int. J. Mol. Sci. 2020, 22, 351. [Google Scholar] [CrossRef]
- Maire, C.L.; Ligon, K.L. Molecular pathologic diagnosis of epidermal growth factor receptor. Neuro. Oncol. 2014, 16 (Suppl. S8), viii1–viii6. [Google Scholar] [CrossRef]
- Gan, H.K.; Cvrljevic, A.N.; Johns, T.G. The epidermal growth factor receptor variant III (EGFRvIII): Where wild things are altered. FEBS J. 2013, 280, 5350–5370. [Google Scholar] [CrossRef]
- Saadeh, F.S.; Mahfouz, R.; Assi, H.I. EGFR as a clinical marker in glioblastomas and other gliomas. Int. J. Biol. Markers 2018, 33, 22–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padfield, E.; Ellis, H.P.; Kurian, K.M. Current Therapeutic Advances Targeting EGFR and EGFRvIII in Glioblastoma. Front Oncol. 2015, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Che, T.F.; Lin, C.W.; Wu, Y.Y.; Chen, Y.J.; Han, C.L.; Chang, Y.L.; Wu, C.; Hsiao, T.; Hong, T.; Yang, P. Mitochondrial translocation of EGFR regulates mitochondria dynamics and promotes metastasis in NSCLC. Oncotarget 2015, 6, 37349–37366. [Google Scholar] [CrossRef]
- Boerner, J.L.; Demory, M.L.; Silva, C.; Parsons, S.J. Phosphorylation of Y845 on the epidermal growth factor receptor mediates binding to the mitochondrial protein cytochrome c oxidase subunit II. Mol. Cell. Biol. 2004, 24, 7059–7071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; You, L.; Xie, J.; Pan, H.; Han, W. The roles of subcellularly located EGFR in autophagy. Cell Signal. 2017, 35, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Cvrljevic, A.N.; Akhavan, D.; Wu, M.; Martinello, P.; Furnari, F.B.; Johnston, A.J.; Guo, D.; Pike, L.; Cavenee, W.K.; Scott, A.M.; et al. Activation of Src induces mitochondrial localisation of de2-7EGFR (EGFRvIII) in glioma cells: Implications for glucose metabolism. J. Cell Sci. 2011, 124 Pt 17, 2938–2950. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Zhu, H.; Ali-Osman, F.; Lo, H.W. EGFR and EGFRvIII undergo stress- and EGFR kinase inhibitor-induced mitochondrial translocalization: A potential mechanism of EGFR-driven antagonism of apoptosis. Mol. Cancer 2011, 10, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Cao, X.; Ali-Osman, F.; Keir, S.; Lo, H.W. EGFR and EGFRvIII interact with PUMA to inhibit mitochondrial translocalization of PUMA and PUMA-mediated apoptosis independent of EGFR kinase activity. Cancer Lett. 2010, 294, 101–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Q.; Ming, L.; Thomas, S.M.; Wang, Y.; Chen, Z.G.; Ferris, R.L.; Grandis, J.R.; Zhang, L.; Yu, J. PUMA mediates EGFR tyrosine kinase inhibitor-induced apoptosis in head and neck cancer cells. Oncogene 2009, 28, 2348–2357. [Google Scholar] [CrossRef] [Green Version]
- Ambroise, G.; Portier, A.; Roders, N.; Arnoult, D.; Vazquez, A. Subcellular localization of PUMA regulates its pro-apoptotic activity in Burkitt’s lymphoma B cells. Oncotarget 2015, 6, 38181–38194. [Google Scholar] [CrossRef] [Green Version]
- Laezza, C.; Notarnicola, M.; Caruso, M.G.; Messa, C.; Macchia, M.; Bertini, S.; Minutolo, F.; Portella, G.; Fiorentino, L.; Stingo, S.; et al. N6-isopentenyladenosine arrests tumor cell proliferation by inhibiting farnesyl diphosphate synthase and protein prenylation. Faseb J. 2006, 20, 412–418. [Google Scholar] [CrossRef]
- Pisanti, S.; Picardi, P.; Ciaglia, E.; Margarucci, L.; Ronca, R.; Giacomini, A.; Malfitano, A.M.; Casapullo, A.; Laezza, C.; Gazzerro, P.; et al. Antiangiogenic effects of N6-isopentenyladenosine, an endogenous isoprenoid end product, mediated by AMPK activation. Faseb J. 2014, 28, 1132–1144. [Google Scholar] [CrossRef] [PubMed]
- Ciaglia, E.; Pisanti, S.; Picardi, P.; Laezza, C.; Sosa, S.; Tubaro, A.; Vitale, M.; Gazzerro, P.; Malfitano, A.M.; Bifulc, M. N6-isopentenyladenosine affects cytotoxic activity and cytokines production by IL-2 activated NK cells and exerts topical anti-inflammatory activity in mice. Pharmacol. Res. 2014, 89, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Navarra, G.; Pagano, C.; Pacelli, R.; Crescenzi, E.; Longobardi, E.; Gazzerro, P.; Fiore, D.; Pastorino, O.; Pentimalli, F.; Laezza, C.; et al. N(6)-Isopentenyladenosine Enhances the Radiosensitivity of Glioblastoma Cells by Inhibiting the Homologous Recombination Repair Protein RAD51 Expression. Front. Oncol. 2019, 9, 1498. [Google Scholar] [CrossRef] [PubMed]
- Pagano, C.; Navarra, G.; Pastorino, O.; Avilia, G.; Coppola, L.; Della Monica, R.; Chiariotti, L.; Florio, T.; Corsaro, A.; Torelli, G.; et al. N6-Isopentenyladenosine Hinders the Vasculogenic Mimicry in Human Glioblastoma Cells through Src-120 Catenin Pathway Modulation and RhoA Activity Inhibition. Int. J. Mol. Sci. 2021, 22, 10530. [Google Scholar] [CrossRef] [PubMed]
- Ciaglia, E.; Abate, M.; Laezza, C.; Pisanti, S.; Vitale, M.; Seneca, V.; Torelli, G.; Franceschelli, S.; Catapano, G.; Gazzerro, P.; et al. Antiglioma effects of N6-isopentenyladenosine, an endogenous isoprenoid end product, through the downregulation of epidermal growth factor receptor. Int. J. Cancer 2017, 140, 959–972. [Google Scholar] [CrossRef] [Green Version]
- Zanca, C.; Villa, G.R.; Benitez, J.A.; Thorne, A.H.; Koga, T.; D’Antonio, M.; Ikegami, S.; Ma, J.; Boyer, A.D.; Banisadr, A.; et al. Glioblastoma cellular cross-talk converges on NF-κB to attenuate EGFR inhibitor sensitivity. Genes Dev. 2017, 31, 1212–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiestler, B.; Capper, D.; Sill, M.; Jones, D.T.; Hovestadt, V.; Sturm, D.; Koelsche, C.; Bertoni, A.; Schweizer, L.; Korshunov, A.; et al. Integrated DNA methylation and copy-number profiling identify three clinically and biologically relevant groups of anaplastic glioma. Acta Neuropathol. 2014, 128, 561–571. [Google Scholar] [CrossRef] [Green Version]
- Esteller, M.; Corn, P.G.; Baylin, S.B.; Herman, J.G. A gene hypermethylation profile of human cancer. Cancer Res. 2001, 61, 3225–3229. [Google Scholar] [PubMed]
- Carter, R.E.; Sorkin, A. Endocytosis of functional epidermal growth factor receptor-green fluorescent protein chimera. J. Biol. Chem. 1998, 273, 35000–35007. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Zhang, L.; Hwang, P.M.; Kinzler, K.W.; Vogelstein, B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol. Cell 2001, 7, 673–682. [Google Scholar] [CrossRef]
- Latha, K.; Li, M.; Chumbalkar, V.; Gururaj, A.; Hwang, Y.; Dakeng, S.; Sawaya, R.; Aldape, K.; Cavenee, W.K.; Bogler, O.; et al. Nuclear EGFRvIII-STAT5b complex contributes to glioblastoma cell survival by direct activation of the Bcl-XL promoter. Int. J. Cancer 2013, 132, 509–520. [Google Scholar] [CrossRef]
- Pagano, C.; Navarra, G.; Coppola, L.; Avilia, G.; Pastorino, O.; Della Monica, R.; Buonaiuto, M.; Torelli, G.; Caiazzo, P.; Bifulco, M.; et al. N6-isopentenyladenosine induces cell death through necroptosis in human glioblastoma cells. Cell Death Discov. 2022, 8, 173. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, N.; Liu, J.; Liu, Y.; Zhang, C.; Long, S.; Luo, G.; Zhang, L.; Zhang, Y. Mutant p53 drives cancer chemotherapy resistance due to loss of function on activating transcription of PUMA. Cell Cycle 2019, 18, 3442–3455. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Neufeld, A.H. Activation of epidermal growth factor receptors in astrocytes: From development to neural injury. J. Neurosci. Res. 2007, 85, 3523–3529. [Google Scholar] [CrossRef] [PubMed]
- Sato, K. Cellular functions regulated by phosphorylation of EGFR on Tyr845. Int. J. Mol. Sci. 2013, 14, 10761–10790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Yu, L.; Chen, W.; Xu, Y.; Wu, M.; Todorova, D.; Tang, Q.; Feng, B.; Jiang, L.; He, J.; et al. Wild-Type p53 Promotes Cancer Metabolic Switch by Inducing PUMA-Dependent Suppression of Oxidative Phosphorylation. Cancer Cell 2019, 35, 191–203.e8. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Tong, J.; Yang, L.; Wei, L.; Stolz, D.B.; Yu, J.; Zhang, J.; Zhang, L. PUMA amplifies necroptosis signaling by activating cytosolic DNA sensors. Proc. Natl. Acad. Sci. USA 2018, 115, 3930–3935. [Google Scholar] [CrossRef] [Green Version]
- Suen, D.F.; Norris, K.L.; Youle, R.J. Mitochondrial dynamics and apoptosis. Genes Dev. 2008, 22, 1577–1590. [Google Scholar] [CrossRef] [Green Version]
- Ranieri, R.; Ciaglia, E.; Amodio, G.; Picardi, P.; Proto, M.C.; Gazzerro, P.; Laezza, C.; Remondelli, P.; Bifulco, M.; Pisanti, S. N6-isopentenyladenosine dual targeting of AMPK and Rab7 prenylation inhibits melanoma growth through the impairment of autophagic flux. Cell Death Differ. 2018, 25, 353–367. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.; Fujimura, A.; Wei, F.Y.; Shinojima, N.; Kuroda, J.I.; Mukasa, A.; Tomizawa, K. 2-Methylthio Conversion of N6-Isopentenyladenosine in Mitochondrial tRNAs by CDK5RAP1 Promotes the Maintenance of Glioma-Initiating Cells. iScience 2019, 21, 42–56. [Google Scholar] [CrossRef] [Green Version]
- An, Z.; Aksoy, O.; Zheng, T.; Fan, Q.W.; Weiss, W.A. Epidermal growth factor receptor and EGFRvIII in glioblastoma: Signaling pathways and targeted therapies. Oncogene 2018, 37, 1561–1575. [Google Scholar] [CrossRef]
- Bosc, C.; Selak, M.A.; Sarry, J.E. Resistance Is Futile: Targeting Mitochondrial Energetics and Metabolism to Overcome Drug Resistance in Cancer Treatment. Cell Metab. 2017, 26, 705–707. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yuan, X.; Wang, S.; Ou, Y.; Zheng, X.; Wang, Q. The relationship between mitochondrial fusion/fission and apoptosis in the process of adipose-derived stromal cells differentiation into astrocytes. Neurosci. Lett. 2014, 575, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Patient | Type | MGMT Methylation | IDH1/IDH2 Mutation | Co-del-1p-19q | Grading | EGFR Amplification | Epigenetic Subclass | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Age | Gender | Biopsy | Primary Cell-Line | Biopsy | Primary Cell-Line | Biopsy | Biopsy | Biopsy | Biopsy | ||
| GBM4 | 64 | M | Primitive | Un-meth | Meth | Wt | Wt | No | GBM (WHO IV) | no | Mesenchymal |
| GBM12 | 60 | F | Primitive | Meth | Un-Meth | Wt | Wt | No | GBM (WHO IV) | yes | RTKII |
| GBM21 | 66 | M | Primitive | Meth | Un-Meth | Wt | Wt | No | GBM (WHO IV) | yes | RTKI |
| GBM22 | 70 | M | Primitive | Un-Meth | Un-Meth | Wt | Wt | No | GBM (WHO IV) | weak | RTKI |
| GBM26 | 50 | M | Primitive | Meth | Meth | Wt | Wt | No | GBM (WHO IV) | weak | Mesenchymal |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pagano, C.; Coppola, L.; Navarra, G.; Avilia, G.; Bruzzaniti, S.; Piemonte, E.; Galgani, M.; Della Monica, R.; Chiariotti, L.; Cuomo, M.; et al. N6-Isopentenyladenosine Impairs Mitochondrial Metabolism through Inhibition of EGFR Translocation on Mitochondria in Glioblastoma Cells. Cancers 2022, 14, 6044. https://doi.org/10.3390/cancers14246044
Pagano C, Coppola L, Navarra G, Avilia G, Bruzzaniti S, Piemonte E, Galgani M, Della Monica R, Chiariotti L, Cuomo M, et al. N6-Isopentenyladenosine Impairs Mitochondrial Metabolism through Inhibition of EGFR Translocation on Mitochondria in Glioblastoma Cells. Cancers. 2022; 14(24):6044. https://doi.org/10.3390/cancers14246044
Chicago/Turabian StylePagano, Cristina, Laura Coppola, Giovanna Navarra, Giorgio Avilia, Sara Bruzzaniti, Erica Piemonte, Mario Galgani, Rosa Della Monica, Lorenzo Chiariotti, Mariella Cuomo, and et al. 2022. "N6-Isopentenyladenosine Impairs Mitochondrial Metabolism through Inhibition of EGFR Translocation on Mitochondria in Glioblastoma Cells" Cancers 14, no. 24: 6044. https://doi.org/10.3390/cancers14246044