

Silibinin Overcomes EMT-Driven Lung Cancer Resistance to New-Generation ALK Inhibitors
 , , ,
, , ,  ,
,  , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Lines
2.3. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.4. Immunoblotting Analyses
2.5. Cell Viability Assay
2.6. Colony Formation Assays
2.7. SMAD-Binding Element Reporter Assays
2.8. Human TGFβ Array
2.9. TGFβ-Related Secretome
2.10. Docking Calculations, Molecular Dynamics Simulations, and Binding Free Energy Analysis
2.11. LanthaScreen Eu Kinase Binding Assays
2.12. Statistical Analysis
3. Results
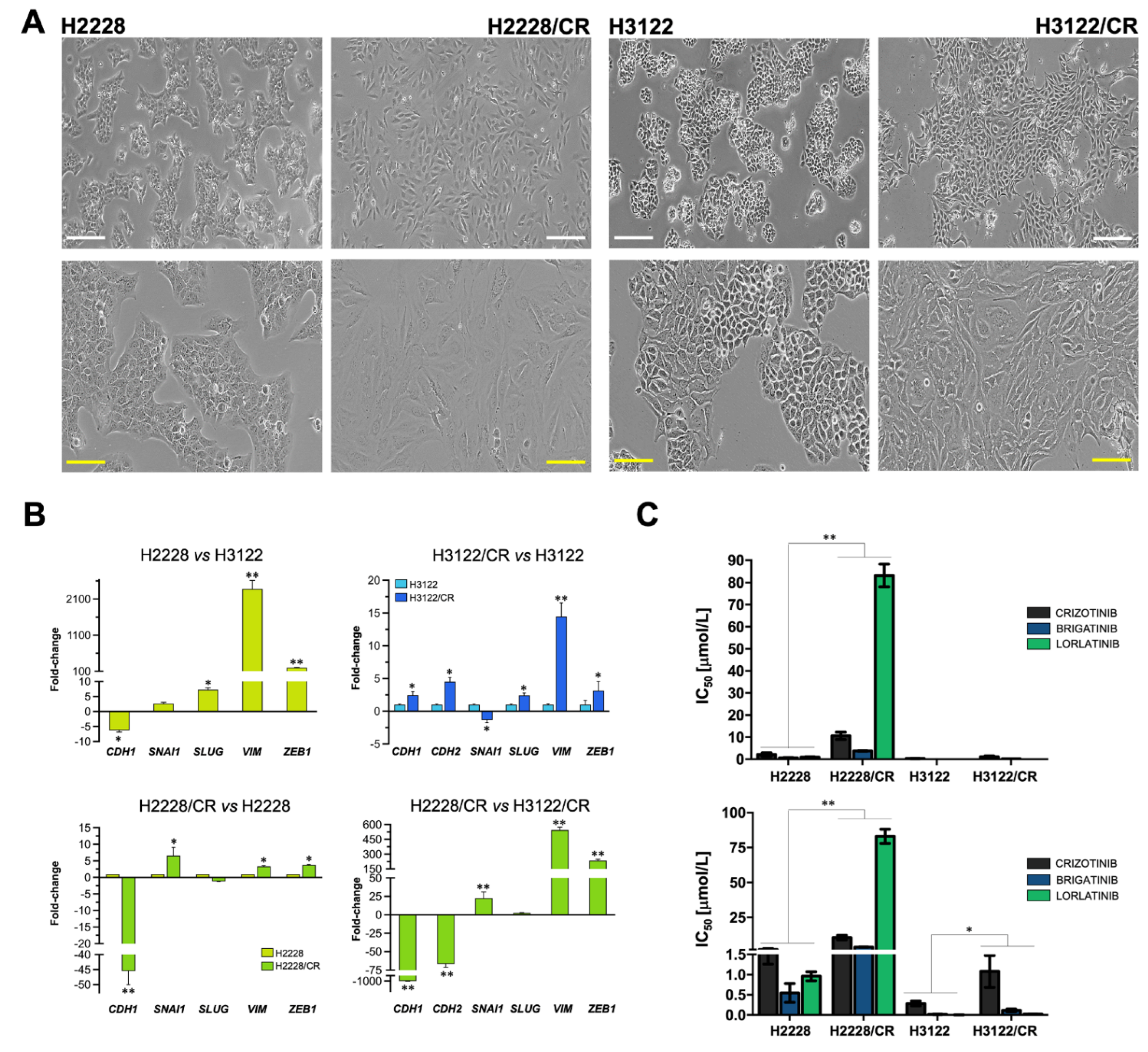
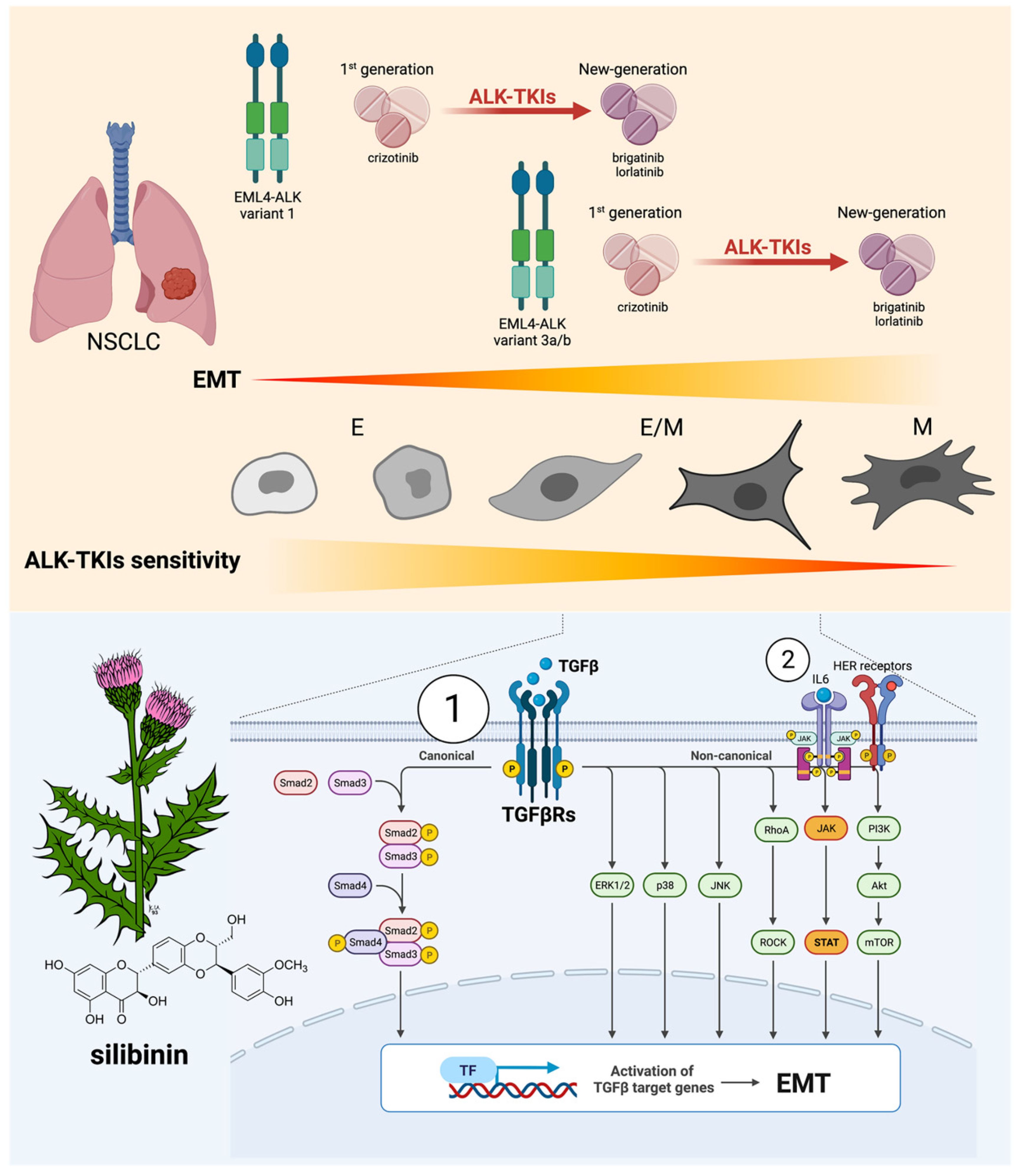
3.1. Acquisition of a Mesenchymal-Like Phenotype Promotes Cross-Resistance to First-, Second-, and Third-Generation ALK–TKIs in ALK-Rearranged NSCLC Cells
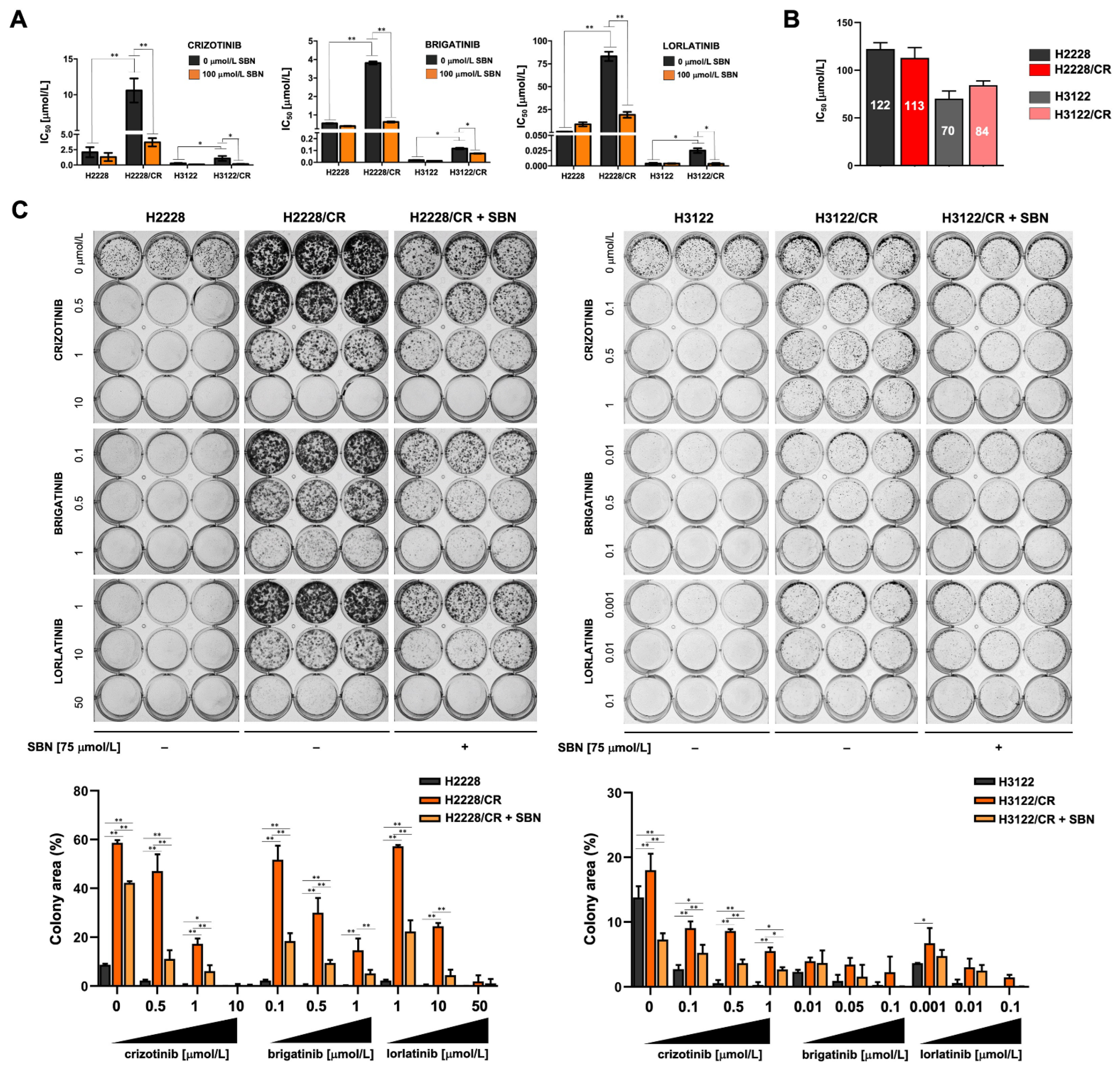
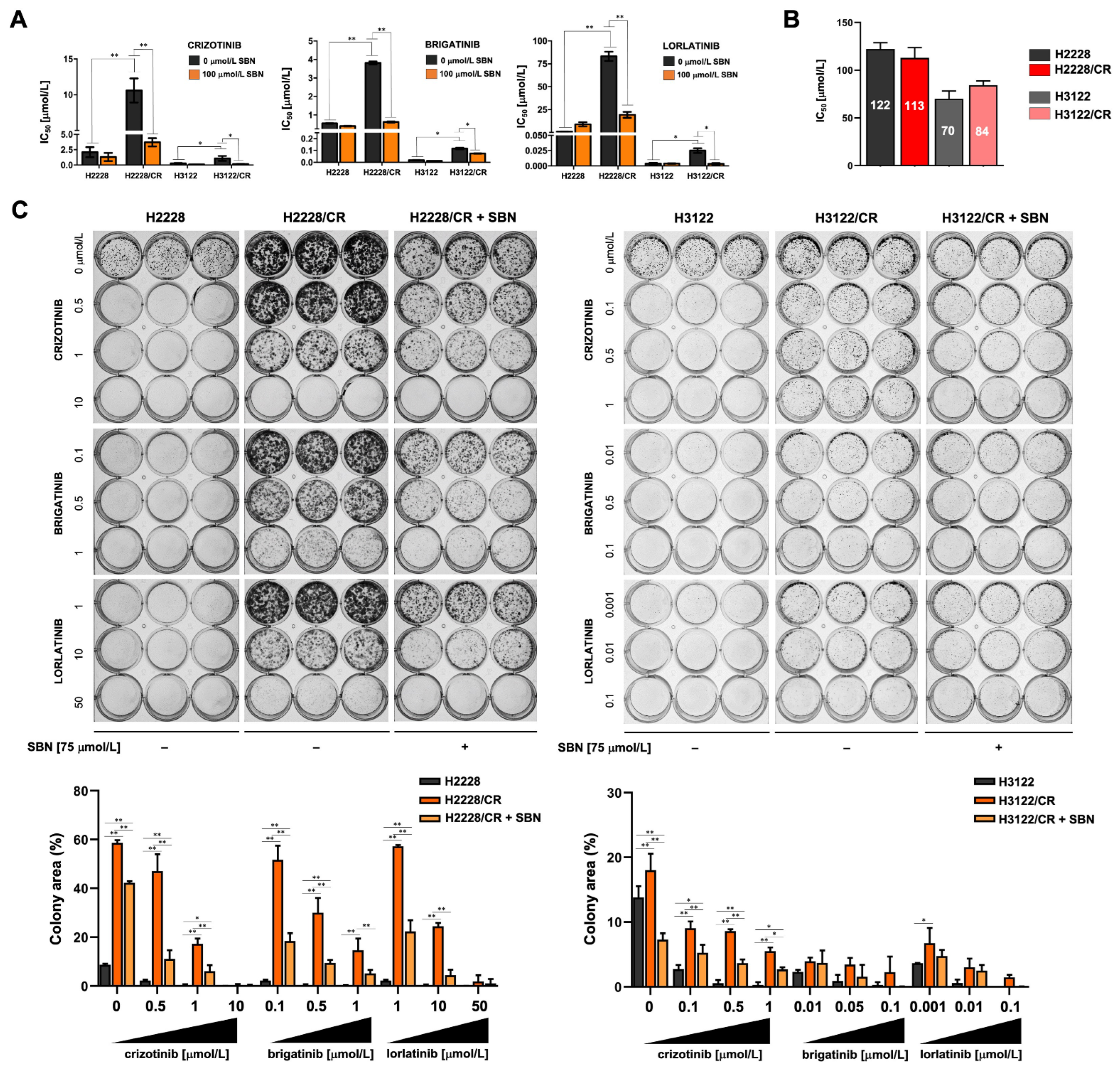
3.2. Silibinin Re-Sensitizes Mesenchymal NSCLC Cells to ALK–TKIs
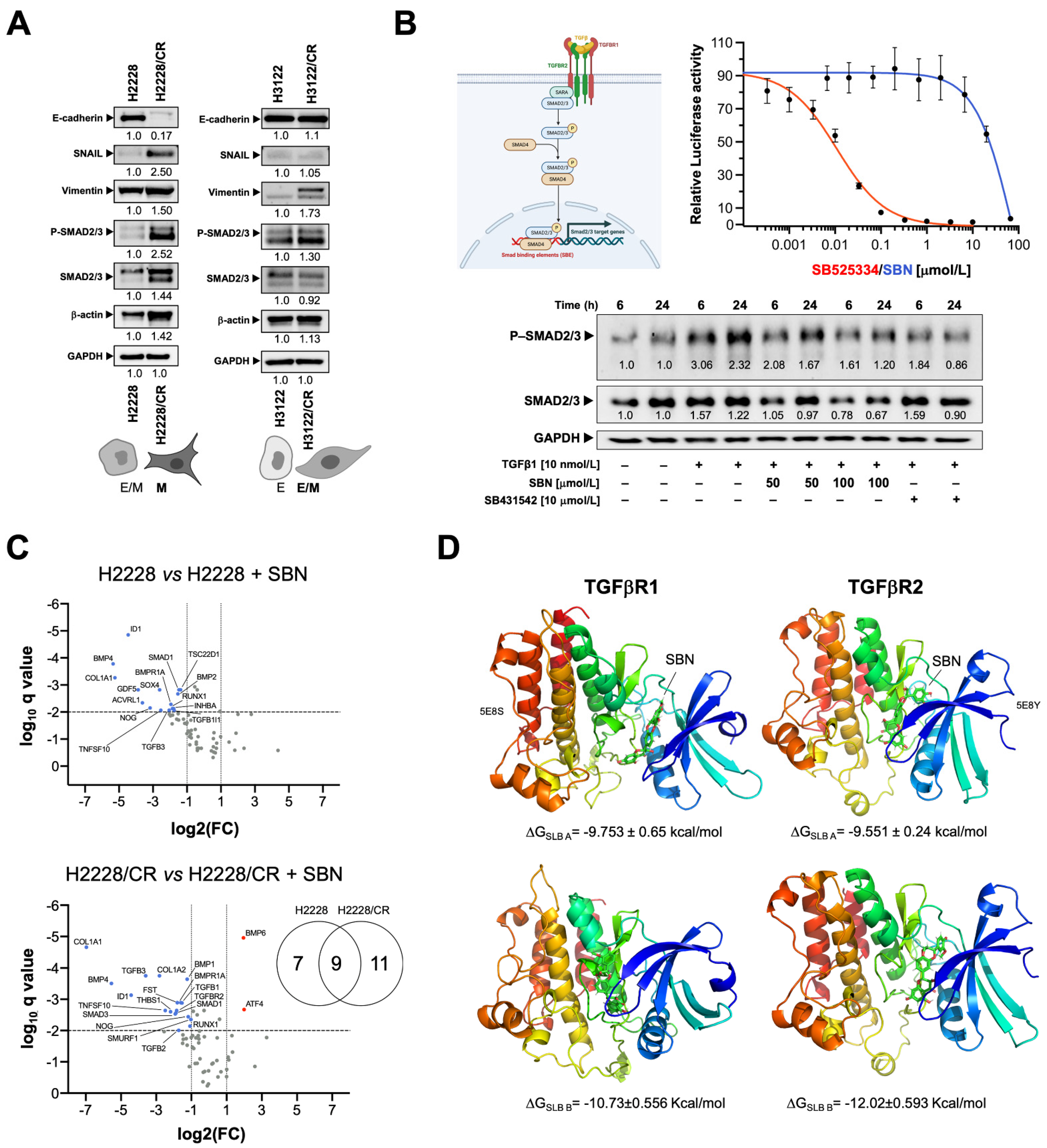
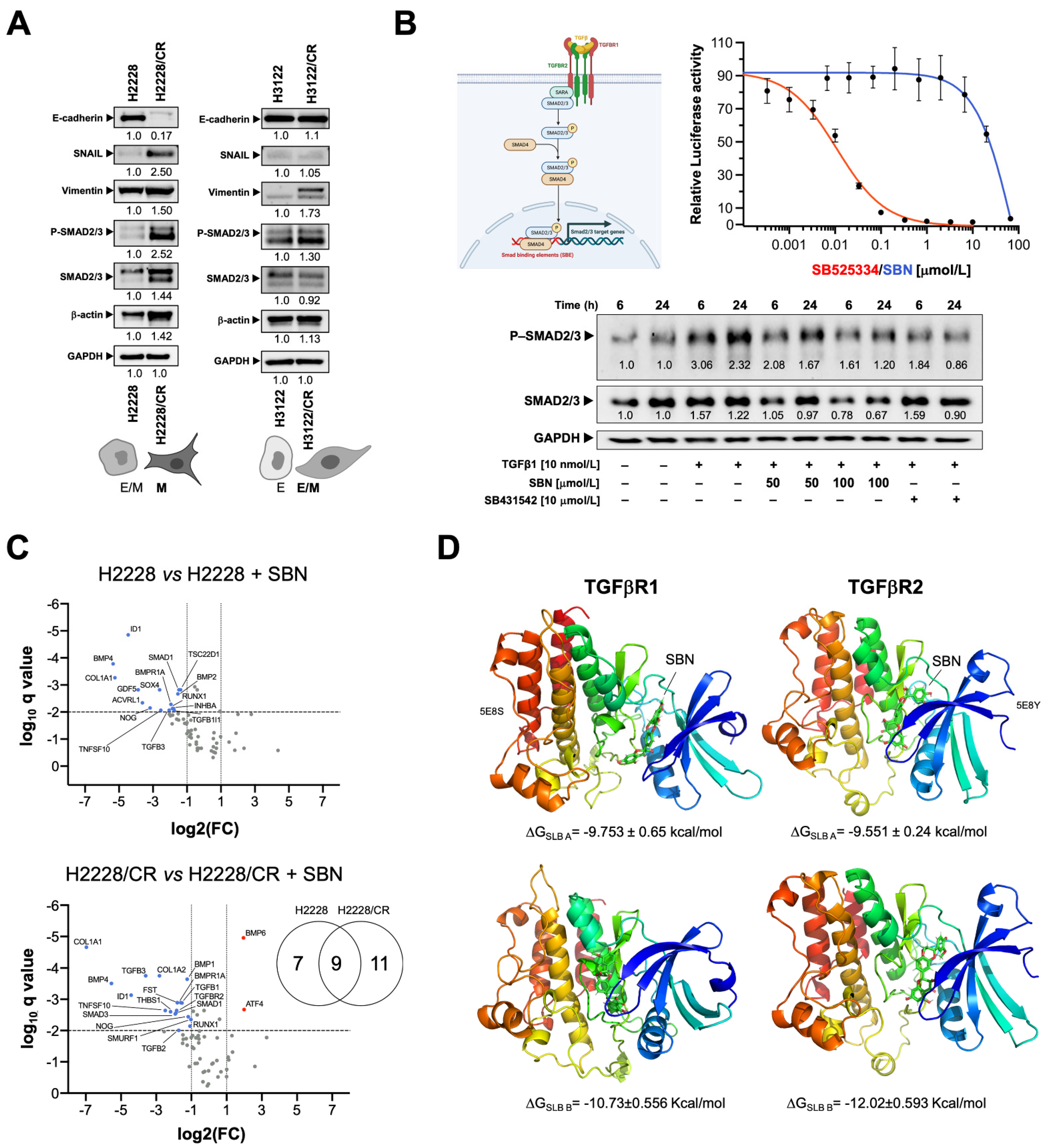
3.3. Silibinin Suppresses the TGFβ/SMAD Signaling Pathway
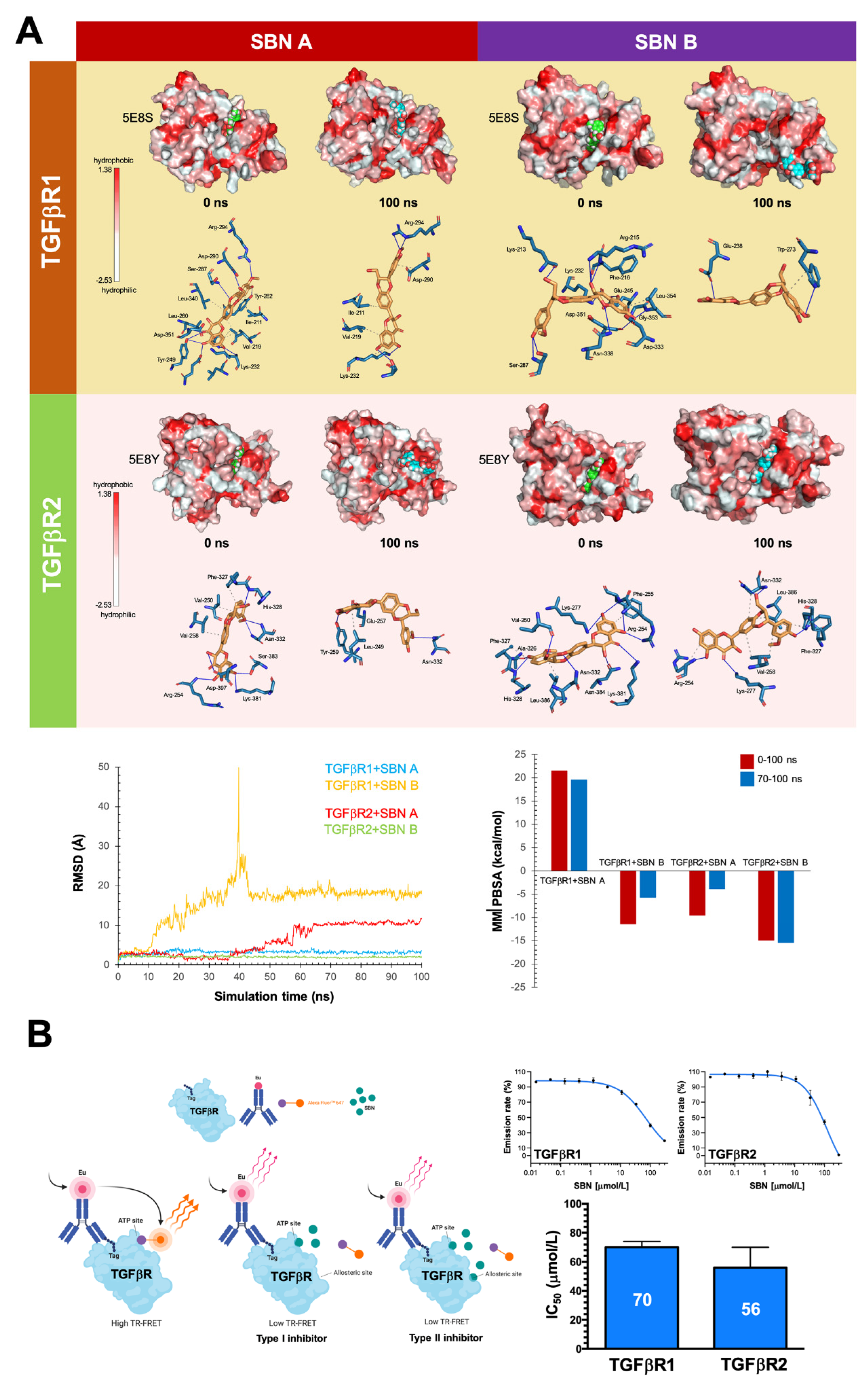
3.4. Silibinin Is Predicted to Directly Inhibit the Kinase Activity of TGFβR1/2
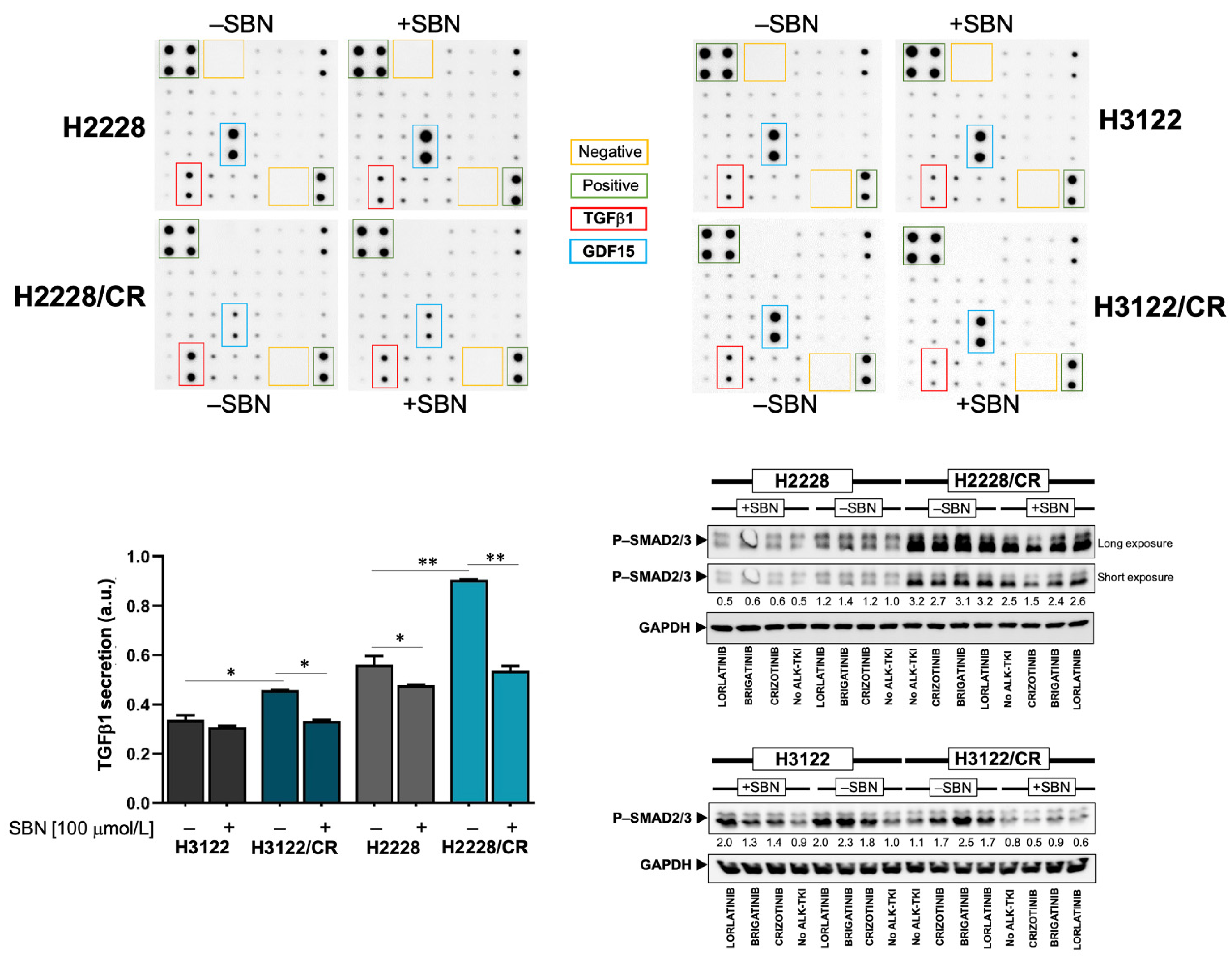
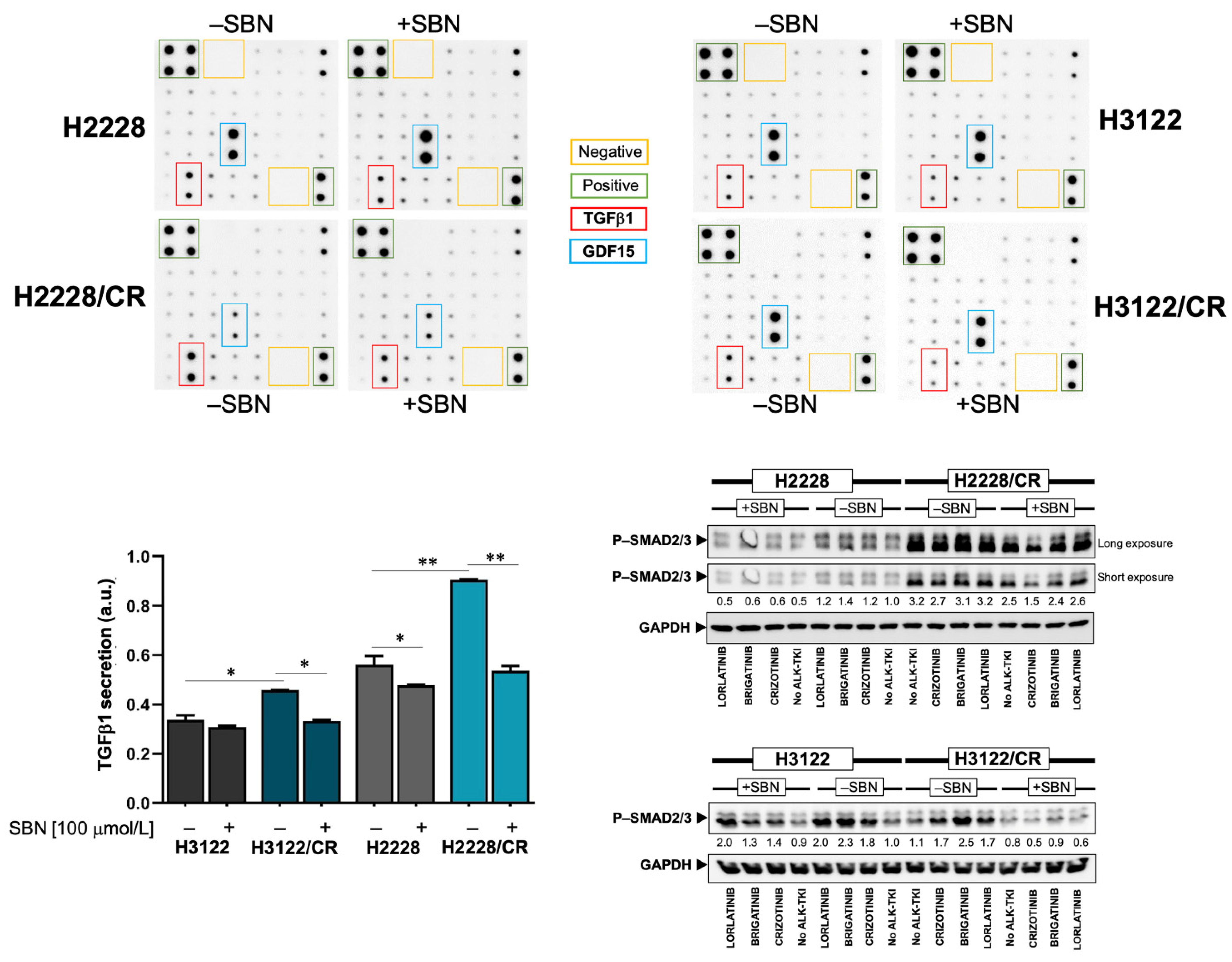
3.5. Silibinin Normalizes TGFβ Oversecretion and SMAD2/3 Hyperactivation in ALK–TKI-Resistant NSCLC Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007, 131, 1190–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, A.T.; Yeap, B.Y.; Solomon, B.J.; Riely, G.J.; Gainor, J.; Engelman, J.A.; Shapiro, G.I.; Costa, D.B.; Ou, S.H.; Butaney, M.; et al. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: A retrospective analysis. Lancet Oncol. 2011, 12, 1004–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, A.T.; Kim, D.W.; Nakagawa, K.; Seto, T.; Crinó, L.; Ahn, M.J.; De Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.; et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N. Engl. J. Med. 2013, 368, 2385–2394. [Google Scholar] [CrossRef] [Green Version]
- Malik, S.M.; Maher, V.E.; Bijwaard, K.E.; Becker, R.L.; Zhang, L.; Tang, S.W.; Song, P.; Liu, Q.; Marathe, A.; Gehrke, B.; et al. U.S. Food and Drug Administration approval: Crizotinib for treatment of advanced or metastatic non-small cell lung cancer that is anaplastic lymphoma kinase positive. Clin. Cancer Res 2014, 20, 2029–2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, B.J.; Mok, T.; Kim, D.W.; Wu, Y.L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N. Engl. J. Med. 2014, 371, 2167–2177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacono, D.; Chiari, R.; Metro, G.; Bennati, C.; Bellezza, G.; Cenci, M.; Ricciuti, B.; Sidoni, A.; Baglivo, S.; Minotti, V.; et al. Future options for ALK-positive non-small cell lung cancer. Lung Cancer 2015, 87, 211–219. [Google Scholar] [CrossRef]
- van der Wekken, A.J.; Pelgrim, R.; Hart, N.; Werner, N.; Mastik, M.F.; Hendriks, L.; van der Heijden, E.H.F.M.; Looijen-Salamon, M.; de Langen, A.J.; Staal-van den Brekel, J.; et al. Dichotomous ALK-IHC Is a Better Predictor for ALK Inhibition Outcome than Traditional ALK-FISH in Advanced Non-Small Cell Lung Cancer. Clin. Cancer Res. 2017, 23, 4251–4258. [Google Scholar] [CrossRef] [Green Version]
- Camidge, D.R.; Doebele, R.C. Treating ALK-positive lung cancer—Early successes and future challenges. Nat. Rev. Clin. Oncol. 2012, 9, 268–277. [Google Scholar] [CrossRef] [Green Version]
- Gainor, J.F.; Dardaei, L.; Yoda, S.; Friboulet, L.; Leshchiner, I.; Katayama, R.; Dagogo-Jack, I.; Gadgeel, S.; Schultz, K.; Singh, M.; et al. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Discov. 2016, 6, 1118–1133. [Google Scholar] [CrossRef]
- Doebele, R.C.; Pilling, A.B.; Aisner, D.L.; Kutateladze, T.G.; Le, A.T.; Weickhardt, A.J.; Kondo, K.L.; Linderman, D.J.; Heasley, L.E.; Franklin, W.A.; et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin. Cancer Res. 2012, 18, 1472–1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katayama, R.; Shaw, A.T.; Khan, T.M.; Mino-Kenudson, M.; Solomon, B.J.; Halmos, B.; Jessop, N.A.; Wain, J.C.; Yeo, A.T.; Benes, C.; et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci. Transl. Med. 2012, 4, 120ra17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.L.; Soda, M.; Yamashita, Y.; Ueno, T.; Takashima, J.; Nakajima, T.; Yatabe, Y.; Takeuchi, K.; Hamada, T.; Haruta, H.; et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N. Engl. J. Med. 2010, 363, 1734–1739. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.; Camidge, D.R.; Shaw, A.T.; Gadgeel, S.; Ahn, J.S.; Kim, D.W.; Ou, S.I.; Pérol, M.; Dziadziuszko, R.; Rosell, R.; et al. Alectinib versus Crizotinib in Untreated ALK-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 829–838. [Google Scholar] [CrossRef]
- Shaw, A.T.; Kim, D.W.; Mehra, R.; Tan, D.S.; Felip, E.; Chow, L.Q.; Camidge, D.R.; Vansteenkiste, J.; Sharma, S.; De Pas, T.; et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N. Engl. J. Med. 2014, 370, 1189–1197. [Google Scholar] [CrossRef] [Green Version]
- Gettinger, S.N.; Bazhenova, L.A.; Langer, C.J.; Salgia, R.; Gold, K.A.; Rosell, R.; Shaw, A.T.; Weiss, G.J.; Tugnait, M.; Narasimhan, N.I.; et al. Activity and safety of brigatinib in ALK-rearranged non-small-cell lung cancer and other malignancies: A single-arm, open-label, phase 1/2 trial. Lancet Oncol. 2016, 17, 1683–1696. [Google Scholar] [CrossRef]
- Zou, H.Y.; Friboulet, L.; Kodack, D.P.; Engstrom, L.D.; Li, Q.; West, M.; Tang, R.W.; Wang, H.; Tsaparikos, K.; Wang, J.; et al. PF-06463922, an ALK/ROS1 Inhibitor, Overcomes Resistance to First and Second Generation ALK Inhibitors in Preclinical Models. Cancer Cell 2015, 28, 70–81. [Google Scholar] [CrossRef] [Green Version]
- Shaw, A.T.; Felip, E.; Bauer, T.M.; Besse, B.; Navarro, A.; Postel-Vinay, S.; Gainor, J.F.; Johnson, M.; Dietrich, J.; James, L.P.; et al. Lorlatinib in non-small-cell lung cancer with ALK or ROS1 rearrangement: An international, multicentre, open-label, single-arm first-in-man phase 1 trial. Lancet Oncol. 2017, 18, 1590–1599. [Google Scholar] [CrossRef]
- Solomon, B.; Shaw, A.; Ou, S.; Besse, B.; Felip, E.; Bauer, T.; Soo, R.; Bearz, A.; Lin, C.; Clancy, J.; et al. Phase 2 study of lorlatinib in patients with advanced ALK+/ROS1+ non-small-cell lung cancer (abstract OA05.06). J. Thorac. Oncol. 2017, 12 (Suppl. 2), S1756. [Google Scholar] [CrossRef]
- Wu, J.; Savooji, J.; Liu, D. Second- and third-generation ALK inhibitors for non-small cell lung cancer. J. Hematol. Oncol. 2016, 9, 19. [Google Scholar] [CrossRef]
- Akamine, T.; Toyokawa, G.; Tagawa, T.; Seto, T. Spotlight on lorlatinib and its potential in the treatment of NSCLC: The evidence to date. OncoTargets Ther. 2018, 11, 5093–5101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Deng, C.; Qiu, Z.; Cao, C.; Wu, F. The Resistance Mechanisms and Treatment Strategies for ALK-Rearranged Non-Small Cell Lung Cancer. Front. Oncol. 2021, 11, 713530. [Google Scholar] [CrossRef] [PubMed]
- Redaelli, S.; Ceccon, M.; Zappa, M.; Sharma, G.G.; Mastini, C.; Mauri, M.; Nigoghossian, M.; Massimino, L.; Cordani, N.; Farina, F.; et al. Lorlatinib Treatment Elicits Multiple On- and Off-Target Mechanisms of Resistance in ALK-Driven Cancer. Cancer Res. 2018, 78, 6866–6880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoda, S.; Lin, J.J.; Lawrence, M.S.; Burke, B.J.; Friboulet, L.; Langenbucher, A.; Dardaei, L.; Prutisto-Chang, K.; Dagogo-Jack, I.; Timofeevski, S.; et al. Sequential ALK Inhibitors Can Select for Lorlatinib-Resistant Compound ALK. Mutations in ALK-Positive Lung Cancer. Cancer Discov. 2018, 8, 714–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyokawa, G.; Seto, T. Updated Evidence on the Mechanisms of Resistance to ALK Inhibitors and Strategies to Overcome Such Resistance: Clinical and Preclinical Data. Oncol. Res. Treat. 2015, 38, 291–298. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Crizotinib resistance: Implications for therapeutic strategies. Ann. Oncol. 2016, 27 (Suppl. 3), iii42–iii50. [Google Scholar] [CrossRef]
- Kim, H.R.; Kim, W.S.; Choi, Y.J.; Choi, C.M.; Rho, J.K.; Lee, J.C. Epithelial-mesenchymal transition leads to crizotinib resistance in H2228 lung cancer cells with EML4-ALK translocation. Mol. Oncol. 2013, 7, 1093–1102. [Google Scholar] [CrossRef]
- Gower, A.; Hsu, W.H.; Hsu, S.T.; Wang, Y.; Giaccone, G. EMT is associated with, but does not drive resistance to ALK inhibitors among EML4-ALK non-small cell lung cancer. Mol. Oncol. 2016, 10, 601–609. [Google Scholar] [CrossRef] [Green Version]
- Kogita, A.; Togashi, Y.; Hayashi, H.; Sogabe, S.; Terashima, M.; De Velasco, M.A.; Sakai, K.; Fujita, Y.; Tomida, S.; Takeyama, Y.; et al. Hypoxia induces resistance to ALK inhibitors in the H3122 non-small cell lung cancer cell line with an ALK rearrangement via epithelial-mesenchymal transition. Int. J. Oncol. 2014, 45, 1430–1436. [Google Scholar] [CrossRef] [Green Version]
- Nakamichi, S.; Seike, M.; Miyanaga, A.; Chiba, M.; Zou, F.; Takahashi, A.; Ishikawa, A.; Kunugi, S.; Noro, R.; Kubota, K.; et al. Overcoming drug-tolerant cancer cell subpopulations showing AXL activation and epithelial-mesenchymal transition is critical in conquering ALK-positive lung cancer. Oncotarget 2018, 9, 27242–27255. [Google Scholar] [CrossRef]
- Debruyne, D.N.; Bhatnagar, N.; Sharma, B.; Luther, W.; Moore, N.F.; Cheung, N.K.; Gray, N.S.; George, R.E. ALK inhibitor resistance in ALK(F1174L)-driven neuroblastoma is associated with AXL activation and induction of EMT. Oncogene 2016, 35, 3681–3691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Recondo, G.; Mezquita, L.; Facchinetti, F.; Planchard, D.; Gazzah, A.; Bigot, L.; Rizvi, A.Z.; Frias, R.L.; Thiery, J.P.; Scoazec, J.Y.; et al. Diverse Resistance Mechanisms to the Third-Generation ALK Inhibitor Lorlatinib in ALK-Rearranged Lung Cancer. Clin. Cancer Res. 2020, 26, 242–255. [Google Scholar] [CrossRef] [Green Version]
- Urbanska, E.M.; Sørensen, J.B.; Melchior, L.C.; Costa, J.C.; Santoni-Rugiu, E. Changing ALK-TKI-Resistance Mechanisms in Rebiopsies of ALK-Rearranged NSCLC: ALK- and BRAF-Mutations Followed by Epithelial-Mesenchymal Transition. Int. J. Mol. Sci. 2020, 21, 2847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Jang, S.J.; Chung, D.H.; Yoo, S.B.; Sun, P.; Jin, Y.; Nam, K.H.; Paik, J.H.; Chung, J.H. A comprehensive comparative analysis of the histomorphological features of ALK-rearranged lung adenocarcinoma based on driver oncogene mutations: Frequent expression of epithelial-mesenchymal transition markers than other genotype. PLoS ONE 2013, 8, e76999. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.; Liu, X.; Qing, Q.; Sang, Y.; Feng, C.; Li, X.; Jiang, L.; Su, P.; Wang, Y. EML4-ALK induces epithelial-mesenchymal transition consistent with cancer stem cell properties in H1299 non-small cell lung cancer cells. Biochem. Biophys. Res. Commun. 2015, 459, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, K.; Takeuchi, S.; Arai, S.; Katayama, R.; Nanjo, S.; Tanimoto, A.; Nishiyama, A.; Nakagawa, T.; Taniguchi, H.; Suzuki, T.; et al. Epithelial-to-Mesenchymal Transition Is a Mechanism of ALK Inhibitor Resistance in Lung Cancer Independent of ALK Mutation Status. Cancer Res. 2019, 79, 1658–1670. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.P.; Raina, K.; Sharma, G.; Agarwal, R. Silibinin inhibits established prostate tumor growth, progression, invasion, and metastasis and suppresses tumor angiogenesis and epithelial-mesenchymal transition in transgenic adenocarcinoma of the mouse prostate model mice. Clin. Cancer Res. 2008, 14, 7773–7780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.; Zeng, J.; Li, L.; Fan, J.; Zhang, D.; Xue, Y.; Zhu, G.; Yang, L.; Wang, X.; He, D. Silibinin reverses epithelial-to-mesenchymal transition in metastatic prostate cancer cells by targeting transcription factors. Oncol. Rep. 2010, 23, 1545–1552. [Google Scholar]
- Cufí, S.; Bonavia, R.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Corominas-Faja, B.; Cuyàs, E.; Martin-Castillo, B.; Barrajón-Catalán, E.; Visa, J.; Segura-Carretero, A.; et al. Silibinin suppresses EMT-driven erlotinib resistance by reversing the high miR-21/low miR-200c signature in vivo. Sci. Rep. 2013, 3, 2459. [Google Scholar] [CrossRef] [Green Version]
- Cufí, S.; Bonavia, R.; Vazquez-Martin, A.; Corominas-Faja, B.; Oliveras-Ferraros, C.; Cuyàs, E.; Martin-Castillo, B.; Barrajón-Catalán, E.; Visa, J.; Segura-Carretero, A.; et al. Silibinin meglumine, a water-soluble form of milk thistle silymarin, is an orally active anti-cancer agent that impedes the epithelial-to-mesenchymal transition (EMT) in EGFR-mutant non-small-cell lung carcinoma cells. Food Chem. Toxicol 2013, 60, 360–368. [Google Scholar] [CrossRef]
- Cho, J.W.; Il, K.J.; Lee, K.S. Downregulation of type I collagen expression in silibinin-treated human skin fibroblasts by blocking the activation of Smad2/3-dependent signaling pathways: Potential therapeutic use in the chemoprevention of keloids. Int. J. Mol. Med. 2013, 31, 1148–1152. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Liang, C.M.; Chen, C.L.; Chen, J.T.; Chang, Y.H.; Lu, D.W.; Chien, K.H.; Tai, M.C. Silibinin inhibits myofibroblast transdifferentiation in human tenon fibroblasts and reduces fibrosis in a rabbit trabeculectomy model. Acta Ophthalmol. 2013, 91, e506–e515. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.W.; Shin, N.R.; Park, S.H.; Lee, I.C.; Ryu, J.M.; Kim, H.J.; Cho, Y.K.; Kim, J.C.; Shin, I.S. Silibinin inhibits the fibrotic responses induced by cigarette smoke via suppression of TGF-β1/Smad 2/3 signaling. Food Chem. Toxicol 2017, 106, 424–429. [Google Scholar] [CrossRef]
- Liu, R.; Wang, Q.; Ding, Z.; Zhang, X.; Li, Y.; Zang, Y.; Zhang, G. Silibinin Augments the Antifibrotic Effect of Valsartan Through Inactivation of TGF-β1 Signaling in Kidney. Drug Des. Dev. Ther. 2020, 14, 603–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, N.; Lucena-Araujo, A.R.; Nakayama, S.; de Figueiredo-Pontes, L.L.; Gonzalez, D.A.; Yasuda, H.; Kobayashi, S.; Costa, D.B. Dual ALK and EGFR inhibition targets a mechanism of acquired resistance to the tyrosine kinase inhibitor crizotinib in ALK rearranged lung cancer. Lung Cancer 2014, 83, 37–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDermott, M.; Eustace, A.J.; Busschots, S.; Breen, L.; Crown, J.; Clynes, M.; O'Donovan, N.; Stordal, B. In vitro Development of Chemotherapy and Targeted Therapy Drug-Resistant Cancer Cell Lines: A Practical Guide with Case Studies. Front. Oncol. 2014, 4, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tebben, A.J.; Ruzanov, M.; Gao, M.; Xie, D.; Kiefer, S.E.; Yan, C.; Newitt, J.A.; Zhang, L.; Kim, K.; Lu, H.; et al. Crystal structures of apo and inhibitor-bound TGFβR2 kinase domain: Insights into TGFβR isoform selectivity. Acta Crystallogr. D Struct. Biol. 2016, 72, 658–674. [Google Scholar] [CrossRef]
- Corominas-Faja, B.; Cuyàs, E.; Lozano-Sánchez, J.; Cufí, S.; Verdura, S.; Fernández-Arroyo, S.; Borrás-Linares, I.; Martin-Castillo, B.; Martin, Á.G.; Lupu, R.; et al. Extra-virgin olive oil contains a metabolo-epigenetic inhibitor of cancer stem cells. Carcinogenesis 2018, 39, 601–613. [Google Scholar] [CrossRef] [Green Version]
- Verdura, S.; Cuyàs, E.; Lozano-Sánchez, J.; Bastidas-Velez, C.; Llorach-Parés, L.; Fernández-Arroyo, S.; Hernández-Aguilera, A.; Joven, J.; Nonell-Canals, A.; Bosch-Barrera, J.; et al. An olive oil phenolic is a new chemotype of mutant isocitrate dehydrogenase 1 (IDH1) inhibitors. Carcinogenesis 2019, 40, 27–40. [Google Scholar] [CrossRef]
- Cuyàs, E.; Castillo, D.; Llorach-Parés, L.; Lozano-Sánchez, J.; Verdura, S.; Nonell-Canals, A.; Brunet, J.; Bosch-Barrera, J.; Joven, J.; Valdés, R.; et al. Computational de-orphanization of the olive oil biophenol oleacein: Discovery of new metabolic and epigenetic targets. Food Chem. Toxicol 2019, 131, 110529. [Google Scholar] [CrossRef]
- Encinar, J.A.; Menendez, J.A. Potential Drugs Targeting Early Innate Immune Evasion of SARS-Coronavirus 2 via 2'-O-Methylation of Viral RNA. Viruses 2020, 12, 525. [Google Scholar] [CrossRef] [PubMed]
- Tramonti, A.; Cuyàs, E.; Encinar, J.A.; Pietzke, M.; Paone, A.; Verdura, S.; Arbusà, A.; Martin-Castillo, B.; Giardina, G.; Joven, J.; et al. Metformin Is a Pyridoxal-5'-phosphate (PLP)-Competitive Inhibitor of SHMT2. Cancers 2021, 13, 4009. [Google Scholar] [CrossRef] [PubMed]
- Verdura, S.; Cuyàs, E.; Cortada, E.; Brunet, J.; Lopez-Bonet, E.; Martin-Castillo, B.; Bosch-Barrera, J.; Encinar, J.A.; Menendez, J.A. Resveratrol targets PD-L1 glycosylation and dimerization to enhance antitumor T-cell immunity. Aging 2020, 12, 8–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeliger, D.; de Groot, B.L. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Aided Mol. Des. 2010, 24, 417–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein-ligand interaction profiler. Nucleic. Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef]
- Koivunen, J.P.; Mermel, C.; Zejnullahu, K.; Murphy, C.; Lifshits, E.; Holmes, A.J.; Choi, H.G.; Kim, J.; Chiang, D.; Thomas, R.; et al. EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin. Cancer Res. 2008, 14, 4275–4283. [Google Scholar] [CrossRef] [Green Version]
- Cuyàs, E.; Pérez-Sánchez, A.; Micol, V.; Menendez, J.A.; Bosch-Barrera, J. STAT3-targeted treatment with silibinin overcomes the acquired resistance to crizotinib in ALK-rearranged lung cancer. Cell Cycle 2016, 15, 3413–3418. [Google Scholar] [CrossRef] [Green Version]
- Heuckmann, J.M.; Balke-Want, H.; Malchers, F.; Peifer, M.; Sos, M.L.; Koker, M.; Meder, L.; Lovly, C.M.; Heukamp, L.C.; Pao, W.; et al. Differential protein stability and ALK inhibitor sensitivity of EML4-ALK fusion variants. Clin. Cancer Res. 2012, 18, 4682–4690. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Oya, Y.; Tanaka, K.; Shimizu, J.; Horio, Y.; Kuroda, H.; Sakao, Y.; Hida, T.; Yatabe, Y. Differential Crizotinib Response Duration Among ALK Fusion Variants in ALK-Positive Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2016, 34, 3383–3389. [Google Scholar] [CrossRef] [Green Version]
- Woo, C.G.; Seo, S.; Kim, S.W.; Jang, S.J.; Park, K.S.; Song, J.Y.; Lee, B.; Richards, M.W.; Bayliss, R.; Lee, D.H.; et al. Differential protein stability and clinical responses of EML4-ALK fusion variants to various ALK inhibitors in advanced ALK-rearranged non-small cell lung cancer. Ann. Oncol. 2017, 28, 791–797. [Google Scholar] [CrossRef]
- Li, M.; Hou, X.; Chen, J.; Zhang, B.; Wang, N.; Han, H.; Chen, L. ALK fusion variant 3a/b, concomitant mutations, and high PD-L1 expression were associated with unfavorable clinical response to second-generation ALK TKIs in patients with advanced ALK-rearranged non-small cell lung cancer (GASTO 1061). Lung Cancer 2022, 165, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J.; Gomis, R.R. The logic of TGFbeta signaling. FEBS Lett. 2006, 580, 2811–2820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell. Biol 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seoane, J.; Gomis, R.R. TGF-β Family Signaling in Tumor Suppression and Cancer Progression. Cold Spring Harb. Perspect. Biol. 2017, 9, a022277. [Google Scholar] [CrossRef] [Green Version]
- Wrana, J.L.; Attisano, L.; Wieser, R.; Ventura, F.; Massagué, J. Mechanism of activation of the TGF-beta receptor. Nature 1994, 370, 341–347. [Google Scholar] [CrossRef]
- Voena, C.; Varesio, L.M.; Zhang, L.; Menotti, M.; Poggio, T.; Panizza, E.; Wang, Q.; Minero, V.G.; Fagoonee, S.; Compagno, M.; et al. Oncogenic ALK regulates EMT in non-small cell lung carcinoma through repression of the epithelial splicing regulatory protein 1. Oncotarget 2016, 7, 33316–33330. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Hölzel, M.; Knijnenburg, T.; Schlicker, A.; Roepman, P.; McDermott, U.; Garnett, M.; Grernrum, W.; Sun, C.; Prahallad, A.; et al. MED12 controls the response to multiple cancer drugs through regulation of TGF-β receptor signaling. Cell 2012, 151, 937–950. [Google Scholar] [CrossRef] [Green Version]
- Sang, J.; Acquaviva, J.; Friedland, J.C.; Smith, D.L.; Sequeira, M.; Zhang, C.; Jiang, Q.; Xue, L.; Lovly, C.M.; Jimenez, J.P.; et al. Targeted inhibition of the molecular chaperone Hsp90 overcomes ALK inhibitor resistance in non-small cell lung cancer. Cancer Discov. 2013, 3, 430–443. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Meng, Y.; Wang, K.; Gao, M.; Du, J.; Wang, J.; Li, Z.; Zuo, D.; Wu, Y. EML4-ALK G1202R mutation induces EMT and confers resistance to ceritinib in NSCLC cells via activation of STAT3/Slug signaling. Cell. Signal 2022, 92, 110264. [Google Scholar] [CrossRef]
- Byers, L.A.; Diao, L.; Wang, J.; Saintigny, P.; Girard, L.; Peyton, M.; Shen, L.; Fan, Y.; Giri, U.; Tumula, P.K.; et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin. Cancer Res. 2013, 19, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Tan, T.Z.; Miow, Q.H.; Miki, Y.; Noda, T.; Mori, S.; Huang, R.Y.; Thiery, J.P. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol. Med. 2014, 6, 1279–1293. [Google Scholar] [CrossRef] [PubMed]
- George, J.T.; Jolly, M.K.; Xu, S.; Somarelli, J.A.; Levine, H. Survival Outcomes in Cancer Patients Predicted by a Partial EMT Gene Expression Scoring Metric. Cancer Res. 2017, 77, 6415–6428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, P.; George, J.T.; Tripathi, S.; Levine, H.; Jolly, M.K. Comparative Study of Transcriptomics-Based Scoring Metrics for the Epithelial-Hybrid-Mesenchymal Spectrum. Front. Bioeng. Biotechnol. 2020, 8, 220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bocci, F.; Mandal, S.; Tejaswi, T.; Jolly, M.K. Investigating epithelial-mesenchymal heterogeneity of tumors and circulating tumor cells with transcriptomic analysis and biophysical modeling. Comput. Syst. Oncol. 2021, 1, e1015. [Google Scholar] [CrossRef]
- Verdura, S.; Cuyàs, E.; Llorach-Parés, L.; Pérez-Sánchez, A.; Micol, V.; Nonell-Canals, A.; Joven, J.; Valiente, M.; Sánchez-Martínez, M.; Bosch-Barrera, J.; et al. Silibinin is a direct inhibitor of STAT3. Food Chem. Toxicol. 2018, 116 Pt B, 161–172. [Google Scholar] [CrossRef]
- Rho, J.K.; Choi, Y.J.; Jeon, B.S.; Choi, S.J.; Cheon, G.J.; Woo, S.K.; Kim, H.R.; Kim, C.H.; Choi, C.M.; Lee, J.C. Combined treatment with silibinin and epidermal growth factor receptor tyrosine kinase inhibitors overcomes drug resistance caused by T790M mutation. Mol. Cancer Ther. 2010, 9, 3233–3243. [Google Scholar] [CrossRef] [Green Version]
- Shien, K.; Papadimitrakopoulou, V.A.; Ruder, D.; Behrens, C.; Shen, L.; Kalhor, N.; Song, J.; Lee, J.J.; Wang, J.; Tang, X.; et al. JAK1/STAT3 Activation through a Proinflammatory Cytokine Pathway Leads to Resistance to Molecularly Targeted Therapy in Non-Small Cell Lung Cancer. Mol. Cancer Ther. 2017, 16, 2234–2245. [Google Scholar] [CrossRef] [Green Version]
- Wilson, F.H.; Johannessen, C.M.; Piccioni, F.; Tamayo, P.; Kim, J.W.; Van Allen, E.M.; Corsello, S.M.; Capelletti, M.; Calles, A.; Butaney, M.; et al. A functional landscape of resistance to ALK inhibition in lung cancer. Cancer Cell 2015, 27, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. EMT International Association (TEMTIA). Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [Green Version]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.M.; Weinberg, R.A.; Lees, J.A.; Guen, V.J. Emerging Mechanisms by which EMT Programs Control Stemness. Trends Cancer 2020, 6, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.M.; Stewart, T.A.; Thompson, E.W.; Monteith, G.R. Targeting EMT in cancer: Opportunities for pharmacological intervention. Trends Pharmacol. Sci. 2014, 35, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, F.; Stassi, G.; De Maria, R. Epithelial-mesenchymal transition: A new target in anticancer drug discovery. Nat. Rev. Drug Discov. 2016, 15, 311–325. [Google Scholar] [CrossRef]
- Post-White, J.; Ladas, E.J.; Kelly, K.M. Advances in the use of milk thistle (Silybum marianum). Integr. Cancer Ther. 2007, 6, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Gazák, R.; Walterová, D.; Kren, V. Silybin and silymarin--new and emerging applications in medicine. Curr. Med. Chem. 2007, 14, 315–338. [Google Scholar] [CrossRef]
- Agarwal, R.; Agarwal, C.; Ichikawa, H.; Singh, R.P.; Aggarwal, B.B. Anticancer potential of silymarin: From bench to bed side. Anticancer Res. 2006, 26, 4457–4498. [Google Scholar]
- Singh, R.P.; Agarwal, R. Prostate cancer chemoprevention by silibinin: Bench to bedside. Mol. Carcinog. 2006, 45, 436–442. [Google Scholar] [CrossRef]
- Verdura, S.; Cuyàs, E.; Ruiz-Torres, V.; Micol, V.; Joven, J.; Bosch-Barrera, J.; Menendez, J.A. Lung Cancer Management with Silibinin: A Historical and Translational Perspective. Pharmaceuticals 2021, 14, 559. [Google Scholar] [CrossRef]
- Bosch-Barrera, J.; Menendez, J.A. Silibinin and STAT3: A natural way of targeting transcription factors for cancer therapy. Cancer Treat. Rev. 2015, 41, 540–546. [Google Scholar] [CrossRef]
- Bosch-Barrera, J.; Queralt, B.; Menendez, J.A. Targeting STAT3 with silibinin to improve cancer therapeutics. Cancer Treat. Rev. 2017, 58, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Deep, G.; Gangar, S.C.; Agarwal, C.; Agarwal, R. Role of E-cadherin in antimigratory and antiinvasive efficacy of silibinin in prostate cancer cells. Cancer Prev. Res. 2011, 4, 1222–1232. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Zang, W.; Wang, H.; Wei, X. Silibinin enhances anti-renal fibrosis effect of MK-521 via downregulation of TGF-β signaling pathway. Hum. Cell. 2020, 33, 330–336. [Google Scholar] [CrossRef]
- Křen, V. Chirality Matters: Biological Activity of Optically Pure Silybin and Its Congeners. Int. J. Mol. Sci. 2021, 22, 7885. [Google Scholar] [CrossRef] [PubMed]
- Sciacca, M.F.M.; Romanucci, V.; Zarrelli, A.; Monaco, I.; Lolicato, F.; Spinella, N.; Galati, C.; Grasso, G.; D'Urso, L.; Romeo, M.; et al. Inhibition of Aβ Amyloid Growth and Toxicity by Silybins: The Crucial Role of Stereochemistry. ACS Chem. Neurosci. 2017, 8, 1767–1778. [Google Scholar] [CrossRef] [PubMed]
- Shoichet, B.K. Interpreting steep dose-response curves in early inhibitor discovery. J. Med. Chem. 2006, 49, 7274–7277. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, A.; Nichol, D.; Kinose, F.; Abazeed, M.E.; Marusyk, A.; Haura, E.B.; Scott, J.G. Collateral sensitivity networks reveal evolutionary instability and novel treatment strategies in ALK mutated non-small cell lung cancer. Sci. Rep. 2017, 7, 1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, K.; Liu, H.; Song, Z.; Zhang, C.; Zhang, M.; Yang, H.; Cao, J.; Geng, M.; Shen, A.; Zhang, A. Design, synthesis and pharmacological evaluation of ALK and Hsp90 dual inhibitors bearing resorcinol and 2,4-diaminopyrimidine motifs. Eur. J. Med. Chem. 2018, 152, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Rong, B.; Yang, S. Molecular mechanism and targeted therapy of Hsp90 involved in lung cancer: New discoveries and developments (Review). Int. J. Oncol. 2018, 52, 321–336. [Google Scholar] [CrossRef] [Green Version]
- Riebold, M.; Kozany, C.; Freiburger, L.; Sattler, M.; Buchfelder, M.; Hausch, F.; Stalla, G.K.; Paez-Pereda, M. A C-terminal HSP90 inhibitor restores glucocorticoid sensitivity and relieves a mouse allograft model of Cushing disease. Nat. Med. 2015, 21, 276–280. [Google Scholar] [CrossRef]
- Cuyàs, E.; Verdura, S.; Micol, V.; Joven, J.; Bosch-Barrera, J.; Encinar, J.A.; Menendez, J.A. Revisiting silibinin as a novobiocin-like Hsp90 C-terminal inhibitor: Computational modeling and experimental validation. Food. Chem. Toxicol. 2019, 132, 110645. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Sánchez, A.; Cuyàs, E.; Ruiz-Torres, V.; Agulló-Chazarra, L.; Verdura, S.; González-Álvarez, I.; Bermejo, M.; Joven, J.; Micol, V.; Bosch-Barrera, J.; et al. Intestinal Permeability Study of Clinically Relevant Formulations of Silibinin in Caco-2 Cell Monolayers. Int. J. Mol. Sci. 2019, 20, 1606. [Google Scholar] [CrossRef] [PubMed]
- Bosch-Barrera, J.; Sais, E.; Cañete, N.; Marruecos, J.; Cuyàs, E.; Izquierdo, A.; Porta, R.; Haro, M.; Brunet, J.; Pedraza, S.; et al. Response of brain metastasis from lung cancer patients to an oral nutraceutical product containing silibinin. Oncotarget 2016, 7, 32006–32014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priego, N.; Zhu, L.; Monteiro, C.; Mulders, M.; Wasilewski, D.; Bindeman, W.; Doglio, L.; Martínez, L.; Martínez-Saez, E.; Ramón y Cajal, S.; et al. STAT3 labels a subpopulation of reactive astrocytes required for brain metastasis. Nat. Med. 2018, 24, 1024–1035. [Google Scholar] [CrossRef] [PubMed]
- Bosch-Barrera, J.; Verdura, S.; Ruffinelli, J.C.; Carcereny, E.; Sais, E.; Cuyàs, E.; Palmero, R.; Lopez-Bonet, E.; Hernández-Martínez, A.; Oliveras, G.; et al. Silibinin Suppresses Tumor Cell-Intrinsic Resistance to Nintedanib and Enhances Its Clinical Activity in Lung Cancer. Cancers 2021, 13, 4168. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verdura, S.; Encinar, J.A.; Teixidor, E.; Segura-Carretero, A.; Micol, V.; Cuyàs, E.; Bosch-Barrera, J.; Menendez, J.A. Silibinin Overcomes EMT-Driven Lung Cancer Resistance to New-Generation ALK Inhibitors. Cancers 2022, 14, 6101. https://doi.org/10.3390/cancers14246101
Verdura S, Encinar JA, Teixidor E, Segura-Carretero A, Micol V, Cuyàs E, Bosch-Barrera J, Menendez JA. Silibinin Overcomes EMT-Driven Lung Cancer Resistance to New-Generation ALK Inhibitors. Cancers. 2022; 14(24):6101. https://doi.org/10.3390/cancers14246101
Chicago/Turabian StyleVerdura, Sara, Jose Antonio Encinar, Eduard Teixidor, Antonio Segura-Carretero, Vicente Micol, Elisabet Cuyàs, Joaquim Bosch-Barrera, and Javier A. Menendez. 2022. "Silibinin Overcomes EMT-Driven Lung Cancer Resistance to New-Generation ALK Inhibitors" Cancers 14, no. 24: 6101. https://doi.org/10.3390/cancers14246101
APA StyleVerdura, S., Encinar, J. A., Teixidor, E., Segura-Carretero, A., Micol, V., Cuyàs, E., Bosch-Barrera, J., & Menendez, J. A. (2022). Silibinin Overcomes EMT-Driven Lung Cancer Resistance to New-Generation ALK Inhibitors. Cancers, 14(24), 6101. https://doi.org/10.3390/cancers14246101