
PDX Models: A Versatile Tool for Studying the Role of Myeloid-Derived Suppressor Cells in Breast Cancer

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. In Vivo Models
2.2. Immunohistochemistry
2.3. Purification of Neutrophils and MDSCs
2.4. Human Interferon Gamma (IFNγ) Measurement
2.5. Immunofluorescence
2.6. Tumor Dissociation
2.7. Flow Cytometry
2.8. RNA Sequencing
3. Results
3.1. Establishment of PDX Models for MDSC Studies
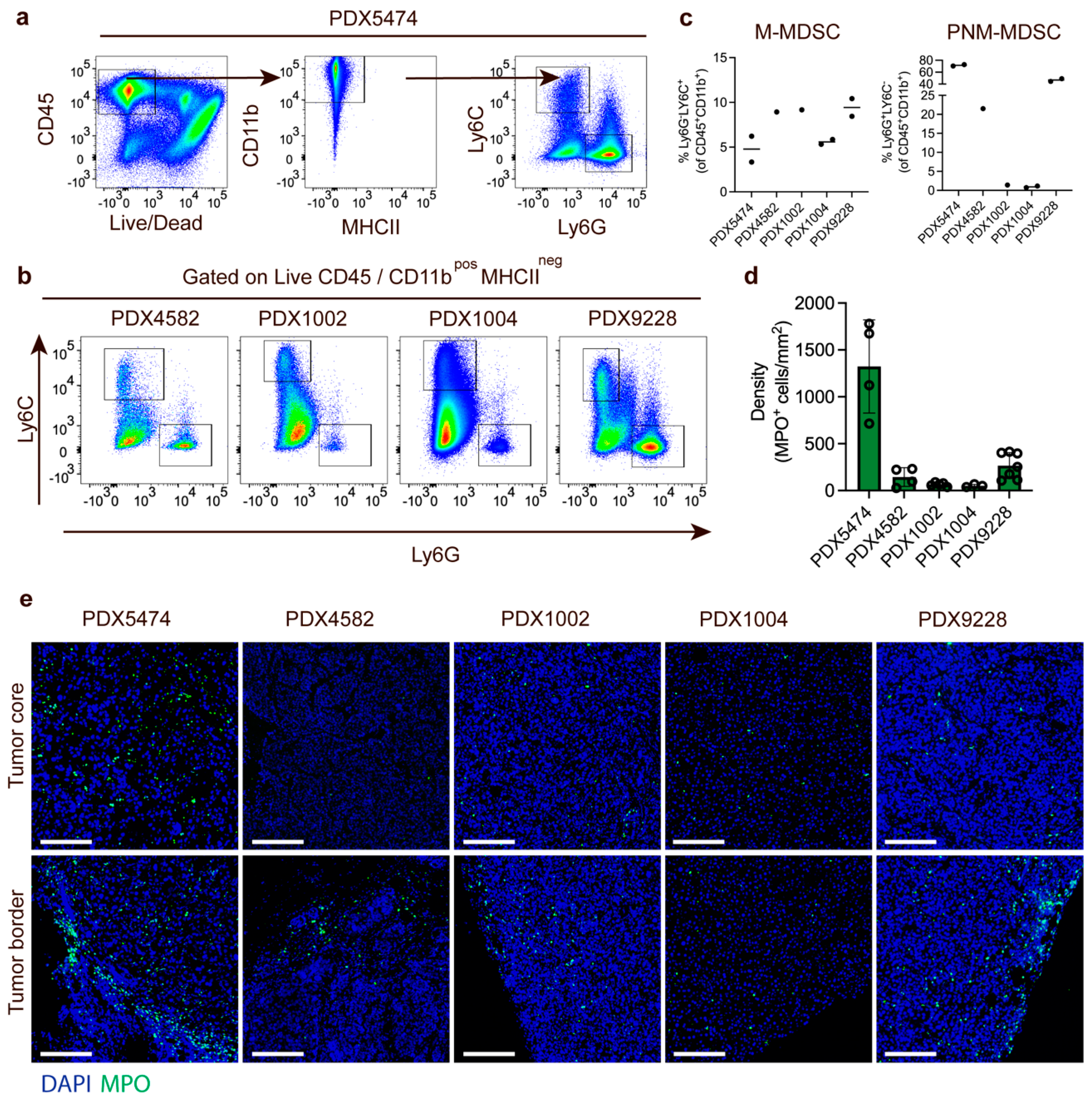
3.2. Characterization of PDX Models Reveals Heterogenic Infiltration of Myeloid Cells
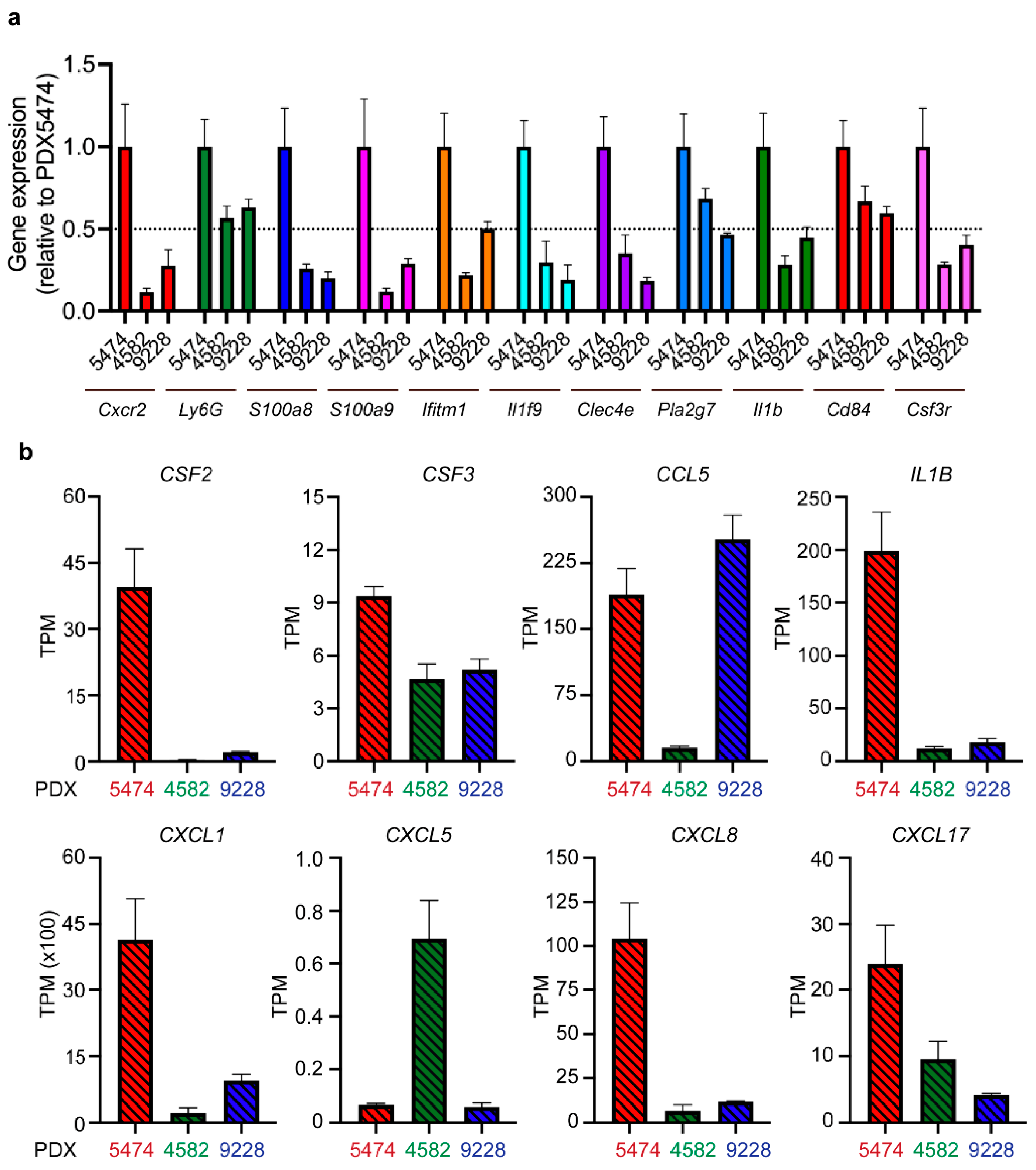
3.3. MDSC Infiltration Correlates with the Expression of Myeloid Cell-Recruiting Chemokines
3.4. Tumor-Associated PMN-MDSC in Breast Cancer PDX Models Display MDSC Expression Profiles
3.5. Recruitment of MDSCs to Spontaneous PDX Lung Metastases
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Condamine, T.; Gabrilovich, D.I. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011, 32, 19–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tcyganov, E.; Mastio, J.; Chen, E.; Gabrilovich, D.I. Plasticity of myeloid-derived suppressor cells in cancer. Curr. Opin. Immunol. 2018, 51, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrand-Rosenberg, S.; Fenselau, C. Myeloid-Derived Suppressor Cells: Immune-Suppressive Cells That Impair Antitumor Immunity and Are Sculpted by Their Environment. J. Immunol. 2018, 200, 422–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Kuang, Z.; Zhang, D.; Gao, Y.; Ying, M.; Wang, T. Reactive oxygen species in immune cells: A new antitumor target. Biomed. Pharm. 2021, 133, 110978. [Google Scholar] [CrossRef]
- Akdis, C.A.; Blaser, K. Mechanisms of interleukin-10-mediated immune suppression. Immunology 2001, 103, 131–136. [Google Scholar] [CrossRef]
- Thomas, D.A.; Massague, J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005, 8, 369–380. [Google Scholar] [CrossRef] [Green Version]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Kim, I.S.; Gao, Y.; Welte, T.; Wang, H.; Liu, J.; Janghorban, M.; Sheng, K.; Niu, Y.; Goldstein, A.; Zhao, N.; et al. Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat. Cell Biol. 2019, 21, 1113–1126. [Google Scholar] [CrossRef]
- Albrengues, J.; Shields, M.A.; Ng, D.; Park, C.G.; Ambrico, A.; Poindexter, M.E.; Upadhyay, P.; Uyeminami, D.L.; Pommier, A.; Kuttner, V.; et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018, 361, eaao4227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonda, K.; Shibata, M.; Ohtake, T.; Matsumoto, Y.; Tachibana, K.; Abe, N.; Ohto, H.; Sakurai, K.; Takenoshita, S. Myeloid-derived suppressor cells are increased and correlated with type 2 immune responses, malnutrition, inflammation, and poor prognosis in patients with breast cancer. Oncol. Lett. 2017, 14, 1766–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez-Cortegana, C.; Galassi, C.; Klapp, V.; Gabrilovich, D.I.; Galluzzi, L. Myeloid-Derived Suppressor Cells and Radiotherapy. Cancer Immunol. Res. 2022, 10, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, S.; Baghdadi, M.; Tsuchikawa, T.; Wada, H.; Nakamura, T.; Abe, H.; Nakanishi, S.; Usui, Y.; Higuchi, K.; Takahashi, M.; et al. Chemotherapy-Derived Inflammatory Responses Accelerate the Formation of Immunosuppressive Myeloid Cells in the Tissue Microenvironment of Human Pancreatic Cancer. Cancer Res. 2015, 75, 2629–2640. [Google Scholar] [CrossRef] [Green Version]
- Highfill, S.L.; Cui, Y.; Giles, A.J.; Smith, J.P.; Zhang, H.; Morse, E.; Kaplan, R.N.; Mackall, C.L. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci. Transl. Med. 2014, 6, 237ra67. [Google Scholar] [CrossRef]
- Steele, C.W.; Karim, S.A.; Leach, J.D.G.; Bailey, P.; Upstill-Goddard, R.; Rishi, L.; Foth, M.; Bryson, S.; McDaid, K.; Wilson, Z.; et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 2016, 29, 832–845. [Google Scholar] [CrossRef] [Green Version]
- Fultang, L.; Panetti, S.; Ng, M.; Collins, P.; Graef, S.; Rizkalla, N.; Booth, S.; Lenton, R.; Noyvert, B.; Shannon-Lowe, C.; et al. MDSC targeting with Gemtuzumab ozogamicin restores T cell immunity and immunotherapy against cancers. EBioMedicine 2019, 47, 235–246. [Google Scholar] [CrossRef] [Green Version]
- Tobin, R.P.; Jordan, K.R.; Robinson, W.A.; Davis, D.; Borges, V.F.; Gonzalez, R.; Lewis, K.D.; McCarter, M.D. Targeting myeloid-derived suppressor cells using all-trans retinoic acid in melanoma patients treated with Ipilimumab. Int. Immunopharmacol. 2018, 63, 282–291. [Google Scholar] [CrossRef]
- Bailur, J.K.; Gueckel, B.; Derhovanessian, E.; Pawelec, G. Presence of circulating Her2-reactive CD8 + T-cells is associated with lower frequencies of myeloid-derived suppressor cells and regulatory T cells, and better survival in older breast cancer patients. Breast Cancer Res. 2015, 17, 34. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Feng, J.; Wu, F.; Cai, J.; Zhang, X.; Wang, H.; Fetahu, I.S.; Iwanicki, I.; Ma, D.; Hu, T.; et al. TET2 promotes anti-tumor immunity by governing G-MDSCs and CD8(+) T-cell numbers. EMBO Rep. 2020, 21, e49425. [Google Scholar] [CrossRef]
- Hou, A.; Hou, K.; Huang, Q.; Lei, Y.; Chen, W. Targeting Myeloid-Derived Suppressor Cell, a Promising Strategy to Overcome Resistance to Immune Checkpoint Inhibitors. Front. Immunol. 2020, 11, 783. [Google Scholar] [CrossRef] [PubMed]
- Terp, M.G.; Gammelgaard, O.L.; Vever, H.; Gjerstorff, M.F.; Ditzel, H.J. Sustained compensatory p38 MAPK signaling following treatment with MAPK inhibitors induces the immunosuppressive protein CD73 in cancer: Combined targeting could improve outcomes. Mol. Oncol. 2021, 15, 3299–3316. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Madsen, J.G.; Schmidt, S.F.; Larsen, B.D.; Loft, A.; Nielsen, R.; Mandrup, S. iRNA-seq: Computational method for genome-wide assessment of acute transcriptional regulation from total RNA-seq data. Nucleic Acids Res. 2015, 43, e40. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alshetaiwi, H.; Pervolarakis, N.; McIntyre, L.L.; Ma, D.; Nguyen, Q.; Rath, J.A.; Nee, K.; Hernandez, G.; Evans, K.; Torosian, L.; et al. Defining the emergence of myeloid-derived suppressor cells in breast cancer using single-cell transcriptomics. Sci. Immunol. 2020, 5, eaay6017. [Google Scholar] [CrossRef]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [Green Version]
- Elkabets, M.; Ribeiro, V.S.; Dinarello, C.A.; Ostrand-Rosenberg, S.; Di Santo, J.P.; Apte, R.N.; Vosshenrich, C.A. IL-1beta regulates a novel myeloid-derived suppressor cell subset that impairs NK cell development and function. Eur. J. Immunol. 2010, 40, 3347–3357. [Google Scholar] [CrossRef]
- Meher, A.K.; Spinosa, M.; Davis, J.P.; Pope, N.; Laubach, V.E.; Su, G.; Serbulea, V.; Leitinger, N.; Ailawadi, G.; Upchurch, G.R., Jr. Novel Role of IL (Interleukin)-1beta in Neutrophil Extracellular Trap Formation and Abdominal Aortic Aneurysms. Arterioscler. Thromb. Vasc Biol. 2018, 38, 843–853. [Google Scholar] [CrossRef] [Green Version]
- Cali, B.; Agnellini, A.H.R.; Cioccarelli, C.; Sanchez-Rodriguez, R.; Predonzani, A.; Toffolo, G.I.; Viola, A.; Bronte, V.; Arrigoni, G.; Zonta, F.; et al. GM-CSF Nitration Is a New Driver of Myeloid Suppressor Cell Activity in Tumors. Front. Immunol. 2021, 12, 718098. [Google Scholar] [CrossRef]
- Yanez, A.; Coetzee, S.G.; Olsson, A.; Muench, D.E.; Berman, B.P.; Hazelett, D.J.; Salomonis, N.; Grimes, H.L.; Goodridge, H.S. Granulocyte-Monocyte Progenitors and Monocyte-Dendritic Cell Progenitors Independently Produce Functionally Distinct Monocytes. Immunity 2017, 47, 890–902 e894. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Xia, X.; Mao, L.; Wang, S. The CCAAT/Enhancer-Binding Protein Family: Its Roles in MDSC Expansion and Function. Front. Immunol. 2019, 10, 1804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theivanthiran, B.; Evans, K.S.; DeVito, N.C.; Plebanek, M.; Sturdivant, M.; Wachsmuth, L.P.; Salama, A.K.; Kang, Y.; Hsu, D.; Balko, J.M.; et al. A tumor-intrinsic PD-L1/NLRP3 inflammasome signaling pathway drives resistance to anti-PD-1 immunotherapy. J. Clin. Investig. 2020, 130, 2570–2586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.; Huang, G.; Cheng, L.; Li, Z.; Xiao, Y.; Deng, Q.; Jiang, Y.; Li, B.; Lin, S.; Wang, S.; et al. Establishment of peripheral blood mononuclear cell-derived humanized lung cancer mouse models for studying efficacy of PD-L1/PD-1 targeted immunotherapy. MAbs 2018, 10, 1301–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinska, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Maelandsmo, G.M.; et al. Patient-derived xenograft models: An emerging platform for translational cancer research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Zhao, Y.; Wei, L.; Li, S.; Liu, J. Tumor-infiltrating Treg, MDSC, and IDO expression associated with outcomes of neoadjuvant chemotherapy of breast cancer. Cancer Biol. Ther. 2018, 19, 695–705. [Google Scholar] [CrossRef] [Green Version]
- Toor, S.M.; Syed Khaja, A.S.; El Salhat, H.; Faour, I.; Kanbar, J.; Quadri, A.A.; Albashir, M.; Elkord, E. Myeloid cells in circulation and tumor microenvironment of breast cancer patients. Cancer Immunol. Immunother. 2017, 66, 753–764. [Google Scholar] [CrossRef] [Green Version]
- Hawila, E.; Razon, H.; Wildbaum, G.; Blattner, C.; Sapir, Y.; Shaked, Y.; Umansky, V.; Karin, N. CCR5 Directs the Mobilization of CD11b(+)Gr1(+)Ly6C(low) Polymorphonuclear Myeloid Cells from the Bone Marrow to the Blood to Support Tumor Development. Cell Rep. 2017, 21, 2212–2222. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, C.; McCampbell, K.K.; David, J.M.; Palena, C. Neutralization of IL-8 decreases tumor PMN-MDSCs and reduces mesenchymalization of claudin-low triple-negative breast cancer. JCI Insight 2017, 2, e94296. [Google Scholar] [CrossRef] [Green Version]
- Mackert, J.R.; Qu, P.; Min, Y.; Johnson, P.F.; Yang, L.; Lin, P.C. Dual negative roles of C/EBPalpha in the expansion and pro-tumor functions of MDSCs. Sci. Rep. 2017, 7, 14048. [Google Scholar] [CrossRef]
- Millrud, C.R.; Bergenfelz, C.; Leandersson, K. On the origin of myeloid-derived suppressor cells. Oncotarget 2017, 8, 3649–3665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traynor, S.; Terp, M.G.; Nielsen, A.Y.; Guldberg, P.; Jakobsen, M.; Pedersen, P.G.; Gammelgaard, O.L.; Pedersen, C.B.; Pedersen, M.T.; Rattenborg, S.; et al. DNA methyltransferase inhibition promotes recruitment of myeloid-derived suppressor cells to the tumor microenvironment through induction of tumor cell-intrinsic interleukin-1. Cancer Lett. 2023, 552, 215982. [Google Scholar] [CrossRef] [PubMed]
- Danilin, S.; Merkel, A.R.; Johnson, J.R.; Johnson, R.W.; Edwards, J.R.; Sterling, J.A. Myeloid-derived suppressor cells expand during breast cancer progression and promote tumor-induced bone destruction. Oncoimmunology 2012, 1, 1484–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mabuchi, S.; Matsumoto, Y.; Kawano, M.; Minami, K.; Seo, Y.; Sasano, T.; Takahashi, R.; Kuroda, H.; Hisamatsu, T.; Kakigano, A.; et al. Uterine cervical cancer displaying tumor-related leukocytosis: A distinct clinical entity with radioresistant feature. J. Natl. Cancer Inst. 2014, 106, dju147. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gjerstorff, M.F.; Traynor, S.; Gammelgaard, O.L.; Johansen, S.; Pedersen, C.B.; Ditzel, H.J.; Terp, M.G. PDX Models: A Versatile Tool for Studying the Role of Myeloid-Derived Suppressor Cells in Breast Cancer. Cancers 2022, 14, 6153. https://doi.org/10.3390/cancers14246153
Gjerstorff MF, Traynor S, Gammelgaard OL, Johansen S, Pedersen CB, Ditzel HJ, Terp MG. PDX Models: A Versatile Tool for Studying the Role of Myeloid-Derived Suppressor Cells in Breast Cancer. Cancers. 2022; 14(24):6153. https://doi.org/10.3390/cancers14246153
Chicago/Turabian StyleGjerstorff, Morten F., Sofie Traynor, Odd L. Gammelgaard, Simone Johansen, Christina B. Pedersen, Henrik J. Ditzel, and Mikkel G. Terp. 2022. "PDX Models: A Versatile Tool for Studying the Role of Myeloid-Derived Suppressor Cells in Breast Cancer" Cancers 14, no. 24: 6153. https://doi.org/10.3390/cancers14246153