
Aberrant Methylation of Somatostatin Receptor 2 Gene Is Initiated in Aged Gastric Mucosa Infected with Helicobacter pylori and Consequential Gene Silencing Is Associated with Establishment of Inflammatory Microenvironment In Vitro Study

 , , , , , , , , and
, , , , , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Human GC Cell Lines
2.2. Human Materials
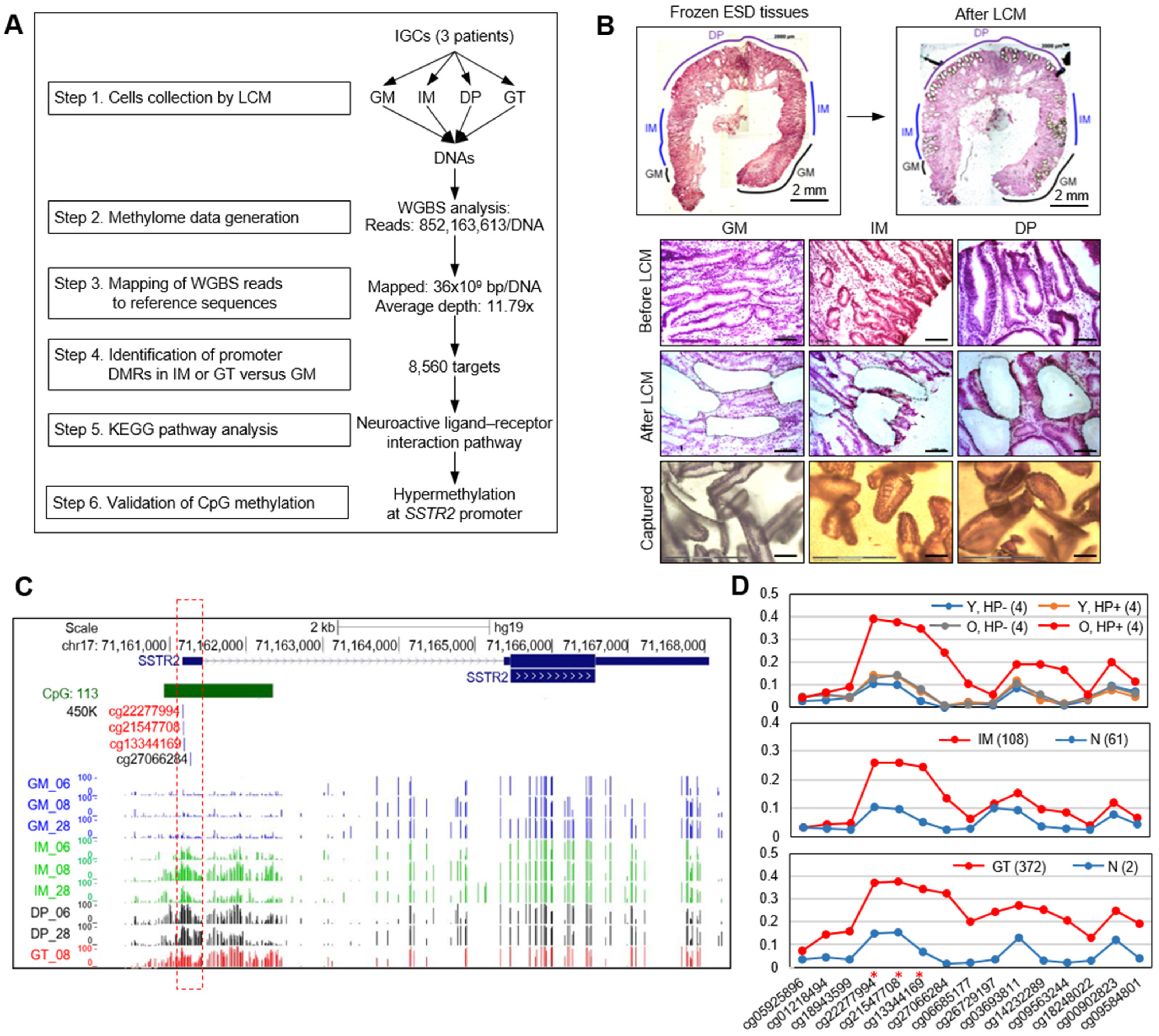
2.3. Laser Capture Microdissection (LCM)
2.4. Whole-Genome Bisulfite Sequencing (WGBS) Analysis
2.5. Bisulfite Sequencing
2.6. Pyrosequencing
2.7. Reverse-Transcription PCR (RT-PCR) and Real-Time Quantitative PCR (RT-qPCR)
2.8. Cell Treatment with 5-aza-2-deoxycytidine (5-aza-dC) and Trichostatin A (TSA)
2.9. Chromatin Immunoprecipitation (ChIP)-PCR Assay
2.10. Immunohistochemical Analysis of Tissue Microarrays of Human GTs
2.11. Establishment of SSTR2-KO Cells using CRISPR/Cas9 System
2.12. Establishment of SSTR2-OVER Cells using Lentivirus Infection
2.13. Cell Viability Assay
2.14. Colony Forming Assay
2.15. Cell Migration Assay
2.16. Wound-Healing Assay
2.17. Tumorigenesis Assay in Nude Mice
2.18. RNA Sequencing (RNA-seq)
2.19. Statistical Analysis
3. Results
3.1. WGBS Analysis Show SSTR2 Promoter to Be Hypermethylated in Primary GT and Premalignant Tissues
3.2. Abnormal Methylation of SSTR2 Promoter in Normal Mucosae Is Associated with H. pylori Infection and Aging
3.3. Abnormal Methylation of SSTR2 Promoter Is Associated with Gene Silencing in Clinical Tissues
3.4. Immunohistochemical Analysis Reveals the Loss-of-Function of SSTR2 in Primary GT and Premalignant Tissues
3.5. Promoter Methylation of SSTR2 Is Associated with Gene Silencing in GC Cell Lines and Drug Treatment Restores SSTR2 Expression in GC Cell Lines
3.6. SSTR2 has Tumor Suppressor Activity In Vitro and In Vivo
3.7. Inflammatory Microenvironment Is Established by the Loss of SSTR2 In Vitro
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Lauren, P. The Two Histological Main Types of Gastric Carcinoma: Diffuse and So-Called Intestinal-Type Carcinoma. An Attempt at a Histo-Clinical Classification. Acta Pathol. Microbiol. Scand. 1965, 64, 31–49. [Google Scholar] [CrossRef] [PubMed]
- Correa, P. Human gastric carcinogenesis: A multistep and multifactorial process--First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 1992, 52, 6735–6740. [Google Scholar] [PubMed]
- Schubert, M.L.; Peura, D.A. Control of gastric acid secretion in health and disease. Gastroenterology 2008, 134, 1842–1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, F.S.; Golodner, E.H.; Wang, J.; Chen, M.C.; Avedian, D.; Calam, J.; Walsh, J.H.; Dubinett, S.; Soll, A.H. Mononuclear cells and cytokines stimulate gastrin release from canine antral cells in primary culture. Am. J. Physiol. 1996, 270, G783–G788. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.; Hellmich, M.R. Gastrin, inflammation, and carcinogenesis. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 33–39. [Google Scholar] [CrossRef] [Green Version]
- Issa, J.P. Aging and epigenetic drift: A vicious cycle. J. Clin. Investig. 2014, 124, 24–29. [Google Scholar] [CrossRef] [Green Version]
- Watson, S.A.; Grabowska, A.M.; El-Zaatari, M.; Takhar, A. Gastrin—Active participant or bystander in gastric carcinogenesis? Nat. Rev. Cancer 2006, 6, 936–946. [Google Scholar] [CrossRef]
- Piqueras, L.; Tache, Y.; Martinez, V. Somatostatin receptor type 2 mediates bombesin-induced inhibition of gastric acid secretion in mice. J. Physiol. 2003, 549, 889–901. [Google Scholar] [CrossRef]
- Van Op den Bosch, J.; Adriaensen, D.; Van Nassauw, L.; Timmermans, J.P. The role(s) of somatostatin, structurally related peptides and somatostatin receptors in the gastrointestinal tract: A review. Regul. Pept. 2009, 156, 1–8. [Google Scholar] [CrossRef]
- Torrisani, J.; Hanoun, N.; Laurell, H.; Lopez, F.; Maoret, J.J.; Souque, A.; Susini, C.; Cordelier, P.; Buscail, L. Identification of an Upstream Promoter of the Human Somatostatin Receptor, Hsstr2, Which is Controlled by Epigenetic Modifications. Endocrinology 2008, 149, 3137–3147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Chen, C.; Bi, X.; Zhou, C.; Huang, T.; Ni, C.; Yang, P.; Chen, S.; Ye, M.; Duan, S. DNA methylation of CMTM3, SSTR2, and MDFI genes in colorectal cancer. Gene 2017, 630, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kang, T.W.; Haam, K.; Kim, M.; Kim, S.K.; Kim, S.Y.; Lee, S.I.; Song, K.S.; Jeong, H.Y.; Kim, Y.S. Whole genome MBD-seq and RRBS analyses reveal that hypermethylation of gastrointestinal hormone receptors is associated with gastric carcinogenesis. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emanuel, O.; Liu, J.; Schartinger, V.H.; Nei, W.L.; Chan, Y.Y.; Tsang, C.M.; Riechelmann, H.; Masterson, L.; Haybaeck, J.; Oppermann, U.; et al. SSTR2 in Nasopharyngeal Carcinoma: Relationship with Latent EBV Infection and Potential as a Therapeutic Target. Cancers 2021, 13, 4944. [Google Scholar] [CrossRef] [PubMed]
- Kotzerke, J.; Buesser, D.; Naumann, A.; Runge, R.; Huebinger, L.; Kliewer, A.; Freudenberg, R.; Brogsitter, C. Epigenetic-Like Stimulation of Receptor Expression in SSTR2 Transfected HEK293 Cells as a New Therapeutic Strategy. Cancers 2022, 14, 2513. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Alcalde, F.; Okonechnikov, K.; Carbonell, J.; Cruz, L.M.; Gotz, S.; Tarazona, S.; Dopazo, J.; Meyer, T.F.; Conesa, A. Qualimap: Evaluating next-generation sequencing alignment data. Bioinformatics 2012, 28, 2678–2679. [Google Scholar] [CrossRef] [Green Version]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. methylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Kim, J.H.; Jang, H.R.; Kim, H.M.; Lee, C.W.; Noh, S.M.; Song, K.S.; Cho, J.S.; Jeong, H.Y.; Hahn, Y.; et al. LRRC3B, encoding a leucine-rich repeat-containing protein, is a putative tumor suppressor gene in gastric cancer. Cancer Res. 2008, 68, 7147–7155. [Google Scholar] [CrossRef] [Green Version]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Han, D.C.; Lee, M.Y.; Shin, K.D.; Jeon, S.B.; Kim, J.M.; Son, K.H.; Kim, H.C.; Kim, H.M.; Kwon, B.M. 2′-benzoyloxycinnamaldehyde induces apoptosis in human carcinoma via reactive oxygen species. J. Biol. Chem. 2004, 279, 6911–6920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, S.; Nanjo, S.; Rehnberg, E.; Iida, N.; Takeshima, H.; Ando, T.; Maekita, T.; Sugiyama, T.; Ushijima, T. Distinct DNA methylation targets by aging and chronic inflammation: A pilot study using gastric mucosa infected with Helicobacter pylori. Clin. Epigenet. 2019, 11, 191. [Google Scholar] [CrossRef]
- Huang, K.K.; Ramnarayanan, K.; Zhu, F.; Srivastava, S.; Xu, C.; Tan, A.L.K.; Lee, M.; Tay, S.; Das, K.; Xing, M.; et al. Genomic and Epigenomic Profiling of High-Risk Intestinal Metaplasia Reveals Molecular Determinants of Progression to Gastric Cancer. Cancer Cell 2018, 33, 137–150.e5. [Google Scholar] [CrossRef] [Green Version]
- Allen, J.P.; Canty, A.J.; Schulz, S.; Humphrey, P.P.; Emson, P.C.; Young, H.M. Identification of cells expressing somatostatin receptor 2 in the gastrointestinal tract of Sstr2 knockout/lacZ knockin mice. J. Comp. Neurol. 2002, 454, 329–340. [Google Scholar] [CrossRef]
- Martinez, V.; Curi, A.P.; Torkian, B.; Schaeffer, J.M.; Wilkinson, H.A.; Walsh, J.H.; Tache, Y. High basal gastric acid secretion in somatostatin receptor subtype 2 knockout mice. Gastroenterology 1998, 114, 1125–1132. [Google Scholar] [CrossRef]
- Schubert, M.L.; Edwards, N.F.; Arimura, A.; Makhlouf, G.M. Paracrine regulation of gastric acid secretion by fundic somatostatin. Am. J. Physiol. 1987, 252, G485–G490. [Google Scholar] [CrossRef]
- Yang, H.; Wong, H.; Wu, V.; Walsh, J.H.; Tache, Y. Somatostatin monoclonal antibody immunoneutralization increases gastrin and gastric acid secretion in urethane-anesthetized rats. Gastroenterology 1990, 99, 659–665. [Google Scholar] [CrossRef]
- Hoyer, D.; Bell, G.I.; Berelowitz, M.; Epelbaum, J.; Feniuk, W.; Humphrey, P.P.; O’Carroll, A.M.; Patel, Y.C.; Schonbrunn, A.; Taylor, J.E.; et al. Classification and nomenclature of somatostatin receptors. Trends Pharmacol. Sci. 1995, 16, 86–88. [Google Scholar] [CrossRef]
- Lloyd, K.C.; Amirmoazzami, S.; Friedik, F.; Chew, P.; Walsh, J.H. Somatostatin inhibits gastrin release and acid secretion by activating sst2 in dogs. Am. J. Physiol. 1997, 272, G1481–G1488. [Google Scholar] [CrossRef] [PubMed]
- Samuelson, L.C.; Hinkle, K.L. Insights into the regulation of gastric acid secretion through analysis of genetically engineered mice. Annu. Rev. Physiol. 2003, 65, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Pfeifer, G.P. Aging and DNA methylation. BMC Biol. 2015, 13, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niwa, T.; Tsukamoto, T.; Toyoda, T.; Mori, A.; Tanaka, H.; Maekita, T.; Ichinose, M.; Tatematsu, M.; Ushijima, T. Inflammatory processes triggered by Helicobacter pylori infection cause aberrant DNA methylation in gastric epithelial cells. Cancer Res. 2010, 70, 1430–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, S.C.; Widschwendter, M.; Teschendorff, A.E. Epigenetic drift, epigenetic clocks and cancer risk. Epigenomics 2016, 8, 705–719. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Blaser, M.J.; Perez-Perez, G.I.; Kleanthous, H.; Cover, T.L.; Peek, R.M.; Chyou, P.H.; Stemmermann, G.N.; Nomura, A. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res. 1995, 55, 2111–2115. [Google Scholar]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.-J.; Park, J.-L.; Yoon, B.-H.; Haam, K.; Heo, H.; Kim, J.-H.; Kim, S.-Y.; Kim, M.; Kim, W.-H.; Lee, S.-I.; et al. Aberrant Methylation of Somatostatin Receptor 2 Gene Is Initiated in Aged Gastric Mucosa Infected with Helicobacter pylori and Consequential Gene Silencing Is Associated with Establishment of Inflammatory Microenvironment In Vitro Study. Cancers 2022, 14, 6183. https://doi.org/10.3390/cancers14246183
Kim H-J, Park J-L, Yoon B-H, Haam K, Heo H, Kim J-H, Kim S-Y, Kim M, Kim W-H, Lee S-I, et al. Aberrant Methylation of Somatostatin Receptor 2 Gene Is Initiated in Aged Gastric Mucosa Infected with Helicobacter pylori and Consequential Gene Silencing Is Associated with Establishment of Inflammatory Microenvironment In Vitro Study. Cancers. 2022; 14(24):6183. https://doi.org/10.3390/cancers14246183
Chicago/Turabian StyleKim, Hee-Jin, Jong-Lyul Park, Byoung-Ha Yoon, Keeok Haam, Haejeong Heo, Jong-Hwan Kim, Seon-Young Kim, Mirang Kim, Woo-Ho Kim, Sang-Il Lee, and et al. 2022. "Aberrant Methylation of Somatostatin Receptor 2 Gene Is Initiated in Aged Gastric Mucosa Infected with Helicobacter pylori and Consequential Gene Silencing Is Associated with Establishment of Inflammatory Microenvironment In Vitro Study" Cancers 14, no. 24: 6183. https://doi.org/10.3390/cancers14246183