
Progestin Resistance and Corresponding Management of Abnormal Endometrial Hyperplasia and Endometrial Carcinoma


Abstract
:Simple Summary
Abstract
1. Introduction
2. Mechanisms of Progestin Repairing Endometrial Pre-Cancer/Cancer
2.1. Cell Cycle Arrest
2.2. Anti-Angiogenesis
2.3. Induction of Apoptosis
2.4. Induction of Cell Differentiation
2.5. Inhibition of Inflammatory Response
2.6. Inhibition of Epithelial-to-Mesenchymal Transition
2.7. Regulation of Estrogen/Androgen Receptor
2.8. Progestin Induced Paracrine Regulation
3. Mechanisms of Progestin Resistance in Endometrial Pre-Cancer/Cancer
3.1. Aberrant PR Signaling
3.2. Other Abnormal Signaling Pathways
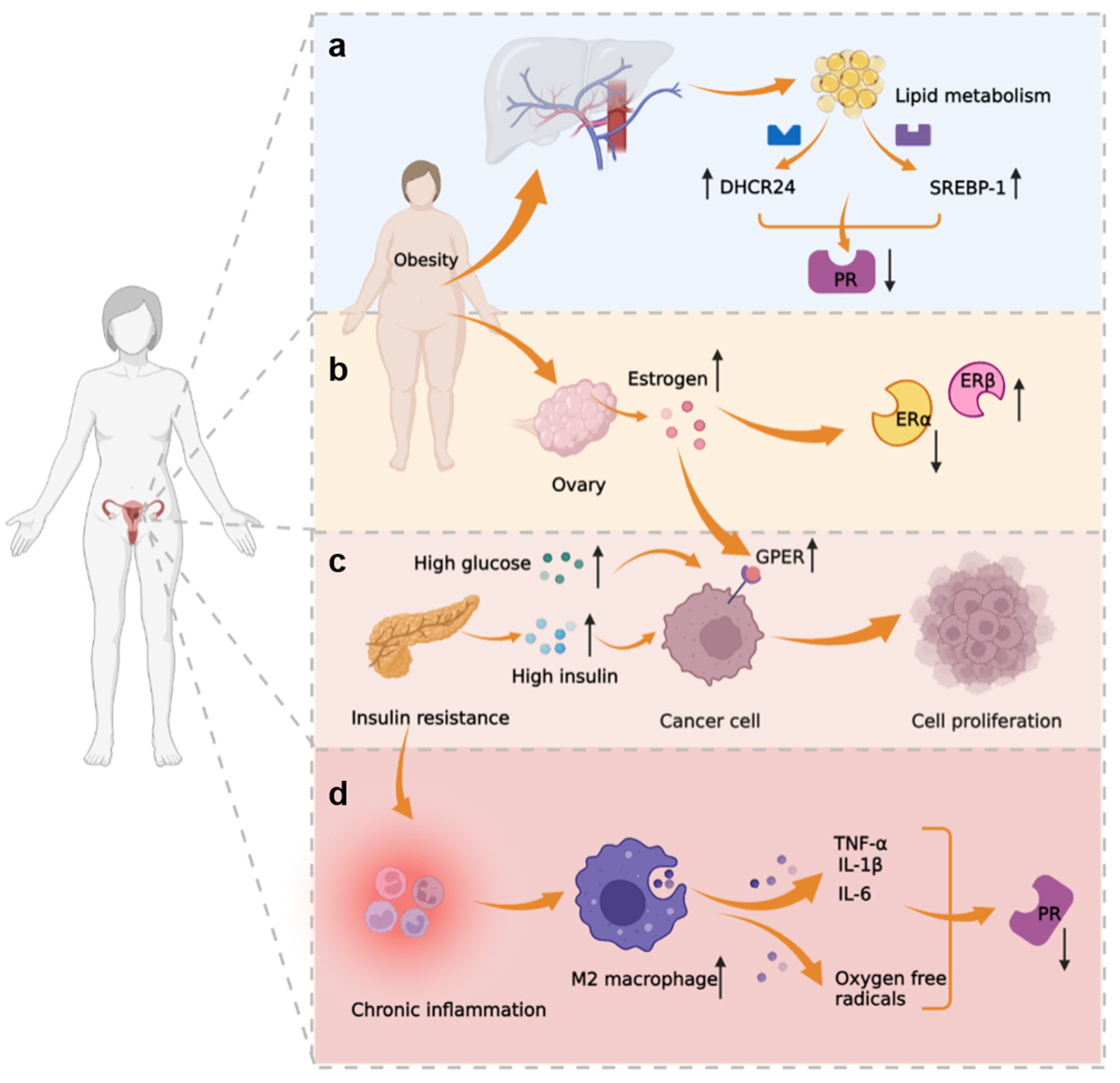
3.3. Metabolic-Immune-Tumor Microenvironment
3.4. Endometrial Cancer Stem Cells
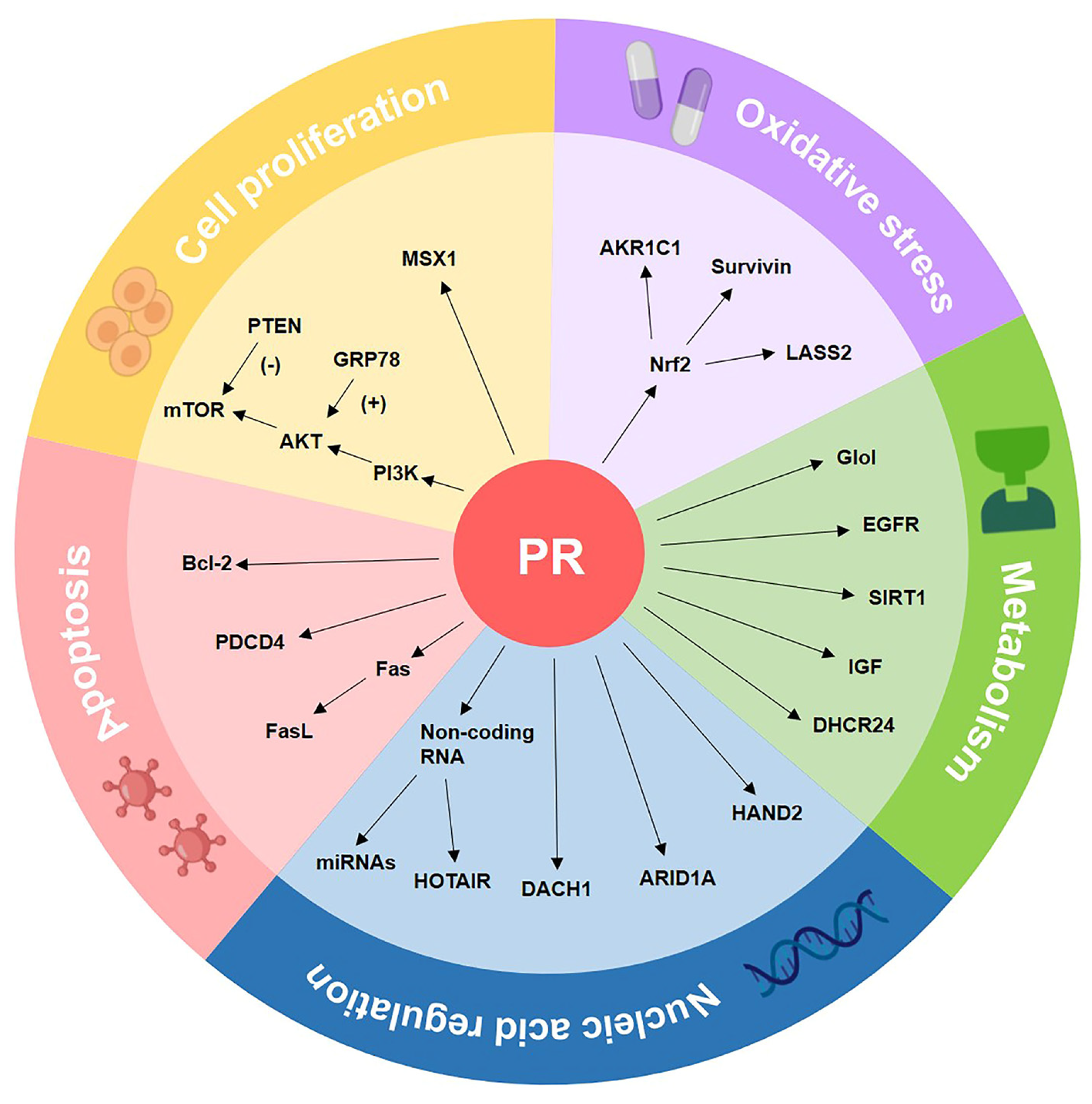
4. The Molecular Biomarkers of Progestin Resistance
4.1. Cell Proliferation-Associated Biomarkers
4.2. Oxidative Stress-Related Biomarkers
4.3. Metabolism-Related Biomarkers
4.4. Apoptosis Pathway-Related Biomarkers
4.5. Nucleic Acid Regulation-Related Biomarkers
4.6. Biomarkers of Endometrial Cancer with Different Molecular Types
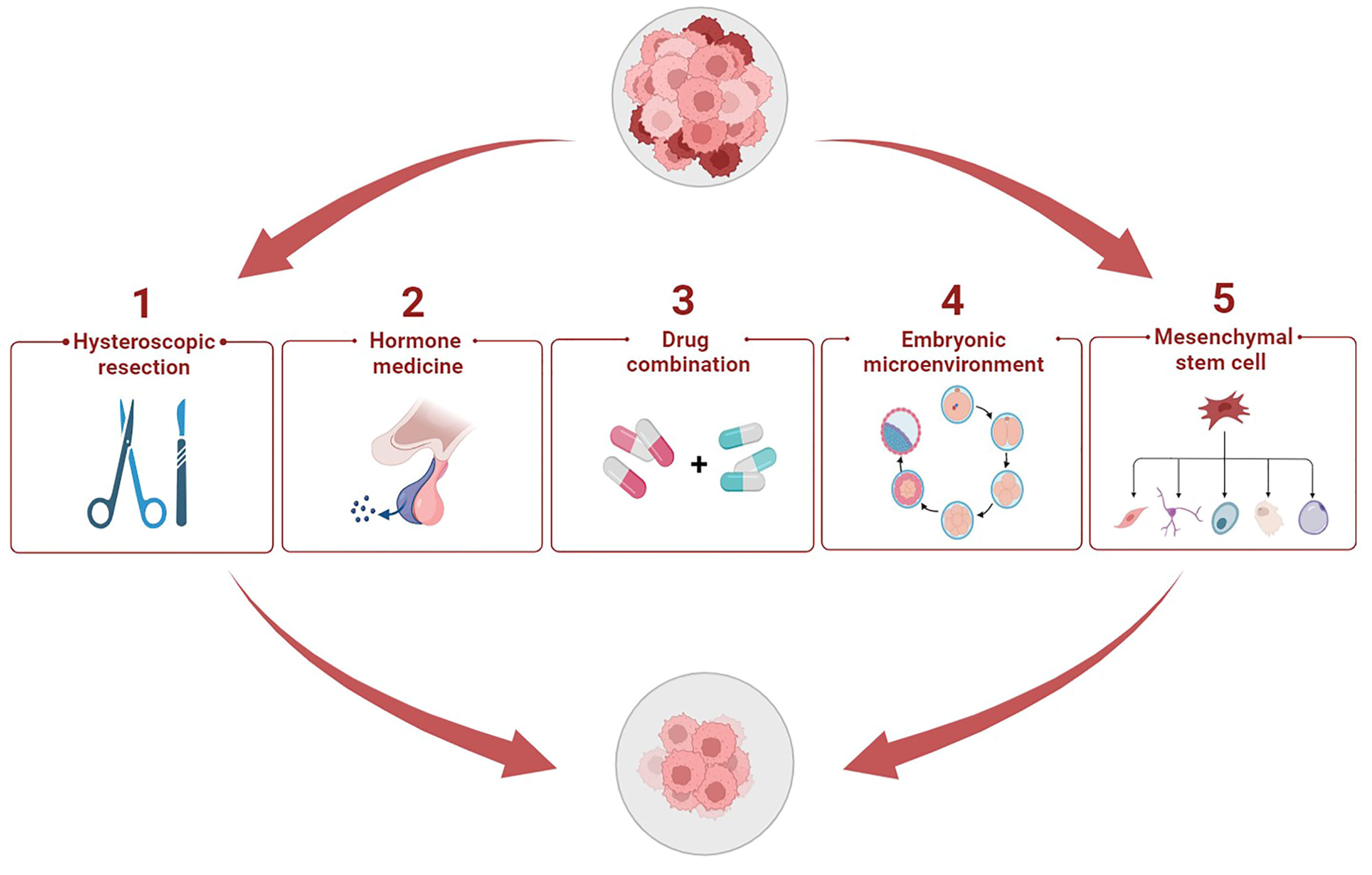
5. Potential Therapeutic Methods to Enhance Progestin Sensitivity
5.1. Hysteroscopic Resection plus Progestin
5.2. Hormone Medicine
5.3. Cocktail Drug Administration
5.4. Cytokines in Embryonic Microenvironment
5.5. Stem Cell Therapy Strategy
6. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Urick, M.E.; Bell, D.W. Clinical actionability of molecular targets in endometrial cancer. Nat. Rev. Cancer 2019, 19, 510–521. [Google Scholar] [CrossRef] [PubMed]
- Kesterson, J.P.; Fanning, J. Fertility-sparing treatment of endometrial cancer: Options, outcomes and pitfalls. J. Gynecol. Oncol. 2012, 23, 120–124. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Zhang, W.; Feng, L.; Gao, W. Comparison of fertility-sparing treatments in patients with early endometrial cancer and atypical complex hyperplasia: A meta-analysis and systematic review. Medicine 2017, 96, e8034. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Zaino, R.J.; Filiaci, V.J.; Leslie, K.K. Relationship of estrogen and progesterone receptors to clinical outcome in metastatic endometrial carcinoma: A Gynecologic Oncology Group Study. Gynecol. Oncol. 2007, 106, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Falcone, F.; Leone Roberti Maggiore, U.; Di Donato, V.; Perrone, A.M.; Frigerio, L.; Bifulco, G.; Polterauer, S.; Casadio, P.; Cormio, G.; Masciullo, V.; et al. Fertility-sparing treatment for intramucous, moderately differentiated, endometrioid endometrial cancer: A Gynecologic Cancer Inter-Group (GCIG) study. J. Gynecol. Oncol. 2020, 31, e74. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.; Obermair, A.; Gebski, V.; Janda, M. Efficacy of oral or intrauterine device-delivered progestin in patients with complex endometrial hyperplasia with atypia or early endometrial adenocarcinoma: A meta-analysis and systematic review of the literature. Gynecol. Oncol. 2012, 125, 263–270. [Google Scholar] [CrossRef] [Green Version]
- Dressing, G.E.; Goldberg, J.E.; Charles, N.J.; Schwertfeger, K.L.; Lange, C.A. Membrane progesterone receptor expression in mammalian tissues: A review of regulation and physiological implications. Steroids 2011, 76, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Anderson, E. The role of oestrogen and progesterone receptors in human mammary development and tumorigenesis. Breast Cancer Res. 2002, 4, 197–201. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Zhong, R.; He, X.; Deng, Q.; Peng, X.; Li, J.; Luo, X. Investigations on the mechanism of progesterone in inhibiting endometrial cancer cell cycle and viability via regulation of long noncoding RNA NEAT1/microRNA-146b-5p mediated Wnt/β-catenin signaling. IUBMB Life 2019, 71, 223–234. [Google Scholar] [CrossRef] [Green Version]
- Saegusa, M.; Hashimura, M.; Kuwata, T.; Hamano, M.; Okayasu, I. Beta-catenin simultaneously induces activation of the p53-p21WAF1 pathway and overexpression of cyclin D1 during squamous differentiation of endometrial carcinoma cells. Am. J. Pathol. 2004, 164, 1739–1749. [Google Scholar] [CrossRef]
- Tang, L.; Zhang, Y.; Pan, H.; Luo, Q.; Zhu, X.M.; Dong, M.Y.; Leung, P.C.; Sheng, J.Z.; Huang, H.F. Involvement of cyclin B1 in progesterone-mediated cell growth inhibition, G2/M cell cycle arrest, and apoptosis in human endometrial cell. Reprod. Biol. Endocrinol. 2009, 7, 144. [Google Scholar] [CrossRef] [Green Version]
- Patel, B.; Elguero, S.; Thakore, S.; Dahoud, W.; Bedaiwy, M.; Mesiano, S. Role of nuclear progesterone receptor isoforms in uterine pathophysiology. Hum. Reprod. Update 2015, 21, 155–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, C.; Zhang, Z.; Yu, Y.; Liu, Y.; Zhao, F.; Yin, L.; Feng, Y.; Chen, X. Inhibiting the PI3K/Akt pathway reversed progestin resistance in endometrial cancer. Cancer Sci. 2011, 102, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, J.; Sakaguchi, H.; Hirose, R.; Ichigo, S.; Tamaya, T. Progestins suppress estrogen-induced expression of vascular endothelial growth factor (VEGF) subtypes in uterine endometrial cancer cells. Cancer Lett. 1999, 141, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Mönckedieck, V.; Sannecke, C.; Husen, B.; Kumbartski, M.; Kimmig, R.; Tötsch, M.; Winterhager, E.; Grümmer, R. Progestins inhibit expression of MMPs and of angiogenic factors in human ectopic endometrial lesions in a mouse model. Mol. Hum. Reprod. 2009, 15, 633–643. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, J.; Toyoki, H.; Jahan, I.; Alam, S.M.; Sakaguchi, H.; Sato, E.; Tamaya, T. Sex steroid-dependent angiogenesis in uterine endometrial cancers. J. Steroid Biochem. Mol. Biol. 2005, 93, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Hyder, S.M.; Liang, Y.; Wu, J.; Welbern, V. Regulation of thrombospondin-1 by natural and synthetic progestins in human breast cancer cells. Endocr. Relat. Cancer 2009, 16, 809–817. [Google Scholar] [CrossRef]
- Amezcua, C.A.; Lu, J.J.; Felix, J.C.; Stanczyk, F.Z.; Zheng, W. Apoptosis may be an early event of progestin therapy for endometrial hyperplasia. Gynecol. Oncol. 2000, 79, 169–176. [Google Scholar] [CrossRef]
- McGlorthan, L.; Paucarmayta, A.; Casablanca, Y.; Maxwell, G.L.; Syed, V. Progesterone induces apoptosis by activation of caspase-8 and calcitriol via activation of caspase-9 pathways in ovarian and endometrial cancer cells in vitro. Apoptosis Int. J. Program. Cell Death 2021, 26, 184–194. [Google Scholar] [CrossRef]
- Ahola, T.M.; Alkio, N.; Manninen, T.; Ylikomi, T. Progestin and G protein-coupled receptor 30 inhibit mitogen-activated protein kinase activity in MCF-7 breast cancer cells. Endocrinology 2002, 143, 4620–4626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, X.; Li, M.; Shao, K.; Yang, Y.; Wang, Q.; Cai, M. Progesterone induces cell apoptosis via the CACNA2D3/Ca2+/p38 MAPK pathway in endometrial cancer. Oncol. Rep. 2020, 43, 121–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bravo, R.; Parra, V.; Gatica, D.; Rodriguez, A.E.; Torrealba, N.; Paredes, F.; Wang, Z.V.; Zorzano, A.; Hill, J.A.; Jaimovich, E.; et al. Endoplasmic reticulum and the unfolded protein response: Dynamics and metabolic integration. Int. Rev. Cell Mol. Biol. 2013, 301, 215–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.Y.; Jo, M.W.; Lee, E.Y.; Lee, D.Y.; Choi, D.S. Ovarian steroid dependence of endoplasmic reticulum stress involvement in endometrial cell apoptosis during the human endometrial cycle. Reproduction 2018, 155, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Varga, A.; Henriksen, E. Clinical and histopathologic evaluation of the effect of 17-alpha-hydroxyprogesterone-17-n-caproate on endometral carcinoma. Obstet. Gynecol. 1961, 18, 658–672. [Google Scholar]
- Wheeler, D.T.; Bristow, R.E.; Kurman, R.J. Histologic alterations in endometrial hyperplasia and well-differentiated carcinoma treated with progestins. Am. J. Surg. Pathol. 2007, 31, 988–998. [Google Scholar] [CrossRef]
- Saegusa, M.; Okayasu, I. Down-regulation of bcl-2 expression is closely related to squamous differentiation and progesterone therapy in endometrial carcinomas. J. Pathol. 1997, 182, 429–436. [Google Scholar] [CrossRef]
- Yoshino, O.; Osuga, Y.; Hirota, Y.; Koga, K.; Hirata, T.; Yano, T.; Ayabe, T.; Tsutsumi, O.; Taketani, Y. Endometrial stromal cells undergoing decidualization down-regulate their properties to produce proinflammatory cytokines in response to interleukin-1 beta via reduced p38 mitogen-activated protein kinase phosphorylation. J. Clin. Endocrinol. Metab. 2003, 88, 2236–2241. [Google Scholar] [CrossRef] [Green Version]
- Witkiewicz, A.K.; McConnell, T.; Potoczek, M.; Emmons, R.V.; Kurman, R.J. Increased natural killer cells and decreased regulatory T cells are seen in complex atypical endometrial hyperplasia and well-differentiated carcinoma treated with progestins. Hum. Pathol. 2010, 41, 26–32. [Google Scholar] [CrossRef]
- Karin, M. Nuclear factor-kappaB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef]
- Davies, S.; Dai, D.; Feldman, I.; Pickett, G.; Leslie, K.K. Identification of a novel mechanism of NF-kappaB inactivation by progesterone through progesterone receptors in Hec50co poorly differentiated endometrial cancer cells: Induction of A20 and ABIN-2. Gynecol. Oncol. 2004, 94, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Di Nezza, L.A.; Jobling, T.; Salamonsen, L.A. Progestin suppresses matrix metalloproteinase production in endometrial cancer. Gynecol. Oncol. 2003, 89, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- van der Horst, P.H.; Wang, Y.; Vandenput, I.; Kühne, L.C.; Ewing, P.C.; van Ijcken, W.F.; van der Zee, M.; Amant, F.; Burger, C.W.; Blok, L.J. Progesterone inhibits epithelial-to-mesenchymal transition in endometrial cancer. PLoS ONE 2012, 7, e30840. [Google Scholar] [CrossRef] [Green Version]
- Mehdinejadiani, S.; Amidi, F.; Mehdizadeh, M.; Barati, M.; Pazhohan, A.; Alyasin, A.; Mehdinejadiani, K.; Sobhani, A. Effects of letrozole and clomiphene citrate on Wnt signaling pathway in endometrium of polycystic ovarian syndrome and healthy women. Biol. Reprod. 2019, 100, 641–648. [Google Scholar] [CrossRef]
- Bokhari, A.A.; Lee, L.R.; Raboteau, D.; Hamilton, C.A.; Maxwell, G.L.; Rodriguez, G.C.; Syed, V. Progesterone inhibits endometrial cancer invasiveness by inhibiting the TGFβ pathway. Cancer Prev. Res. 2014, 7, 1045–1055. [Google Scholar] [CrossRef] [Green Version]
- Graham, J.D.; Clarke, C.L. Physiological action of progesterone in target tissues. Endocr. Rev. 1997, 18, 502–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, A.; Leavitt, W.W. Progestin-induced down regulation of nuclear estrogen receptor in uterine decidual cells: Analysis of receptor synthesis and turnover by the density-shift method. Biochem. Biophys. Res. Commun. 1986, 135, 98–104. [Google Scholar] [CrossRef]
- Kashima, H.; Horiuchi, A.; Uchikawa, J.; Miyamoto, T.; Suzuki, A.; Ashida, T.; Konishi, I.; Shiozawa, T. Up-regulation of nuclear receptor corepressor (NCoR) in progestin-induced growth suppression of endometrial hyperplasia and carcinoma. Anticancer Res. 2009, 29, 1023–1029. [Google Scholar]
- Luu-The, V.; Dufort, I.; Pelletier, G.; Labrie, F. Type 5 17beta-hydroxysteroid dehydrogenase: Its role in the formation of androgens in women. Mol. Cell. Endocrinol. 2001, 171, 77–82. [Google Scholar] [CrossRef]
- Yang, S.; Fang, Z.; Gurates, B.; Tamura, M.; Miller, J.; Ferrer, K.; Bulun, S.E. Stromal PRs mediate induction of 17beta-hydroxysteroid dehydrogenase type 2 expression in human endometrial epithelium: A paracrine mechanism for inactivation of E2. Mol. Endocrinol. 2001, 15, 2093–2105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utsunomiya, H.; Suzuki, T.; Ito, K.; Moriya, T.; Konno, R.; Sato, S.; Yaegashi, N.; Okamura, K.; Sasano, H. The correlation between the response to progestogen treatment and the expression of progesterone receptor B and 17beta-hydroxysteroid dehydrogenase type 2 in human endometrial carcinoma. Clin. Endocrinol. 2003, 58, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Sasagawa, S.; Shimizu, Y.; Kami, H.; Takeuchi, T.; Mita, S.; Imada, K.; Kato, S.; Mizuguchi, K. Dienogest is a selective progesterone receptor agonist in transactivation analysis with potent oral endometrial activity due to its efficient pharmacokinetic profile. Steroids 2008, 73, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Brenner, R.M.; Slayden, O.D. Progesterone receptor antagonists and the endometrial antiproliferative effect. Semin. Reprod. Med. 2005, 23, 74–81. [Google Scholar] [CrossRef]
- Hackenberg, R.; Beck, S.; Filmer, A.; Hushmand Nia, A.; Kunzmann, R.; Koch, M.; Slater, E.P.; Schulz, K.D. Androgen responsiveness of the new human endometrial cancer cell line MFE-296. Int. J. Cancer 1994, 57, 117–122. [Google Scholar] [CrossRef]
- Goddard, L.M.; Murphy, T.J.; Org, T.; Enciso, J.M.; Hashimoto-Partyka, M.K.; Warren, C.M.; Domigan, C.K.; McDonald, A.I.; He, H.; Sanchez, L.A.; et al. Progesterone receptor in the vascular endothelium triggers physiological uterine permeability preimplantation. Cell 2014, 156, 549–562. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Zhang, H.; Li, M.; Xue, J.; Fu, Y.; Yan, L.; Zhao, X. Normal endometrial stromal cells regulate survival and apoptosis signaling through PI3K/AKt/Survivin pathway in endometrial adenocarcinoma cells in vitro. Gynecol. Oncol. 2011, 123, 387–392. [Google Scholar] [CrossRef]
- Arnold, J.T.; Lessey, B.A.; Seppälä, M.; Kaufman, D.G. Effect of normal endometrial stroma on growth and differentiation in Ishikawa endometrial adenocarcinoma cells. Cancer Res. 2002, 62, 79–88. [Google Scholar]
- Kurita, T.; Young, P.; Brody, J.R.; Lydon, J.P.; O’Malley, B.W.; Cunha, G.R. Stromal Progesterone Receptors Mediate the Inhibitory Effects of Progesterone on Estrogen-Induced Uterine Epithelial Cell Deoxyribonucleic Acid Synthesis. Endocrinology 1998, 139, 4708–4713. [Google Scholar] [CrossRef]
- Li, Q.; Kannan, A.; DeMayo, F.J.; Lydon, J.P.; Cooke, P.S.; Yamagishi, H.; Srivastava, D.; Bagchi, M.K.; Bagchi, I.C. The antiproliferative action of progesterone in uterine epithelium is mediated by Hand2. Science 2011, 331, 912–916. [Google Scholar] [CrossRef] [Green Version]
- Jones, A.; Teschendorff, A.E.; Li, Q.; Hayward, J.D.; Kannan, A.; Mould, T.; West, J.; Zikan, M.; Cibula, D.; Fiegl, H.; et al. Role of DNA methylation and epigenetic silencing of HAND2 in endometrial cancer development. PLoS Med. 2013, 10, e1001551. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, C.E.; Young, P.C.; Stehman, F.B.; Sutton, G.P.; Alford, W.M. Steroid receptors and clinical outcome in patients with adenocarcinoma of the endometrium. Am. J. Obstet. Gynecol. 1988, 158, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Smid-Koopman, E.; Blok, L.J.; Kühne, L.C.; Burger, C.W.; Helmerhorst, T.J.; Brinkmann, A.O.; Huikeshoven, F.J. Distinct functional differences of human progesterone receptors A and B on gene expression and growth regulation in two endometrial carcinoma cell lines. J. Soc. Gynecol. Investig. 2003, 10, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Upson, K.; Allison, K.H.; Reed, S.D.; Jordan, C.D.; Newton, K.M.; Swisher, E.M.; Doherty, J.A.; Garcia, R.L. Biomarkers of progestin therapy resistance and endometrial hyperplasia progression. Am. J. Obstet. Gynecol. 2012, 207, 36.e1–36.e8. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Huang, C.; Kavlashvili, T.; Fronk, A.; Zhang, Y.; Wei, Y.; Dai, D.; Devor, E.J.; Meng, X.; Thiel, K.W.; et al. Loss of progesterone receptor through epigenetic regulation is associated with poor prognosis in solid tumors. Am. J. Cancer Res. 2020, 10, 1827–1843. [Google Scholar]
- Sasaki, M.; Dharia, A.; Oh, B.R.; Tanaka, Y.; Fujimoto, S.; Dahiya, R. Progesterone receptor B gene inactivation and CpG hypermethylation in human uterine endometrial cancer. Cancer Res. 2001, 61, 97–102. [Google Scholar]
- Qiu, M.; Lange, C.A. MAP kinases couple multiple functions of human progesterone receptors: Degradation, transcriptional synergy, and nuclear association. J. Steroid Biochem. Mol. Biol. 2003, 85, 147–157. [Google Scholar] [CrossRef]
- Lange, C.A.; Shen, T.; Horwitz, K.B. Phosphorylation of human progesterone receptors at serine-294 by mitogen-activated protein kinase signals their degradation by the 26S proteasome. Proc. Natl. Acad. Sci. USA 2000, 97, 1032–1037. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Jia, Y.; Liu, X.; Winters, C.; Wang, X.; Zhang, Y.; Devor, E.J.; Hovey, A.M.; Reyes, H.D.; Xiao, X.; et al. Systematic dissection of the mechanisms underlying progesterone receptor downregulation in endometrial cancer. Oncotarget 2014, 5, 9783–9797. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Hafiz, H.A.; Horwitz, K.B. Control of progesterone receptor transcriptional synergy by SUMOylation and deSUMOylation. BMC Mol. Biol. 2012, 13, 10. [Google Scholar] [CrossRef] [Green Version]
- Marquardt, R.M.; Kim, T.H.; Shin, J.H.; Jeong, J.W. Progesterone and Estrogen Signaling in the Endometrium: What Goes Wrong in Endometriosis? Int. J. Mol. Sci. 2019, 20, 3822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szwarc, M.M.; Lydon, J.P.; O’Malley, B.W. Steroid receptor coactivators as therapeutic targets in the female reproductive system. J. Steroid Biochem. Mol. Biol. 2015, 154, 32–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, A.R.E.; Sze, S.K.; Chung, H.H.; Lin, V.C. Delineation of critical amino acids in activation function 1 of progesterone receptor for recruitment of transcription coregulators. Biochim. Et Biophys. Acta. Gene Regul. Mech. 2019, 1862, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Ai, Z.; Wang, J.; Wang, Y.; Lu, L.; Tong, J.; Teng, Y. Overexpressed epidermal growth factor receptor (EGFR)-induced progestin insensitivity in human endometrial carcinoma cells by the EGFR/mitogen-activated protein kinase signaling pathway. Cancer 2010, 116, 3603–3613. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, T.; Watanabe, J.; Hata, H.; Jobo, T.; Kawaguchi, M.; Hattori, M.; Saito, M.; Kuramoto, H. Significance of progesterone receptor-A and -B expressions in endometrial adenocarcinoma. J. Steroid Biochem. Mol. Biol. 2004, 92, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Chen, H.; Zhang, M.; Xiong, H.; Jiang, Q. Knocking down FAM83B inhibits endometrial cancer cell proliferation and metastasis by silencing the PI3K/AKT/mTOR pathway. Biomed. Pharmacother. 2019, 115, 108939. [Google Scholar] [CrossRef]
- Xu, Y.; Tong, J.; Ai, Z.; Wang, J.; Teng, Y. Epidermal growth factor receptor signaling pathway involved in progestin-resistance of human endometrial carcinoma: In a mouse model. J. Obstet. Gynaecol. Res. 2012, 38, 1358–1366. [Google Scholar] [CrossRef]
- Dong, J.; Jiao, Y.; Mu, W.; Lu, B.; Wei, M.; Sun, L.; Hu, S.; Cui, B.; Liu, X.; Chen, Z.; et al. FKBP51 decreases cell proliferation and increases progestin sensitivity of human endometrial adenocarcinomas by inhibiting Akt. Oncotarget 2017, 8, 80405–80415. [Google Scholar] [CrossRef] [Green Version]
- Temkin, S.M.; Fleming, G. Current treatment of metastatic endometrial cancer. Cancer Control 2009, 16, 38–45. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Pudney, J.; Song, J.; Mor, G.; Schwartz, P.E.; Zheng, W. Mechanisms involved in the evolution of progestin resistance in human endometrial hyperplasia--precursor of endometrial cancer. Gynecol. Oncol. 2003, 88, 108–117. [Google Scholar] [CrossRef]
- Amezcua, C.A.; Zheng, W.; Muderspach, L.I.; Felix, J.C. Down-regulation of bcl-2 is a potential marker of the efficacy of progestin therapy in the treatment of endometrial hyperplasia. Gynecol. Oncol. 1999, 73, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef] [PubMed]
- Niture, S.K.; Kaspar, J.W.; Shen, J.; Jaiswal, A.K. Nrf2 signaling and cell survival. Toxicol. Appl. Pharmacol. 2010, 244, 37–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Chen, N.; Zhao, F.; Wang, X.J.; Kong, B.; Zheng, W.; Zhang, D.D. High levels of Nrf2 determine chemoresistance in type II endometrial cancer. Cancer Res. 2010, 70, 5486–5496. [Google Scholar] [CrossRef] [Green Version]
- Al-Sabbagh, M.; Lam, E.W.; Brosens, J.J. Mechanisms of endometrial progesterone resistance. Mol. Cell. Endocrinol. 2012, 358, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Wheelock, M.J.; Shintani, Y.; Maeda, M.; Fukumoto, Y.; Johnson, K.R. Cadherin switching. J. Cell Sci. 2008, 121, 727–735. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Li, W.; Kong, D.; Liu, Z.; Shi, Z.; Ma, X.; Li, Y.; Jiang, J. DACH1 suppresses epithelial to mesenchymal transition (EMT) through Notch1 pathway and reverses progestin resistance in endometrial carcinoma. Cancer Med. 2019, 8, 4380–4388. [Google Scholar] [CrossRef] [Green Version]
- Ali, S.H.; O’Donnell, A.L.; Balu, D.; Pohl, M.B.; Seyler, M.J.; Mohamed, S.; Mousa, S.; Dandona, P. Estrogen receptor-alpha in the inhibition of cancer growth and angiogenesis. Cancer Res. 2000, 60, 7094–7098. [Google Scholar]
- Zhao, S.; Li, G.; Yang, L.; Li, L.; Li, H. Response-specific progestin resistance in a newly characterized Ishikawa human endometrial cancer subcell line resulting from long-term exposure to medroxyprogesterone acetate. Oncol. Lett. 2013, 5, 139–144. [Google Scholar] [CrossRef] [Green Version]
- Simó, R.; Sáez-López, C.; Barbosa-Desongles, A.; Hernández, C.; Selva, D.M. Novel insights in SHBG regulation and clinical implications. Trends Endocrinol. Metab. 2015, 26, 376–383. [Google Scholar] [CrossRef]
- Gu, C.J.; Xie, F.; Zhang, B.; Yang, H.L.; Cheng, J.; He, Y.Y.; Zhu, X.Y.; Li, D.J.; Li, M.Q. High Glucose Promotes Epithelial-Mesenchymal Transition of Uterus Endometrial Cancer Cells by Increasing ER/GLUT4-Mediated VEGF Secretion. Cell. Physiol. Biochem. 2018, 50, 706–720. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.Y.; Xie, B.Y.; Yang, B.Y.; Ning, C.C.; Shan, W.W.; Gu, C.; Luo, X.Z.; Chen, X.J.; Zhang, Z.B.; Feng, Y.J. Increased TET1 Expression in Inflammatory Microenvironment of Hyperinsulinemia Enhances the Response of Endometrial Cancer to Estrogen by Epigenetic Modulation of GPER. J. Cancer 2017, 8, 894–902. [Google Scholar] [CrossRef]
- Yilmaz, E.; Melekoglu, R.; Taskapan, C.; Olmez Budak, F.; Toprak, S. The investigation of serum levels of ADAMTS 5 and 8 (the A disintegrin and metalloproteinase with thrombospondin motifs) in the etiology of endometrial cancer. J. Obstet. Gynaecol. 2020, 40, 856–859. [Google Scholar] [CrossRef]
- Li, W.; Wang, S.; Qiu, C.; Liu, Z.; Zhou, Q.; Kong, D.; Ma, X.; Jiang, J. Comprehensive bioinformatics analysis of acquired progesterone resistance in endometrial cancer cell line. J. Transl. Med. 2019, 17, 58. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Xie, L.; Zhang, H.; Zhu, Q.; Du, Y.; Luo, X.; Chen, X. Insulin resistance and overweight prolonged fertility-sparing treatment duration in endometrial atypical hyperplasia patients. J. Gynecol. Oncol. 2018, 29, e35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Zhou, D.; Lai, Y.; Liu, Y.; Tao, X.; Wang, Q.; Zhao, G.; Gu, H.; Liao, H.; Zhu, Y.; et al. Estrogen induces endometrial cancer cell proliferation and invasion by regulating the fat mass and obesity-associated gene via PI3K/AKT and MAPK signaling pathways. Cancer Lett. 2012, 319, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Seong, S.J.; Kim, T.J.; Kim, J.W.; Bae, D.S.; Nam, J.H. Significance of body weight change during fertility-sparing progestin therapy in young women with early endometrial cancer. Gynecol. Oncol. 2017, 146, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, L.; Che, X.; Li, W.; Liu, Z.; Jiang, J. Roles of SIRT1/FoxO1/SREBP-1 in the development of progestin resistance in endometrial cancer. Arch. Gynecol. Obstet. 2018, 298, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.Y.; Gulinazi, Y.; Du, Y.; Ning, C.C.; Cheng, Y.L.; Shan, W.W.; Luo, X.Z.; Zhang, H.W.; Zhu, Q.; Ma, F.H.; et al. Metformin plus megestrol acetate compared with megestrol acetate alone as fertility-sparing treatment in patients with atypical endometrial hyperplasia and well-differentiated endometrial cancer: A randomised controlled trial. BJOG Int. J. Obstet. Gynaecol. 2020, 127, 848–857. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Wang, Y.L.; Yu, L.; Hu, Q.; Ji, L.; Zhang, Y.; Liao, Q.P. Metformin promotes progesterone receptor expression via inhibition of mammalian target of rapamycin (mTOR) in endometrial cancer cells. J. Steroid Biochem. Mol. Biol. 2011, 126, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.; Zhu, X.L.; Liu, F.; Xu, Q.Y.; Ge, Q.L.; Jiang, S.H.; Yang, X.M.; Li, J.; Wang, Y.H.; Wu, Q.K.; et al. Cholesterol Synthetase DHCR24 Induced by Insulin Aggravates Cancer Invasion and Progesterone Resistance in Endometrial Carcinoma. Sci. Rep. 2017, 7, 41404. [Google Scholar] [CrossRef] [PubMed]
- Jordan, C.T.; Guzman, M.L.; Noble, M. Cancer stem cells. N. Engl. J. Med. 2006, 355, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Götte, M.; Wolf, M.; Staebler, A.; Buchweitz, O.; Kelsch, R.; Schüring, A.N.; Kiesel, L. Increased expression of the adult stem cell marker Musashi-1 in endometriosis and endometrial carcinoma. J. Pathol. 2008, 215, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Gargett, B.E.; Chan, R.W. Endometrial stem/progenitor cells and proliferative disorders of the endometrium. Minerva Ginecol. 2006, 58, 511–526. [Google Scholar] [PubMed]
- Sun, D.; Qin, Z.; Xu, Y.; Xiao, Q.; Xu, Y.; Bai, M.; Li, W.; Liu, Y.; Zheng, W.; Zhang, Z. The IVF-generated human embryonic microenvironment reverses progestin resistance in endometrial cancer cells by inducing cancer stem cell differentiation. Cancer Lett. 2022, 526, 311–321. [Google Scholar] [CrossRef]
- Raffone, A.; Travaglino, A.; Flacco, M.E.; Iasevoli, M.; Mollo, A.; Guida, M.; Insabato, L.; Di Spiezio Sardo, A.; Carugno, J.; Zullo, F. Clinical Predictive Factors of Response to Treatment in Patients Undergoing Conservative Management of Atypical Endometrial Hyperplasia and Early Endometrial Cancer. J. Adolesc. Young Adult Oncol. 2021, 10, 193–201. [Google Scholar] [CrossRef]
- Yang, Y.F.; Liao, Y.Y.; Liu, X.L.; Su, S.G.; Li, L.Z.; Peng, N.F. Prognostic factors of regression and relapse of complex atypical hyperplasia and well-differentiated endometrioid carcinoma with conservative treatment. Gynecol. Oncol. 2015, 139, 419–423. [Google Scholar] [CrossRef]
- Zhou, R.; Yang, Y.; Lu, Q.; Wang, J.; Miao, Y.; Wang, S.; Wang, Z.; Zhao, C.; Wei, L. Prognostic factors of oncological and reproductive outcomes in fertility-sparing treatment of complex atypical hyperplasia and low-grade endometrial cancer using oral progestin in Chinese patients. Gynecol. Oncol. 2015, 139, 424–428. [Google Scholar] [CrossRef]
- Dancey, J. mTOR signaling and drug development in cancer. Nat. Rev. Clin. Oncol. 2010, 7, 209–219. [Google Scholar] [CrossRef]
- Salvesen, H.B.; Carter, S.L.; Mannelqvist, M.; Dutt, A.; Getz, G.; Stefansson, I.M.; Raeder, M.B.; Sos, M.L.; Engelsen, I.B.; Trovik, J.; et al. Integrated genomic profiling of endometrial carcinoma associates aggressive tumors with indicators of PI3 kinase activation. Proc. Natl. Acad. Sci. USA 2009, 106, 4834–4839. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Zhang, L.; Zhang, X.; Cui, Z. PI3K/AKT/mTOR pathway promotes progestin resistance in endometrial cancer cells by inhibition of autophagy. OncoTargets Ther. 2017, 10, 2865–2871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.; Dong, S.M.; Kim, B.R.; Park, M.S.; Trink, B.; Byun, H.J.; Rho, S.B. Thioridazine induces apoptosis by targeting the PI3K/Akt/mTOR pathway in cervical and endometrial cancer cells. Apoptosis 2012, 17, 989–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granville, C.A.; Memmott, R.M.; Gills, J.J.; Dennis, P.A. Handicapping the race to develop inhibitors of the phosphoinositide 3-kinase/Akt/mammalian target of rapamycin pathway. Clin. Cancer Res. 2006, 12, 679–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutter, G.L.; Lin, M.C.; Fitzgerald, J.T.; Kum, J.B.; Baak, J.P.; Lees, J.A.; Weng, L.P.; Eng, C. Altered PTEN expression as a diagnostic marker for the earliest endometrial precancers. J. Natl. Cancer Inst. 2000, 92, 924–930. [Google Scholar] [CrossRef]
- Milam, M.R.; Soliman, P.T.; Chung, L.H.; Schmeler, K.M.; Bassett, R.L., Jr.; Broaddus, R.R.; Lu, K.H. Loss of phosphatase and tensin homologue deleted on chromosome 10 and phosphorylation of mammalian target of rapamycin are associated with progesterone refractory endometrial hyperplasia. Int. J. Gynecol. Cancer 2008, 18, 146–151. [Google Scholar] [CrossRef]
- Guertin, D.A.; Sabatini, D.M. An expanding role for mTOR in cancer. Trends Mol. Med. 2005, 11, 353–361. [Google Scholar] [CrossRef]
- Ni, M.; Lee, A.S. ER chaperones in mammalian development and human diseases. FEBS Lett. 2007, 581, 3641–3651. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, K.; Gray, M.J.; Yang, D.Y.; Srivastava, S.A.; Tripathi, P.B.; Sonoda, L.A.; Yoo, E.J.; Dubeau, L.; Lee, A.S.; Lin, Y.G. The endoplasmic reticulum stress marker, glucose-regulated protein-78 (GRP78) in visceral adipocytes predicts endometrial cancer progression and patient survival. Gynecol. Oncol. 2013, 128, 552–559. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.G.; Shen, J.; Yoo, E.; Liu, R.; Yen, H.Y.; Mehta, A.; Rajaei, A.; Yang, W.; Mhawech-Fauceglia, P.; DeMayo, F.J.; et al. Targeting the glucose-regulated protein-78 abrogates Pten-null driven AKT activation and endometrioid tumorigenesis. Oncogene 2015, 34, 5418–5426. [Google Scholar] [CrossRef] [Green Version]
- Yue, Y.; Zhou, K.; Li, J.; Jiang, S.; Li, C.; Men, H. MSX1 induces G0/G1 arrest and apoptosis by suppressing Notch signaling and is frequently methylated in cervical cancer. OncoTargets Ther. 2018, 11, 4769–4780. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Cui, Y.; Huang, T.; Sun, X.; Wang, Y. Identification and Validation of MSX1 as a Key Candidate for Progestin Resistance in Endometrial Cancer. OncoTargets Ther. 2020, 13, 11669–11688. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.; Yi, X.; Abushahin, N.; Pang, S.; Zhang, D.; Kong, B.; Zheng, W. Nrf2 expression in endometrial serous carcinomas and its precancers. Int. J. Clin. Exp. Pathol. 2010, 4, 85–96. [Google Scholar]
- Yang, B.; Hu, M.; Fu, Y.; Sun, D.; Zheng, W.; Liao, H.; Zhang, Z.; Chen, X. LASS2 mediates Nrf2-driven progestin resistance in endometrial cancer. Am. J. Transl. Res. 2021, 13, 1280–1289. [Google Scholar] [PubMed]
- Fan, R.; Wang, Y.; Wang, Y.; Wei, L.; Zheng, W. Mechanism of progestin resistance in endometrial precancer/cancer through Nrf2-survivin pathway. Am J Transl Res 2017, 9, 1483–1491. [Google Scholar] [PubMed]
- Wang, Y.; Wang, Y.; Zhang, Z.; Park, J.Y.; Guo, D.; Liao, H.; Yi, X.; Zheng, Y.; Zhang, D.; Chambers, S.K.; et al. Mechanism of progestin resistance in endometrial precancer/cancer through Nrf2-AKR1C1 pathway. Oncotarget 2016, 7, 10363–10372. [Google Scholar] [CrossRef] [Green Version]
- Rizner, T.L.; Smuc, T.; Rupreht, R.; Sinkovec, J.; Penning, T.M. AKR1C1 and AKR1C3 may determine progesterone and estrogen ratios in endometrial cancer. Mol. Cell. Endocrinol. 2006, 248, 126–135. [Google Scholar] [CrossRef]
- Ji, Q.; Aoyama, C.; Nien, Y.D.; Liu, P.I.; Chen, P.K.; Chang, L.; Stanczyk, F.Z.; Stolz, A. Selective loss of AKR1C1 and AKR1C2 in breast cancer and their potential effect on progesterone signaling. Cancer Res. 2004, 64, 7610–7617. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zhang, Z.; Feng, Y.; Fadare, O.; Wang, J.; Ai, Z.; Jin, H.; Gu, C.; Zheng, W. Aberrant survivin expression in endometrial hyperplasia: Another mechanism of progestin resistance. Mod. Pathol. 2009, 22, 699–708. [Google Scholar] [CrossRef] [Green Version]
- Laviad, E.L.; Albee, L.; Pankova-Kholmyansky, I.; Epstein, S.; Park, H.; Merrill, A.H., Jr.; Futerman, A.H. Characterization of ceramide synthase 2: Tissue distribution, substrate specificity, and inhibition by sphingosine 1-phosphate. J. Biol. Chem. 2008, 283, 5677–5684. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Dong, L.; Sui, L.; Yang, Y.; Liu, X.; Yu, Y.; Zhu, Y.; Feng, Y. Metformin reverses progestin resistance in endometrial cancer cells by downregulating GloI expression. Int. J. Gynecol. Cancer 2011, 21, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zhang, Y.; Yang, X.; Lu, P.; Yan, X.; Xiao, F.; Zhou, H.; Wen, C.; Shi, M.; Lu, J.; et al. Effects of methylglyoxal and glyoxalase I inhibition on breast cancer cells proliferation, invasion, and apoptosis through modulation of MAPKs, MMP9, and Bcl-2. Cancer Biol. Ther. 2016, 17, 169–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Chen, X.; Wei, Y.; Feng, Y.; Zheng, W.; Zhang, Z. Metformin sensitizes endometrial cancer cells to progestin by targeting TET1 to downregulate glyoxalase I expression. Biomed. Pharmacother. 2019, 113, 108712. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S. Review of epidermal growth factor receptor biology. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Chen, X.; Lu, X.; Yu, Y.; Feng, Y. Epidermal growth factor receptor signaling enhanced by long-term medroxyprogesterone acetate treatment in endometrial carcinoma. Gynecol. Oncol. 2007, 105, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Olayioye, M.A.; Neve, R.M.; Lane, H.A.; Hynes, N.E. The ErbB signaling network: Receptor heterodimerization in development and cancer. EMBO J. 2000, 19, 3159–3167. [Google Scholar] [CrossRef] [Green Version]
- Konecny, G.E.; Santos, L.; Winterhoff, B.; Hatmal, M.; Keeney, G.L.; Mariani, A.; Jones, M.; Neuper, C.; Thomas, B.; Muderspach, L.; et al. HER2 gene amplification and EGFR expression in a large cohort of surgically staged patients with nonendometrioid (type II) endometrial cancer. Br. J. Cancer 2009, 100, 89–95. [Google Scholar] [CrossRef]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [Green Version]
- de Boer, V.C.; de Goffau, M.C.; Arts, I.C.; Hollman, P.C.; Keijer, J. SIRT1 stimulation by polyphenols is affected by their stability and metabolism. Mech. Ageing Dev. 2006, 127, 618–627. [Google Scholar] [CrossRef]
- Fang, Y.; Nicholl, M.B. Sirtuin 1 in malignant transformation: Friend or foe? Cancer Lett. 2011, 306, 10–14. [Google Scholar] [CrossRef]
- Lin, L.; Zheng, X.; Qiu, C.; Dongol, S.; Lv, Q.; Jiang, J.; Kong, B.; Wang, C. SIRT1 promotes endometrial tumor growth by targeting SREBP1 and lipogenesis. Oncol. Rep. 2014, 32, 2831–2835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luu, W.; Zerenturk, E.J.; Kristiana, I.; Bucknall, M.P.; Sharpe, L.J.; Brown, A.J. Signaling regulates activity of DHCR24, the final enzyme in cholesterol synthesis. J. Lipid Res. 2014, 55, 410–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zerenturk, E.J.; Sharpe, L.J.; Ikonen, E.; Brown, A.J. Desmosterol and DHCR24: Unexpected new directions for a terminal step in cholesterol synthesis. Prog. Lipid Res. 2013, 52, 666–680. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.N.; Gerulath, A.H.; Cecutti, A.; Bhavnani, B.R. Discordant expression of insulin-like growth factors and their receptor messenger ribonucleic acids in endometrial carcinomas relative to normal endometrium. Mol. Cell. Endocrinol. 1999, 153, 19–27. [Google Scholar] [CrossRef]
- Navarro, M.; Baserga, R. Limited redundancy of survival signals from the type 1 insulin-like growth factor receptor. Endocrinology 2001, 142, 1073–1081. [Google Scholar] [CrossRef]
- Suda, T.; Takahashi, T.; Golstein, P.; Nagata, S. Molecular cloning and expression of the Fas ligand, a novel member of the tumor necrosis factor family. Cell 1993, 75, 1169–1178. [Google Scholar] [CrossRef]
- Song, J.; Rutherford, T.; Naftolin, F.; Brown, S.; Mor, G. Hormonal regulation of apoptosis and the Fas and Fas ligand system in human endometrial cells. Mol. Hum. Reprod. 2002, 8, 447–455. [Google Scholar] [CrossRef]
- Schenk, R.L.; Strasser, A.; Dewson, G. BCL-2: Long and winding path from discovery to therapeutic target. Biochem. Biophys. Res. Commun. 2017, 482, 459–469. [Google Scholar] [CrossRef]
- Vereide, A.B.; Kaino, T.; Sager, G.; Ørbo, A. Bcl-2, BAX, and apoptosis in endometrial hyperplasia after high dose gestagen therapy: A comparison of responses in patients treated with intrauterine levonorgestrel and systemic medroxyprogesterone. Gynecol. Oncol. 2005, 97, 740–750. [Google Scholar] [CrossRef]
- Ding, L.; Zhang, X.; Zhao, M.; Qu, Z.; Huang, S.; Dong, M.; Gao, F. An essential role of PDCD4 in progression and malignant proliferation of gastrointestinal stromal tumors. Med. Oncol. 2012, 29, 1758–1764. [Google Scholar] [CrossRef]
- Lu, K.; Chen, Q.; Li, M.; He, L.; Riaz, F.; Zhang, T.; Li, D. Programmed cell death factor 4 (PDCD4), a novel therapy target for metabolic diseases besides cancer. Free Radic. Biol. Med. 2020, 159, 150–163. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, Y.; Wan, L.; Liu, Y.; Sun, Y.; Liu, Y.; Shi, Y.; Zhang, L.; Zhou, H.; Wang, J.; et al. Downregulation of PDCD4 induced by progesterone is mediated by the PI3K/AKT signaling pathway in human endometrial cancer cells. Oncol. Rep. 2019, 42, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Umene, K.; Banno, K.; Kisu, I.; Yanokura, M.; Nogami, Y.; Tsuji, K.; Masuda, K.; Ueki, A.; Kobayashi, Y.; Yamagami, W.; et al. New candidate therapeutic agents for endometrial cancer: Potential for clinical practice (review). Oncol. Rep. 2013, 29, 855–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cochrane, D.R.; Spoelstra, N.S.; Richer, J.K. The role of miRNAs in progesterone action. Mol. Cell. Endocrinol. 2012, 357, 50–59. [Google Scholar] [CrossRef]
- Woo, C.J.; Kingston, R.E. HOTAIR lifts noncoding RNAs to new levels. Cell 2007, 129, 1257–1259. [Google Scholar] [CrossRef] [Green Version]
- Chi, S.; Liu, Y.; Zhou, X.; Feng, D.; Xiao, X.; Li, W.; Zhao, Y.; Wang, H. Knockdown of long non-coding HOTAIR enhances the sensitivity to progesterone in endometrial cancer by epigenetic regulation of progesterone receptor isoform B. Cancer Chemother. Pharmacol. 2019, 83, 277–287. [Google Scholar] [CrossRef]
- Popov, V.M.; Zhou, J.; Shirley, L.A.; Quong, J.; Yeow, W.S.; Wright, J.A.; Wu, K.; Rui, H.; Vadlamudi, R.K.; Jiang, J.; et al. The cell fate determination factor DACH1 is expressed in estrogen receptor-alpha-positive breast cancer and represses estrogen receptor-alpha signaling. Cancer Res. 2009, 69, 5752–5760. [Google Scholar] [CrossRef]
- Wu, K.; Katiyar, S.; Witkiewicz, A.; Li, A.; McCue, P.; Song, L.N.; Tian, L.; Jin, M.; Pestell, R.G. The cell fate determination factor dachshund inhibits androgen receptor signaling and prostate cancer cellular growth. Cancer Res. 2009, 69, 3347–3355. [Google Scholar] [CrossRef] [Green Version]
- Nan, F.; Lü, Q.; Zhou, J.; Cheng, L.; Popov, V.M.; Wei, S.; Kong, B.; Pestell, R.G.; Lisanti, M.P.; Jiang, J.; et al. Altered expression of DACH1 and cyclin D1 in endometrial cancer. Cancer Biol. Ther. 2009, 8, 1534–1539. [Google Scholar] [CrossRef] [Green Version]
- Caumanns, J.J.; Wisman, G.B.A.; Berns, K.; van der Zee, A.G.J.; de Jong, S. ARID1A mutant ovarian clear cell carcinoma: A clear target for synthetic lethal strategies. Biochim. Et Biophys. Acta. Rev. Cancer 2018, 1870, 176–184. [Google Scholar] [CrossRef]
- Werner, H.M.; Berg, A.; Wik, E.; Birkeland, E.; Krakstad, C.; Kusonmano, K.; Petersen, K.; Kalland, K.H.; Oyan, A.M.; Akslen, L.A.; et al. ARID1A loss is prevalent in endometrial hyperplasia with atypia and low-grade endometrioid carcinomas. Mod. Pathol. 2013, 26, 428–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Tang, Z.; Li, T.; Liu, M.; Li, Y.; Xing, B. CRISPR/Cas9-Mediated Gene Knockout of ARID1A Promotes Primary Progesterone Resistance by Downregulating Progesterone Receptor B in Endometrial Cancer Cells. Oncol. Res. 2019, 27, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.R.; Reske, J.J.; Koeman, J.; Adams, M.; Joshi, N.R.; Fazleabas, A.T.; Chandler, R.L. SWI/SNF Antagonism of PRC2 Mediates Estrogen-Induced Progesterone Receptor Expression. Cells 2022, 11, 1000. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; Benz, C.C.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Thiel, K.W.; Leslie, K.K. Progesterone: The ultimate endometrial tumor suppressor. Trends Endocrinol. Metab. 2011, 22, 145–152. [Google Scholar] [CrossRef] [Green Version]
- Trojano, G.; Olivieri, C.; Tinelli, R.; Damiani, G.R.; Pellegrino, A.; Cicinelli, E. Conservative treatment in early stage endometrial cancer: A review. Acta Bio-Med. Atenei Parm. 2019, 90, 405–410. [Google Scholar] [CrossRef]
- Zakhour, M.; Cohen, J.G.; Gibson, A.; Walts, A.E.; Karimian, B.; Baltayan, A.; Aoyama, C.; Garcia, L.; Dhaliwal, S.K.; Elashoff, D.; et al. Abnormal mismatch repair and other clinicopathologic predictors of poor response to progestin treatment in young women with endometrial complex atypical hyperplasia and well-differentiated endometrial adenocarcinoma: A consecutive case series. BJOG Int. J. Obstet. Gynaecol. 2017, 124, 1576–1583. [Google Scholar] [CrossRef]
- Raffone, A.; Travaglino, A.; Cerbone, M.; Gencarelli, A.; Mollo, A.; Insabato, L.; Zullo, F. Diagnostic Accuracy of Immunohistochemistry for Mismatch Repair Proteins as Surrogate of Microsatellite Instability Molecular Testing in Endometrial Cancer. Pathol. Oncol. Res. 2020, 26, 1417–1427. [Google Scholar] [CrossRef]
- Chung, Y.S.; Woo, H.Y.; Lee, J.Y.; Park, E.; Nam, E.J.; Kim, S.; Kim, S.W.; Kim, Y.T. Mismatch repair status influences response to fertility-sparing treatment of endometrial cancer. Am. J. Obstet. Gynecol. 2021, 224, 370.e1–370.e13. [Google Scholar] [CrossRef]
- Xi, Y.; Liu, G.; Liu, D.; Jiang, J.; Gong, R. Efficacy and pregnancy outcomes of hysteroscopic surgery combined with progestin as fertility-sparing therapy in patients with early stage endometrial cancer and atypical hyperplasia. Arch. Gynecol. Obstet. 2022, 1–8. [Google Scholar] [CrossRef]
- Yang, B.; Xu, Y.; Zhu, Q.; Xie, L.; Shan, W.; Ning, C.; Xie, B.; Shi, Y.; Luo, X.; Zhang, H.; et al. Treatment efficiency of comprehensive hysteroscopic evaluation and lesion resection combined with progestin therapy in young women with endometrial atypical hyperplasia and endometrial cancer. Gynecol. Oncol. 2019, 153, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giampaolino, P.; Di Spiezio Sardo, A.; Mollo, A.; Raffone, A.; Travaglino, A.; Boccellino, A.; Zizolfi, B.; Insabato, L.; Zullo, F.; De Placido, G.; et al. Hysteroscopic Endometrial Focal Resection followed by Levonorgestrel Intrauterine Device Insertion as a Fertility-Sparing Treatment of Atypical Endometrial Hyperplasia and Early Endometrial Cancer: A Retrospective Study. J. Minim. Invasive Gynecol. 2019, 26, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Satyaswaroop, P.G.; Zaino, R.J.; Mortel, R. Estrogen-like effects of tamoxifen on human endometrial carcinoma transplanted into nude mice. Cancer Res. 1984, 44, 4006–4010. [Google Scholar] [PubMed]
- Whitney, C.W.; Brunetto, V.L.; Zaino, R.J.; Lentz, S.S.; Sorosky, J.; Armstrong, D.K.; Lee, R.B. Phase II study of medroxyprogesterone acetate plus tamoxifen in advanced endometrial carcinoma: A Gynecologic Oncology Group study. Gynecol. Oncol. 2004, 92, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Satyaswaroop, P.G.; Clarke, C.L.; Zaino, R.J.; Mortel, R. Apparent resistance in human endometrial carcinoma during combination treatment with tamoxifen and progestin may result from desensitization following downregulation of tumor progesterone receptor. Cancer Lett. 1992, 62, 107–114. [Google Scholar] [CrossRef]
- Meyer, M.E.; Pornon, A.; Ji, J.W.; Bocquel, M.T.; Chambon, P.; Gronemeyer, H. Agonistic and antagonistic activities of RU486 on the functions of the human progesterone receptor. EMBO J. 1990, 9, 3923–3932. [Google Scholar] [CrossRef]
- Moe, B.G.; Vereide, A.B.; Orbo, A.; Sager, G. High concentrations of progesterone and mifepristone mutually reinforce cell cycle retardation and induction of apoptosis. Anticancer Res. 2009, 29, 1053–1058. [Google Scholar]
- Murphy, A.A.; Zhou, M.H.; Malkapuram, S.; Santanam, N.; Parthasarathy, S.; Sidell, N. RU486-induced growth inhibition of human endometrial cells. Fertil Steril 2000, 74, 1014–1019. [Google Scholar] [CrossRef]
- Han, S.; Sidell, N. RU486-induced growth inhibition of human endometrial cells involves the nuclear factor-kappa B signaling pathway. J. Clin. Endocrinol. Metab. 2003, 88, 713–719. [Google Scholar] [CrossRef] [Green Version]
- Li, C.Z.; Wen, Z.Q.; Lan, S.M.; Wang, J.Y.; Liu, Y. Study on the treatment of high dose mifepristone and progesterone in endometrial carcinoma. Zhonghua Fu Chan Ke Za Zhi 2003, 38, 552–555. [Google Scholar]
- Navo, M.A.; Smith, J.A.; Gaikwad, A.; Burke, T.; Brown, J.; Ramondetta, L.M. In vitro evaluation of the growth inhibition and apoptosis effect of mifepristone (RU486) in human Ishikawa and HEC1A endometrial cancer cell lines. Cancer Chemother. Pharmacol. 2008, 62, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; Cao, D.Y.; Zhou, H.M.; Yu, M.; Yang, J.X.; Wang, J.H.; Zhang, Y.; Cheng, N.H.; Peng, P. GnRH-a combined fertility-sparing re-treatment in women with endometrial carcinoma or atypical endomertial hyperplasia who failed to oral progestin therapy. Zhonghua Fu Chan Ke Za Zhi 2021, 56, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Jiang, S.; Tian, W.; Yan, Y.; Gao, C.; Gao, J.; Sheng, Y.; Wang, Y.; Xue, F. Preliminary clinical application of an aromatase inhibitor and a gonadotropin-releasing hormone agonist combination for inoperable endometrial cancer patients with comorbidities: Case report and literature review. Cancer Biol. Ther. 2018, 19, 956–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Cao, D.; Yang, J.; Yu, M.; Zhou, H.; Cheng, N.; Wang, J.; Zhang, Y.; Peng, P.; Shen, K. Fertility-Sparing Treatment for Endometrial Cancer or Atypical Endometrial Hyperplasia Patients With Obesity. Front. Oncol. 2022, 12, 812346. [Google Scholar] [CrossRef] [PubMed]
- Straubhar, A.; Soisson, A.P.; Dodson, M.; Simons, E. Successful treatment of low-grade endometrial cancer in premenopausal women with an aromatase inhibitor after failure with oral or intrauterine progesterone. Gynecol. Oncol. Rep. 2017, 21, 10–12. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Babayev, S.; Carr, B.R.; Keller, P.W.; Word, R.A.; Bukulmez, O. Androgen regulation of progesterone receptor (PR) expression in endometrium: Implications for endometriosis. Fertil. Steril. 2014, 102, e79–e80. [Google Scholar] [CrossRef]
- Hackenberg, R.; Schulz, K.D. Androgen receptor mediated growth control of breast cancer and endometrial cancer modulated by antiandrogen- and androgen-like steroids. J. Steroid Biochem. Mol. Biol. 1996, 56, 113–117. [Google Scholar] [CrossRef]
- Tangen, I.L.; Onyango, T.B.; Kopperud, R.; Berg, A.; Halle, M.K.; Øyan, A.M.; Werner, H.M.; Trovik, J.; Kalland, K.H.; Salvesen, H.B.; et al. Androgen receptor as potential therapeutic target in metastatic endometrial cancer. Oncotarget 2016, 7, 49289–49298. [Google Scholar] [CrossRef] [Green Version]
- Katsuki, Y.; Shibutani, Y.; Aoki, D.; Nozawa, S. Dienogest, a novel synthetic steroid, overcomes hormone-dependent cancer in a different manner than progestins. Cancer 1997, 79, 169–176. [Google Scholar] [CrossRef]
- Ma, A.Y.; Xie, S.W.; Zhou, J.Y.; Zhu, Y. Nomegestrol Acetate Suppresses Human Endometrial Cancer RL95-2 Cells Proliferation In Vitro and In Vivo Possibly Related to Upregulating Expression of SUFU and Wnt7a. Int. J. Mol. Sci. 2017, 18, 1337. [Google Scholar] [CrossRef] [Green Version]
- Cao, C.; Zhou, J.Y.; Xie, S.W.; Guo, X.J.; Li, G.T.; Gong, Y.J.; Yang, W.J.; Li, Z.; Zhong, R.H.; Shao, H.H.; et al. Metformin Enhances Nomegestrol Acetate Suppressing Growth of Endometrial Cancer Cells and May Correlate to Downregulating mTOR Activity In Vitro and In Vivo. Int. J. Mol. Sci. 2019, 20, 3308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyo, S.; Nakayama, K. Endometrial Cancer as a Metabolic Disease with Dysregulated PI3K Signaling: Shedding Light on Novel Therapeutic Strategies. Int. J. Mol. Sci. 2020, 21, 6073. [Google Scholar] [CrossRef] [PubMed]
- Yates, M.S.; Coletta, A.M.; Zhang, Q.; Schmandt, R.E.; Medepalli, M.; Nebgen, D.; Soletsky, B.; Milbourne, A.; Levy, E.; Fellman, B.; et al. Prospective Randomized Biomarker Study of Metformin and Lifestyle Intervention for Prevention in Obese Women at Increased Risk for Endometrial Cancer. Cancer Prev. Res. 2018, 11, 477–490. [Google Scholar] [CrossRef] [Green Version]
- Mitsuhashi, A.; Sato, Y.; Kiyokawa, T.; Koshizaka, M.; Hanaoka, H.; Shozu, M. Phase II study of medroxyprogesterone acetate plus metformin as a fertility-sparing treatment for atypical endometrial hyperplasia and endometrial cancer. Ann. Oncol. 2016, 27, 262–266. [Google Scholar] [CrossRef]
- Mitsuhashi, A.; Habu, Y.; Kobayashi, T.; Kawarai, Y.; Ishikawa, H.; Usui, H.; Shozu, M. Long-term outcomes of progestin plus metformin as a fertility-sparing treatment for atypical endometrial hyperplasia and endometrial cancer patients. J. Gynecol. Oncol. 2019, 30, e90. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Wu, H.; Yang, L.; Huang, T.; Li, J.; Gong, X.; Li, L.; Sun, X.; Mao, F.; Wang, Y. Chlorpromazine Sensitizes Progestin-Resistant Endometrial Cancer Cells to MPA by Upregulating PRB. Front. Oncol. 2021, 11, 665832. [Google Scholar] [CrossRef]
- Meng, Q.; Sun, X.; Wang, J.; Wang, Y.; Wang, L. The important application of thioridazine in the endometrial cancer. Am. J. Transl. Res. 2016, 8, 2767–2775. [Google Scholar]
- Pant, A.; Lee, I.I.; Lu, Z.; Rueda, B.R.; Schink, J.; Kim, J.J. Inhibition of AKT with the orally active allosteric AKT inhibitor, MK-2206, sensitizes endometrial cancer cells to progestin. PLoS ONE 2012, 7, e41593. [Google Scholar] [CrossRef]
- Neubauer, N.L.; Ward, E.C.; Patel, P.; Lu, Z.; Lee, I.; Blok, L.J.; Hanifi-Moghaddam, P.; Schink, J.; Kim, J.J. Progesterone receptor-B induction of BIRC3 protects endometrial cancer cells from AP1-59-mediated apoptosis. Horm. Cancer 2011, 2, 170–181. [Google Scholar] [CrossRef] [Green Version]
- Oza, A.M.; Elit, L.; Tsao, M.S.; Kamel-Reid, S.; Biagi, J.; Provencher, D.M.; Gotlieb, W.H.; Hoskins, P.J.; Ghatage, P.; Tonkin, K.S.; et al. Phase II study of temsirolimus in women with recurrent or metastatic endometrial cancer: A trial of the NCIC Clinical Trials Group. J. Clin. Oncol. 2011, 29, 3278–3285. [Google Scholar] [CrossRef] [Green Version]
- Slomovitz, B.M.; Lu, K.H.; Johnston, T.; Coleman, R.L.; Munsell, M.; Broaddus, R.R.; Walker, C.; Ramondetta, L.M.; Burke, T.W.; Gershenson, D.M.; et al. A phase 2 study of the oral mammalian target of rapamycin inhibitor, everolimus, in patients with recurrent endometrial carcinoma. Cancer 2010, 116, 5415–5419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oza, A.M.; Pignata, S.; Poveda, A.; McCormack, M.; Clamp, A.; Schwartz, B.; Cheng, J.; Li, X.; Campbell, K.; Dodion, P.; et al. Randomized Phase II Trial of Ridaforolimus in Advanced Endometrial Carcinoma. J. Clin. Oncol. 2015, 33, 3576–3582. [Google Scholar] [CrossRef] [PubMed]
- Bae-Jump, V.L.; Zhou, C.; Boggess, J.F.; Whang, Y.E.; Barroilhet, L.; Gehrig, P.A. Rapamycin inhibits cell proliferation in type I and type II endometrial carcinomas: A search for biomarkers of sensitivity to treatment. Gynecol. Oncol. 2010, 119, 579–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banno, K.; Kisu, I.; Yanokura, M.; Masuda, K.; Ueki, A.; Kobayashi, Y.; Susumu, N.; Aoki, D. Epigenetics and genetics in endometrial cancer: New carcinogenic mechanisms and relationship with clinical practice. Epigenomics 2012, 4, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Dowdy, S.C.; Gonzalez Bosquet, J.; Zhao, Y.; Eberhardt, N.L.; Podratz, K.C.; Jiang, S.W. Epigenetic-mediated upregulation of progesterone receptor B gene in endometrial cancer cell lines. Gynecol. Oncol. 2005, 99, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Xiao, X.; Jia, Y.; Liu, X.; Zhang, Y.; Wang, X.; Winters, C.J.; Devor, E.J.; Meng, X.; Thiel, K.W.; et al. Epigenetic modification restores functional PR expression in endometrial cancer cells. Curr. Pharm. Des. 2014, 20, 1874–1880. [Google Scholar] [CrossRef] [Green Version]
- Boumber, Y.; Issa, J.P. Epigenetics in cancer: What’s the future? Oncology 2011, 25, 220–226, 228. [Google Scholar]
- Ren, Y.; Liu, X.; Ma, D.; Feng, Y.; Zhong, N. Down-regulation of the progesterone receptor by the methylation of progesterone receptor gene in endometrial cancer cells. Cancer Genet. Cytogenet. 2007, 175, 107–116. [Google Scholar] [CrossRef]
- Ihira, K.; Dong, P.; Xiong, Y.; Watari, H.; Konno, Y.; Hanley, S.J.; Noguchi, M.; Hirata, N.; Suizu, F.; Yamada, T.; et al. EZH2 inhibition suppresses endometrial cancer progression via miR-361/Twist axis. Oncotarget 2017, 8, 13509–13520. [Google Scholar] [CrossRef] [Green Version]
- Mintz, B.; Illmensee, K. Normal genetically mosaic mice produced from malignant teratocarcinoma cells. Proc. Natl. Acad. Sci. USA 1975, 72, 3585–3589. [Google Scholar] [CrossRef] [Green Version]
- de Carvalho Rodrigues, D.; Asensi, K.D.; Vairo, L.; Azevedo-Pereira, R.L.; Silva, R.; Rondinelli, E.; Goldenberg, R.C.; Campos de Carvalho, A.C.; Urményi, T.P. Human menstrual blood-derived mesenchymal cells as a cell source of rapid and efficient nuclear reprogramming. Cell Transplant. 2012, 21, 2215–2224. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2013. CA: A Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazaki, K.; Dyson, M.T.; Coon, V.J.; Furukawa, Y.; Yilmaz, B.D.; Maruyama, T.; Bulun, S.E. Generation of Progesterone-Responsive Endometrial Stromal Fibroblasts from Human Induced Pluripotent Stem Cells: Role of the WNT/CTNNB1 Pathway. Stem Cell Rep. 2018, 11, 1136–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, Q.; Wang, L.; Luo, X.; Chen, X. Adult stem cells in endometrial regeneration: Molecular insights and clinical applications. Mol. Reprod. Dev. 2021, 88, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, L.L.; Yao, J.L.; Wang, K.; Ai, H. Human Umbilical Cord Mesenchymal Stem Cell-Derived Extracellular Vesicles Inhibit Endometrial Cancer Cell Proliferation and Migration through Delivery of Exogenous miR-302a. Stem Cells Int. 2019, 2019, 8108576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abal, M.; Planaguma, J.; Gil-Moreno, A.; Monge, M.; Gonzalez, M.; Baro, T.; Garcia, A.; Castellvi, J.; Ramon, Y.C.S.; Xercavins, J.; et al. Molecular pathology of endometrial carcinoma: Transcriptional signature in endometrioid tumors. Histol. Histopathol. 2006, 21, 197–204. [Google Scholar] [CrossRef]
- Scheffer, G.L.; Wijngaard, P.L.; Flens, M.J.; Izquierdo, M.A.; Slovak, M.L.; Pinedo, H.M.; Meijer, C.J.; Clevers, H.C.; Scheper, R.J. The drug resistance-related protein LRP is the human major vault protein. Nat. Med. 1995, 1, 578–582. [Google Scholar] [CrossRef]
- Song, Y.; Wang, M.; Tong, H.; Tan, Y.; Hu, X.; Wang, K.; Wan, X. Plasma exosomes from endometrial cancer patients contain LGALS3BP to promote endometrial cancer progression. Oncogene 2021, 40, 633–646. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification | Marker | Function and Role | References |
|---|---|---|---|
| Cell proliferation | PI3K/AKT/mTOR | Blocking the PI3K/AKT/mTOR pathway induces autophagy and makes progestin-resistant cells more sensitive to progestin. | [99,100,101,102] |
| PTEN | PTEN inhibits the PI3K/AKT/mTOR pathway by dephosphorylating related molecules and decreasing mTOR downstream activity. | [103,104,105,106] | |
| GRP78 | GRP78, as an upstream gene of the PI3K/AKT pathway, indicated a poor response to progestin treatment in CAH samples. | [107,108,109] | |
| MSX1 | MSX1 is reported to control the cell cycle, and its knockdown increased the effectiveness of progestin treatment in progestin-resistant EC cells. | [110,111] | |
| Oxidative stress | Nrf2 | Nrf2 activates genes that protect cells from intrinsic or exogenous oxidative stress and contributes to progestin resistance of EH and EC. | [112,113,114,115,116] |
| AKR1C1 | Overexpression of AKR1C1 may cause increased progestin catabolism, and AKR1C1 facilitates Nrf2-driven progestin resistance as a target gene of Nrf2. | [117,118] | |
| Survivin | Survivin is an inhibitor of apoptosis proteins; it regulates cell division, apoptosis, and angiogenesis, and mediates progestin resistance in EC. | [119] | |
| LASS2 | LASS2 is a synthesizer of ceramides with broad tissue distribution. As the target gene of Nrf2, down-regulated LASS2 can increase the progestin sensitivity of EC. | [120] | |
| Metabolism | GloI | GloI is a component of the glyoxalase system, and metformin reverses progestin resistance by downregulating the expression of GloI in EC. | [121,122,123] |
| EGFR | EGFR promotes tumor growth and metastasis by increasing the PI3K/AKT signal and suppressing PRB, therefore causing EC to be insensitive to progestin. | [124,125,126,127] | |
| SIRT1 | SIRT1 regulates cell proliferation, inflammation, and metabolism; SIRT1 is highly expressed in EC, especially in progestin-resistant EC. | [88,128,129,130,131] | |
| DHCR24 | DHCR24 is an enzyme that mediates cholesterol synthesis. It is highly expressed in EC and is associated with progestin resistance. | [91,132,133] | |
| IGF | Both IGF-I and IGF-II suppress the expression of PR. IGF-II enhances cell proliferation through raising phosphorylation of AKT and p70S6K in EC. | [90,134,135] | |
| Apoptosis | Fas/FasL | Fas-mediated apoptosis is critical for the endometrial cycle, and Fas/FasL dysregulation may play a role in the formation of progestin-resistant cells. | [70,136,137] |
| Bcl-2 | Bcl-2 prevents cells from entering apoptosis, and the expression of Bcl-2 in stromal cells can distinguish progestin responders from non-responders. | [27,71,138,139] | |
| PDCD4 | PDCD4 is a target gene of progestin treatment in EC. Progestin inhibits the protein expression of PDCD4 via the PI3K/AKT signal pathway. | [140,141,142] | |
| Nucleic acid regulation | MicroRNA | Five miRNAs limited the effect of progestin therapy in EC cells, and miR-96 reported to have the most obvious inhibitory effect on PR expression. | [59,143,144] |
| HOTAIR | HOTAIR is a well-known lncRNA that suppresses PRB expression, and HOTAIR knockdown increased PRB transcription by recruiting LSD1 to the PRB promoter. | [145,146] | |
| DACH1 | DACH1 plays a tumor-suppressing role in EC. DACH1 is positively associated with PR, and DACH1 knockdown enhanced progestin resistance. | [77,147,148,149] | |
| ARID1A | ARID1A deletion increased MPA resistance in EC by over-activating the PI3K/AKT signaling pathway and downregulating the PRB expression. | [150,151,152,153] | |
| HAND2 | HAND2 suppresses estrogen-mediated signals in EC. Furthermore, methylation levels of HAND2 can predict patients’ response to progestin treatment. | [50,51] |
| Register number | Conditions | Interventions | Phase | Number of Patients | Group |
|---|---|---|---|---|---|
| Drug combination | |||||
| NCT03077698 | Endometrial Cancer | Drug: Sodium Cridanimod Drug: Progestin therapy | II | 25 | Sodium Cridanimod + Progestin therapy |
| NCT02064725 | Recurrent or Persistent Endometrial Carcinoma | Drug: Sodium cridanimod | II | 8 | Sodium cridanimod + megestrol acetate or MPA |
| NCT04792749 | Endometrial Cancer Stage I | Drug: Metformin | III | 77 | Metformin + MPA |
| NCT05316935 | Endometrial Neoplasms Atypical Endometrial Hyperplasia Progesterone Resistance | Drug: GnRHa Drug: Letrozole 2.5 mg Drug: Diane-35 Drug: Metformin | II–III | 80 | GnRHa + letrozole vs. Ethinylestradiol cyproterone + metformin |
| NCT04046185 | Endometrial Cancer Stage I | Drug: PD-1 inhibitor Drug: Progesterone | I | 60 | PD-1 inhibitor + progesterone |
| NCT04607252 | Atypical Endometrial Hyperplasia | Drug: Metformin plus megestrol acetate Drug: Megestrol Acetate | II–III | 12 | Metformin + megestrol acetate |
| Application of LNG-IUS | |||||
| NCT02990728 | Endometrial Cancer | Drug: Metformin Device: Mirena | II | 120 | Metformin + Mirena |
| NCT03463252 | Endometrial Cancer Atypical Endometrial Hyperplasia | Drug: Progesterone Device: Mirena Drug: GnRH agonist | II–III | 224 | MPA + Mirena vs. progesterone GnRH-a + Mirena vs. Mirena |
| NCT01074892 | Endometrial Hyperplasia | Drug: Provera (medroxyprogesterone/progestin) Device: Mirena (levonorgestrel) | IV | 170 | medroxyprogesterone/progestin vs. Mirena (levonorgestrel) |
| NCT04385667 | Atypical Endometrial Hyperplasia | Device: Levonorgestrel intrauterine system (LNG-IUD) Drug: Oral megesterol 160 mg daily | II–III | 140 | Oral megesterol vs. LNG-IUS |
| NCT04897217 | Endometrial Hyperplasia | Drug: Megestrol acetate Drug: Levonorgestrel drug implant | III | 40 | Megestrol acetate vs. LNG-IUS |
| NCT03992937 | Endometrial Hyperplasia Without Atypia | Drug: Vaginal micronized progesterone Device: Levonorgestrel-intrauterine system | Not Applicable | 132 | Vaginal micronized progesterone vs. LNG-IUS |
| In combination with radiotherapy | |||||
| NCT05255653 (NSMP-ORANGE trial) | Endometrial Cancer | Radiation: Pelvic external beam radiotherapy Drug: Medroxyprogesterone acetate Drug: Megestrol acetate Other: Observation | III | 1611 | Pelvic external beam radiotherapy + oral progestagens |
| In combination with chemotherapy | |||||
| NCT00739830 | Endometrial Cancer | Drug: Ridaforolimus Drug: Medroxyprogesterone acetate tablets OR megestrol acetate Drug: Chemotherapy | II | 130 | Ridaforolimus vs. MPA or megestrol acetate + chemotherapy |
| In combination with surgery | |||||
| NCT04008563 | Endometrial Cancer Atypical Hyperplasia Bariatric Surgery Candidate | Bariatric surgery | Not Applicable | 36 | Bariatric surgery + progestin intrauterine device |
| NCT04362046 | Endometrial Hyperplasia Endometrial Cancer Gynecologic Cancer | Procedure: Hysteroscopic uterine resection | Not Applicable | 30 | Progestin + hysteroscopic uterine resection |
| Drug Class | Agent | Function | References |
|---|---|---|---|
| Hypoglycemia agent | Metformin | Metformin enhanced the progestin sensitivity of EC by decreasing GloI expression and downregulating Nrf2 and survivin expression. | [182,183,184,185] |
| Antipsychotic drugs | Chlorpromazine | CPZ pretreatment may increase PRB expression and CPZ, phosphorylate PI3K/AKT, and downregulate the expression of IGF-IR. | [186] |
| Thioridazine | THIO combined with MPA inhibited the PI3K/AKT/mTOR pathway and enhanced progestin sensitivity by downregulating EGFR and upregulating PRB. | [187] | |
| PI3K/AKT/mTOR pathway inhibitors | PI3K Inhibitors | Pretreatment with the PI3K inhibitor LY294002 caused cell apoptosis, increased PRB expression, and, therefore, enhanced the therapeutic effect of MPA in EC. | [13,152] |
| AKT Inhibitors | MK-2206, an active AKT inhibitor, regulated the expression of progestin-related genes, increased PRB protein levels, and induced apoptosis of EC cells. | [188,189] | |
| mTOR Inhibitors | mTOR Inhibitors increased the expression of PR messenger RNA in patients with recurrent or metastatic EC, and suppressed the growth of EC. | [190,191,192,193] | |
| Epigenetic modulation | Histone deacetylating inhibitors | HDACi therapy restored the protein and mRNA expression of PR in EC cell lines. LBH589 treatment resulted in cell cycle arrest in G1, which was further promoted by progestin. | [194,195,196] |
| DNA methyl transferase inhibitor | 5-aza-deoxycytidine, as one kind of DNMTi, reduced the methylation of the PR promoter and restored functional PR expression in EC cells. | [197,198] | |
| Histone methylation inhibitors | EZH2 caused trimethylation of histone H3, thus silencing the PR expression. EZH2-specific inhibitors reduced EC cell proliferation and invasion. | [199] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lv, M.; Chen, P.; Bai, M.; Huang, Y.; Li, L.; Feng, Y.; Liao, H.; Zheng, W.; Chen, X.; Zhang, Z. Progestin Resistance and Corresponding Management of Abnormal Endometrial Hyperplasia and Endometrial Carcinoma. Cancers 2022, 14, 6210. https://doi.org/10.3390/cancers14246210
Lv M, Chen P, Bai M, Huang Y, Li L, Feng Y, Liao H, Zheng W, Chen X, Zhang Z. Progestin Resistance and Corresponding Management of Abnormal Endometrial Hyperplasia and Endometrial Carcinoma. Cancers. 2022; 14(24):6210. https://doi.org/10.3390/cancers14246210
Chicago/Turabian StyleLv, Mu, Peiqin Chen, Mingzhu Bai, Yan Huang, Linxia Li, Youji Feng, Hong Liao, Wenxin Zheng, Xiaojun Chen, and Zhenbo Zhang. 2022. "Progestin Resistance and Corresponding Management of Abnormal Endometrial Hyperplasia and Endometrial Carcinoma" Cancers 14, no. 24: 6210. https://doi.org/10.3390/cancers14246210
APA StyleLv, M., Chen, P., Bai, M., Huang, Y., Li, L., Feng, Y., Liao, H., Zheng, W., Chen, X., & Zhang, Z. (2022). Progestin Resistance and Corresponding Management of Abnormal Endometrial Hyperplasia and Endometrial Carcinoma. Cancers, 14(24), 6210. https://doi.org/10.3390/cancers14246210