
Ex Vivo Drug Sensitivity Correlates with Clinical Response and Supports Personalized Therapy in Pediatric AML

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
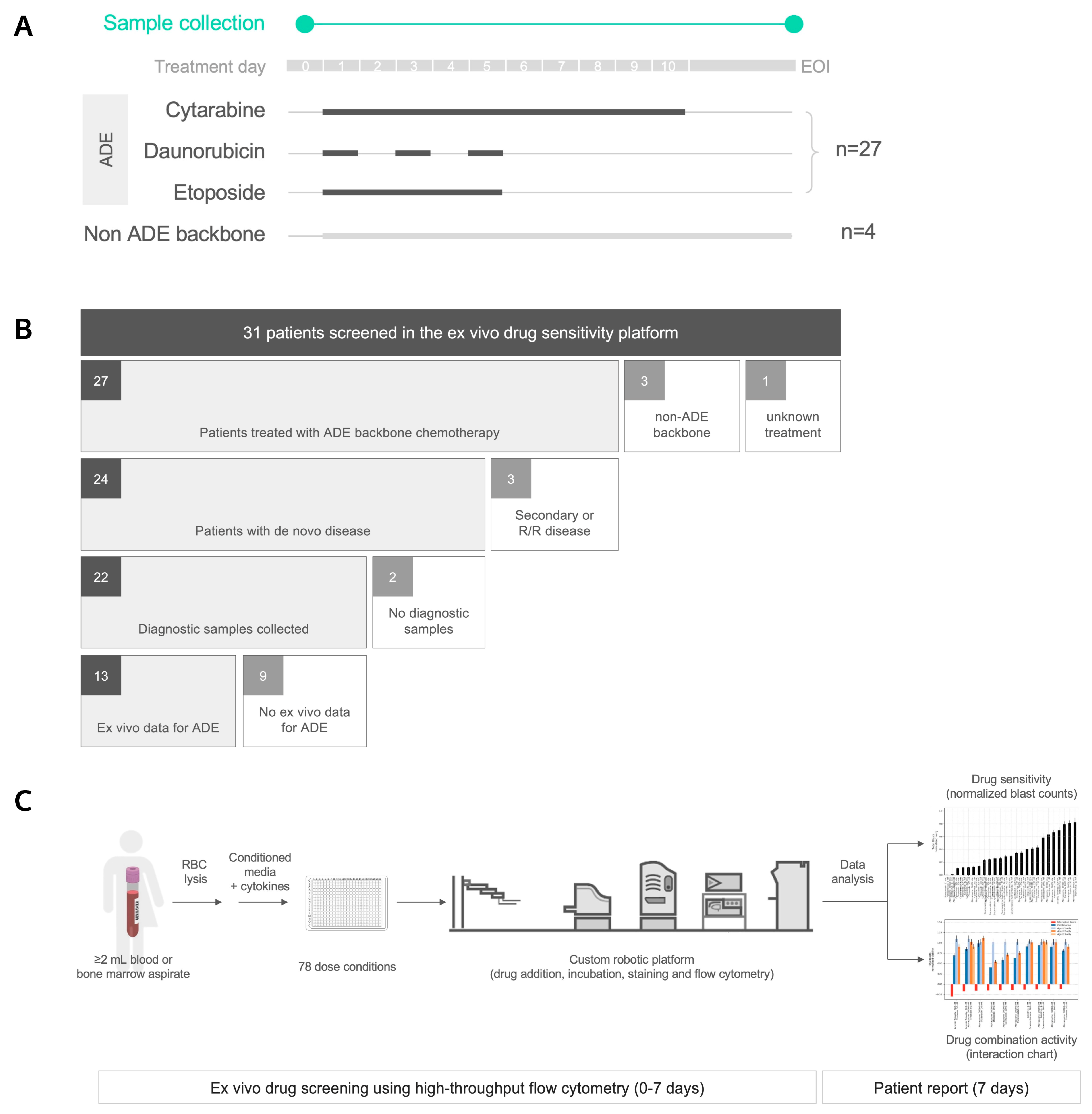
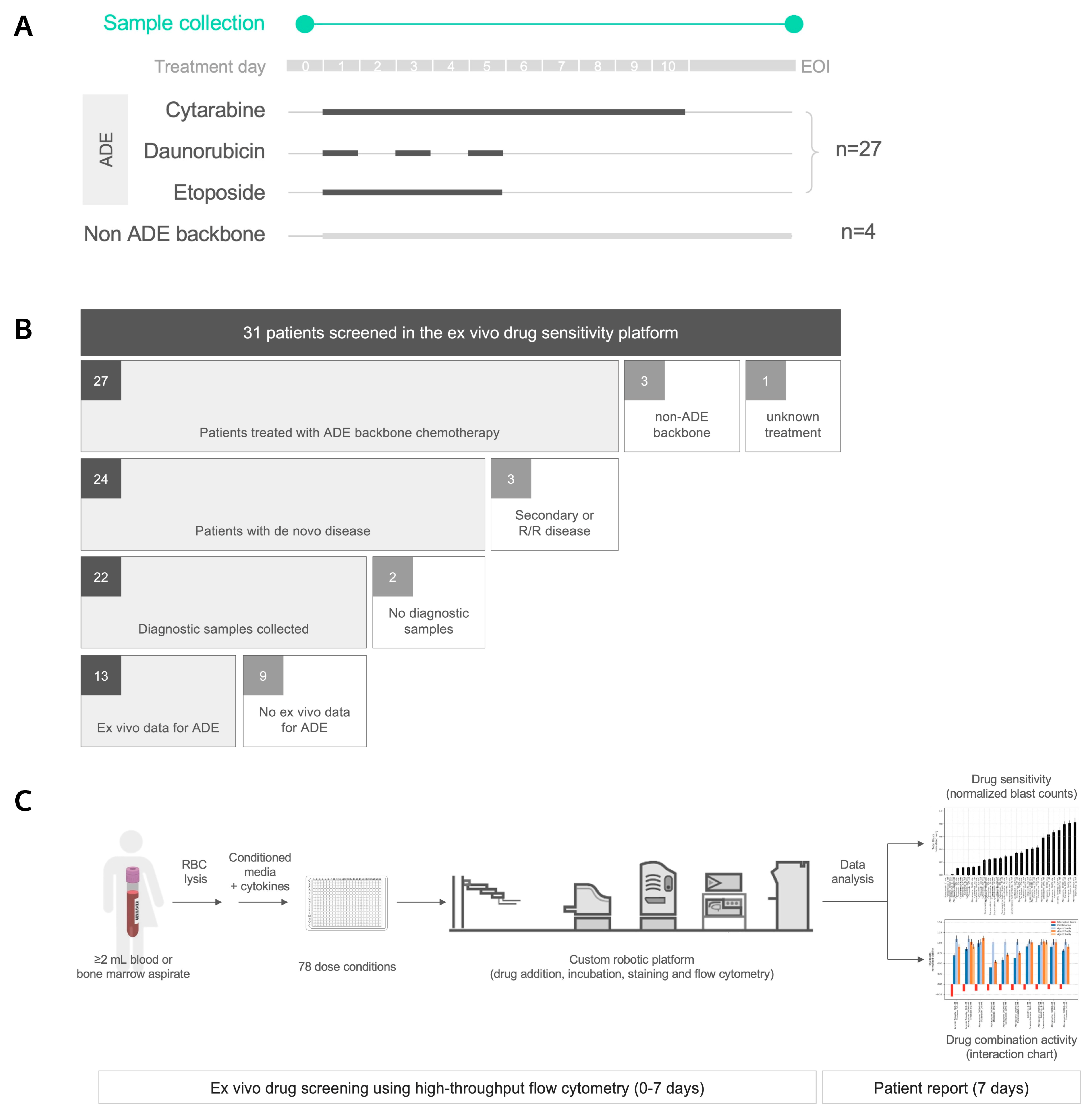
2.1. Patients
2.2. Sample Collection
2.3. Ex Vivo Drug Sensitivity Platform
2.4. Flow Cytometry and Blast Gating
3. Data Analysis
4. Results
4.1. Thirty-One Pediatric AML Patients Were Profiled Using an Ex Vivo Drug Sensitivity Platform
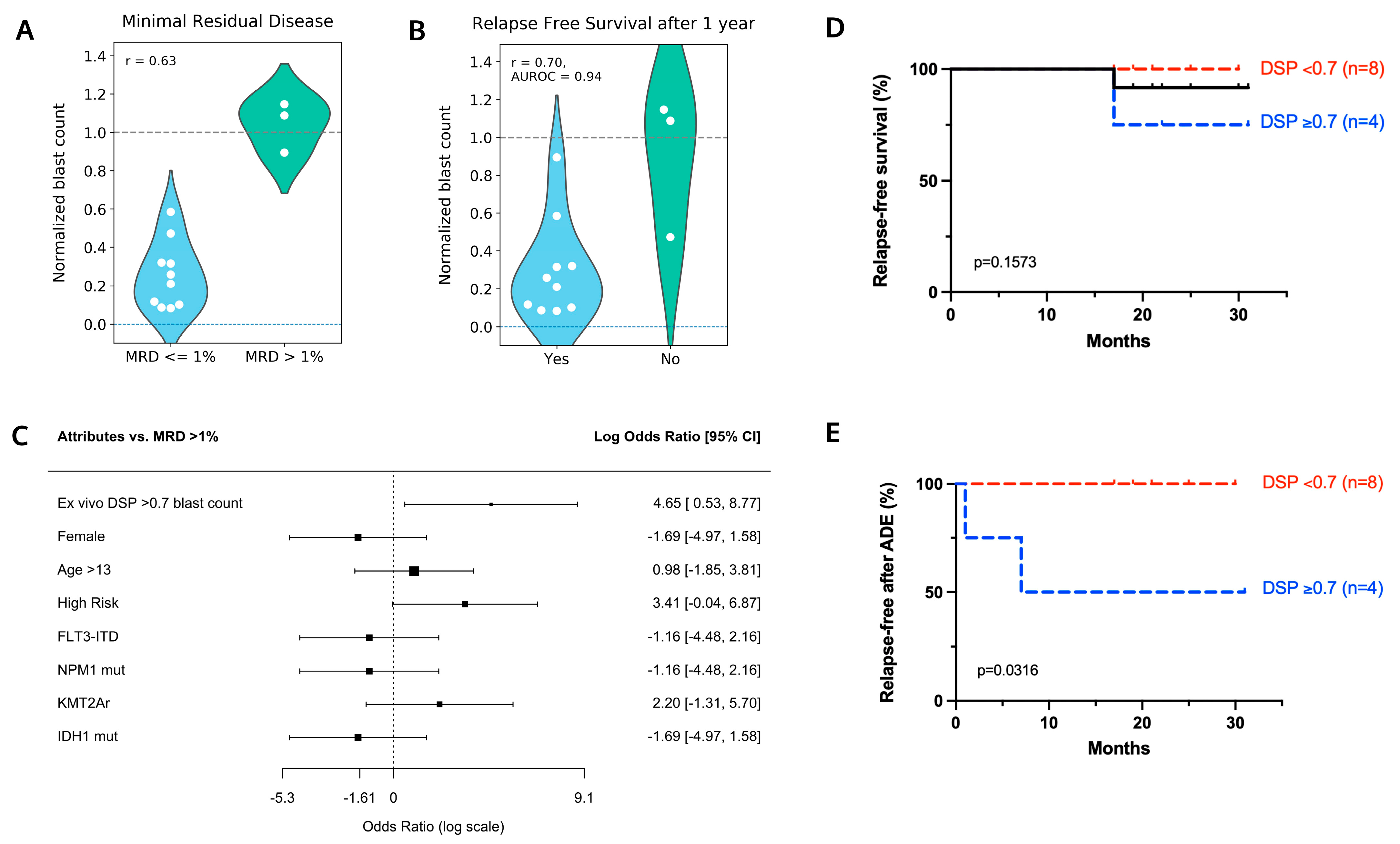
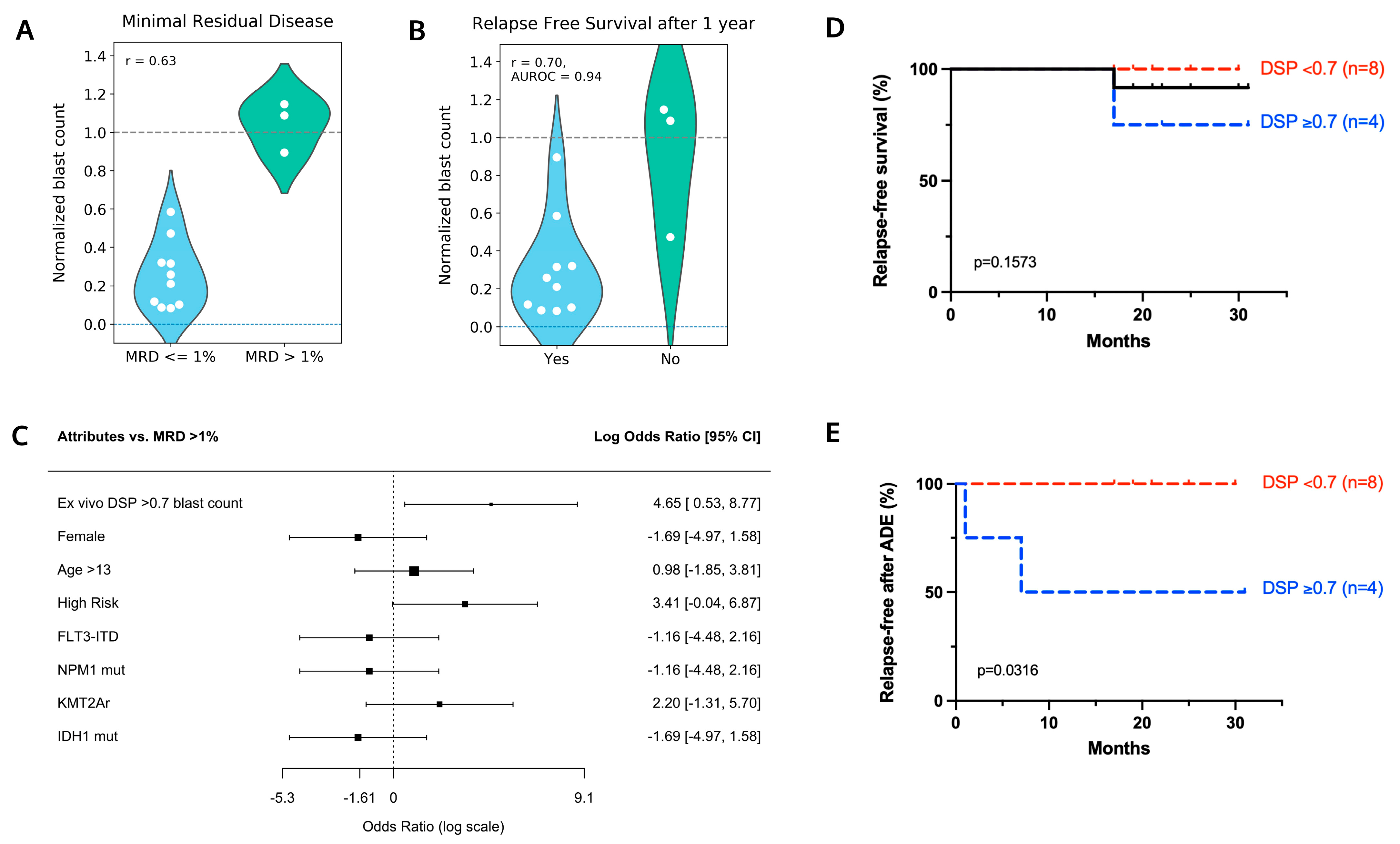
4.2. Ex Vivo Drug Sensitivity in Response to ADE Correlates with Clinical Response
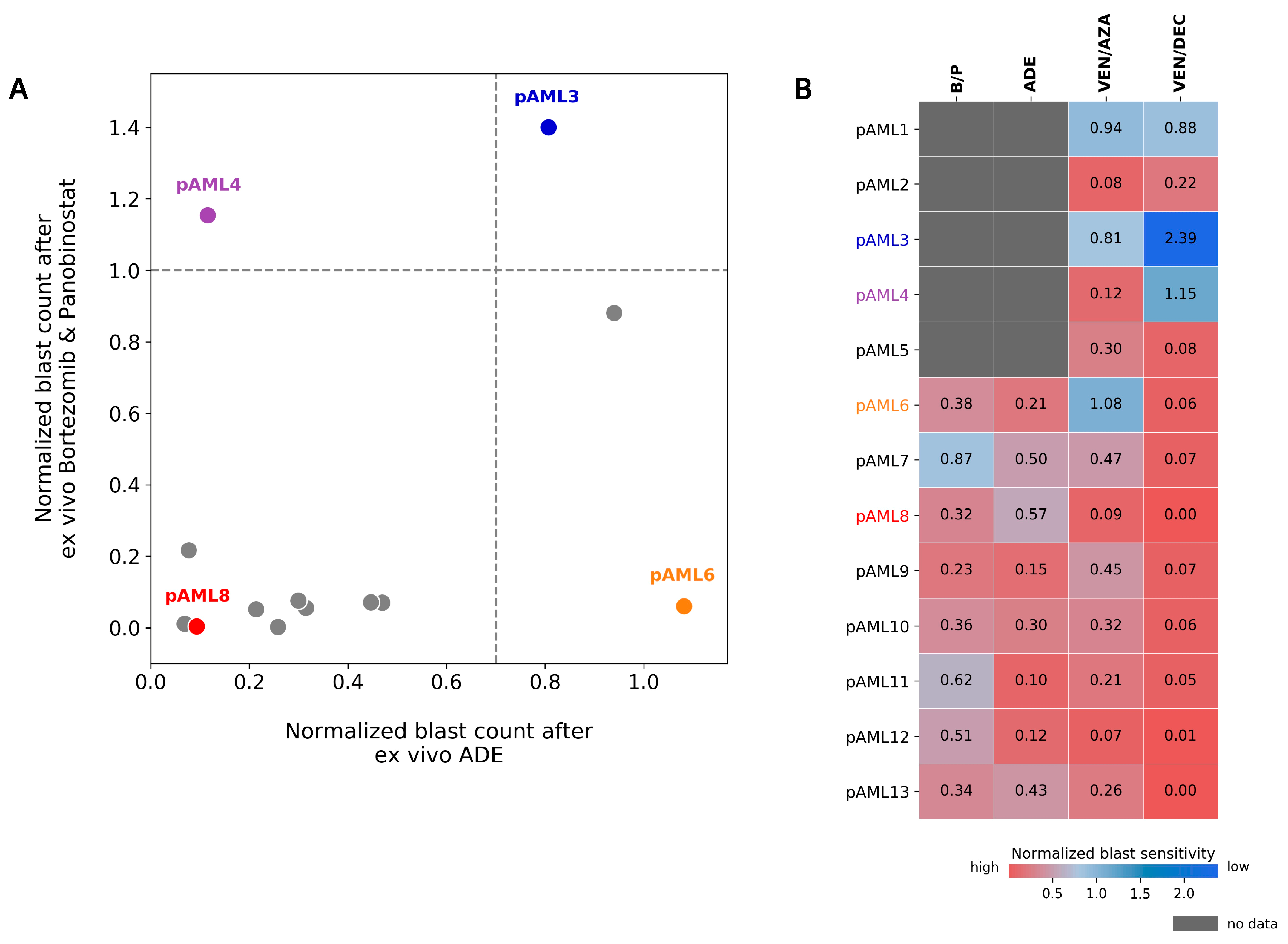
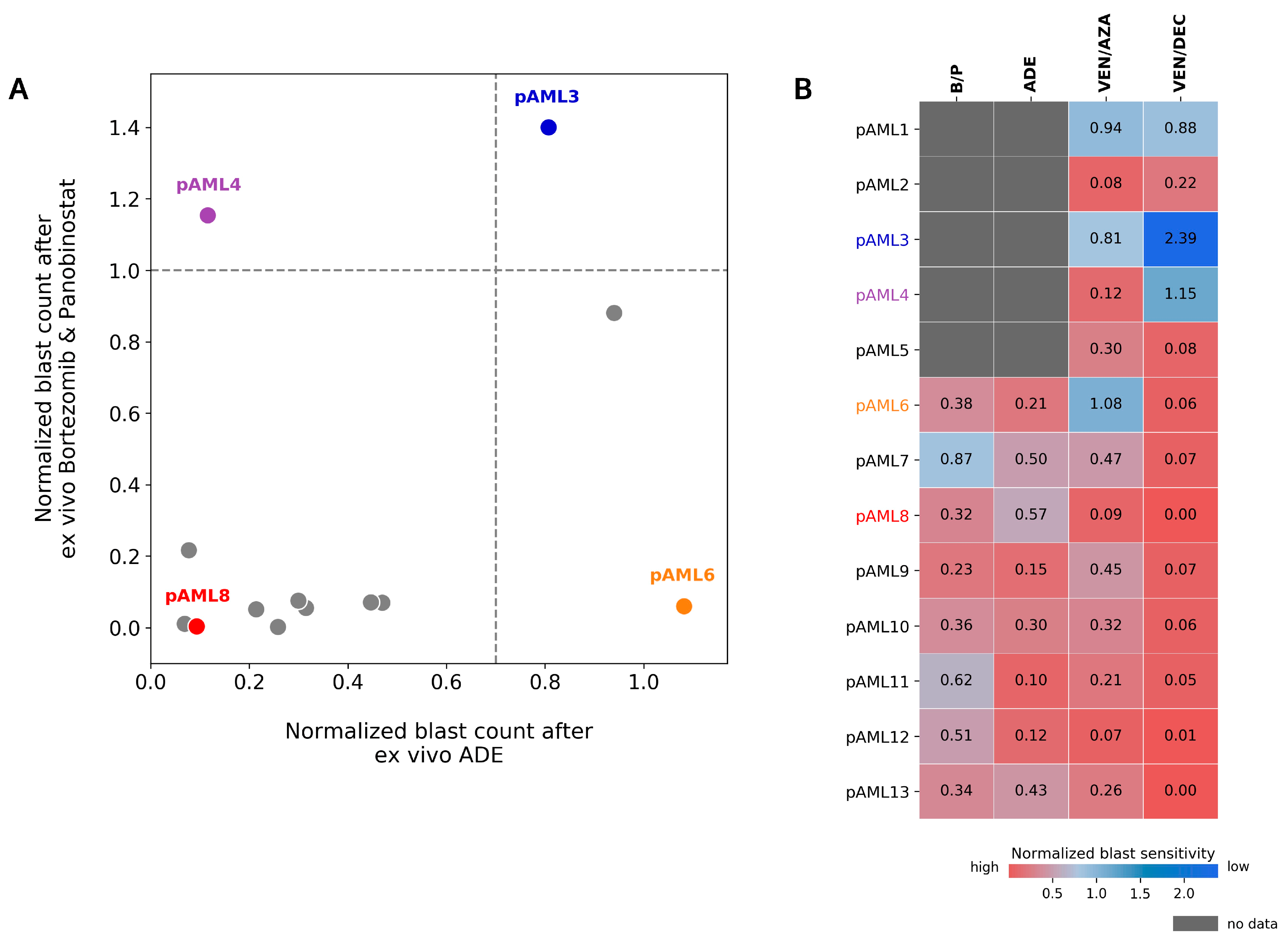
4.3. Patient pAML3 Non-Responder and Patient pAML8 Responder Captures the Range of High and Low Drug Sensitivity across Multiple Conditions Tested Ex Vivo
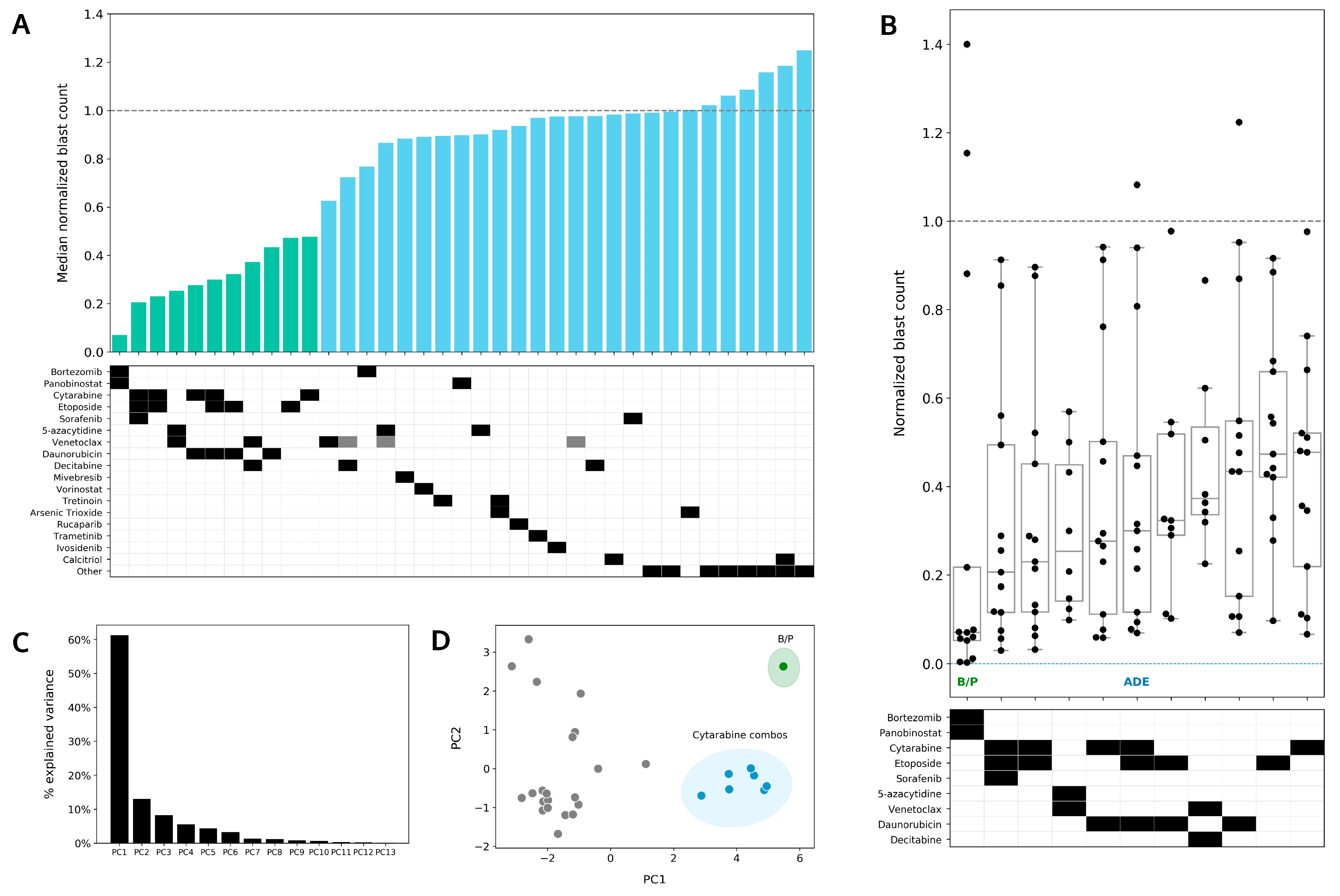
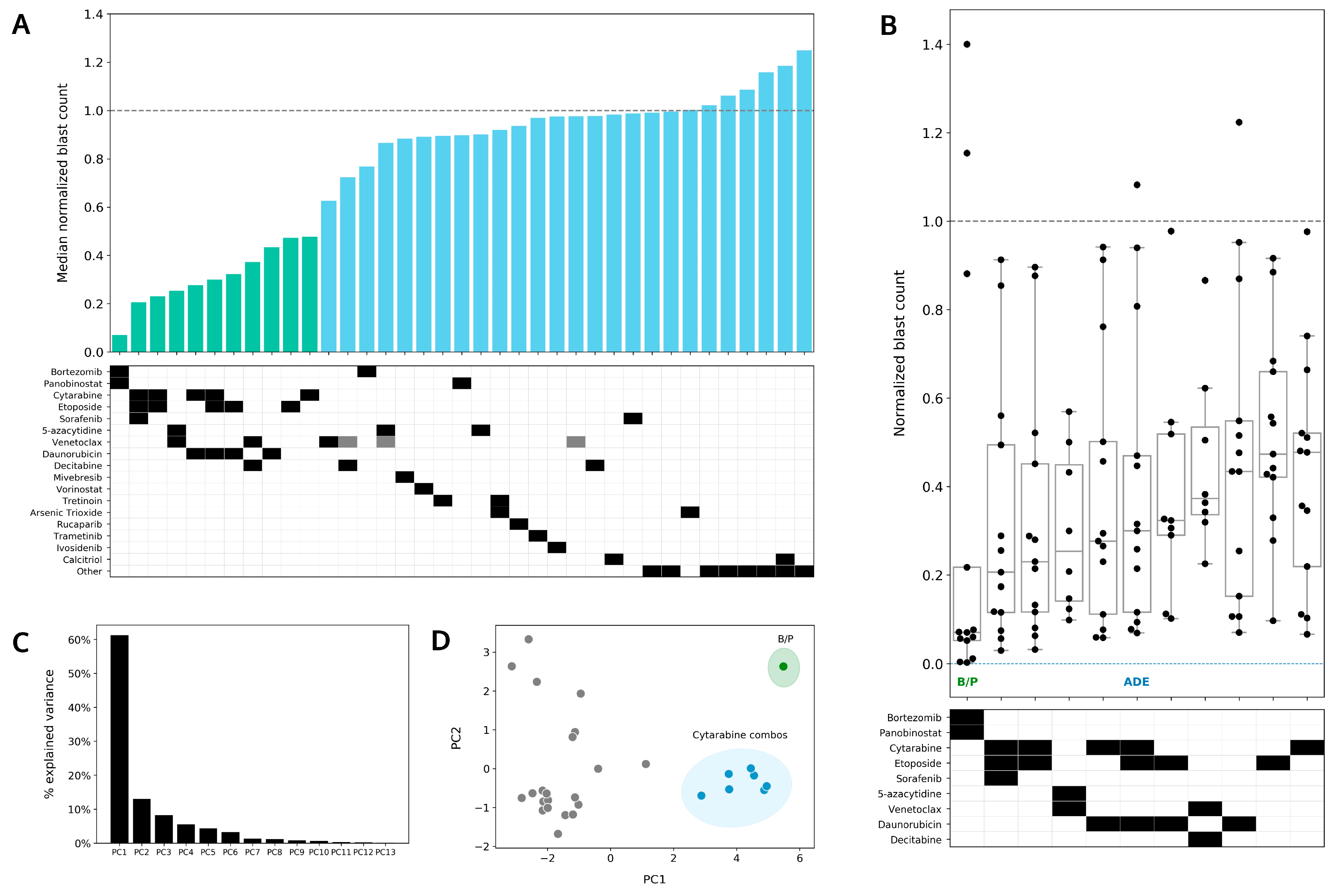
4.4. Bortezomib in Combination with Panobinostat Shows the Highest Median Sensitivity out of the 37 Conditions Tested in the DSP
4.5. Preferential Sensitivity between ADE and Bortezomib/Panobinostat Is Observed in a Subset of Pediatric AML Patients in the DSP
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, S.; Garrett-Bakelman, F.E.; Chung, S.S.; Sanders, M.A.; Hricik, T.; Rapaport, F.; Patel, J.; Dillon, R.; Vijay, P.; Brown, A.L.; et al. Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nat. Med. 2016, 22, 792–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R. (Eds.) SEER Cancer Statistics Review, 1975–2018; National Cancer Institute: Bethesda, MD, USA, 2021. Available online: https://seer.cancer.gov/csr/1975_2018/ (accessed on 5 May 2022).
- Steliarova-Foucher, E.; Colombet, M.; Ries, L.; Moreno, F.; Dolya, A.; Bray, F.; Hesseling, P.; Shin, H.Y.; Stiller, C.A.; IICC-3 Contributors. International incidence of childhood cancer, 2001–2010: A population-based registry study. Lancet Oncol. 2017, 18, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Aplenc, R.; Meshinchi, S.; Sung, L.; Alonzo, T.; Choi, J.; Fisher, B.; Gerbing, R.; Hirsch, B.; Horton, T.; Kahwash, S.; et al. Bortezomib with standard chemotherapy for children with acute myeloid leukemia does not improve treatment outcomes: A report from the Children’s Oncology Group. Haematologica 2020, 105, 1879–1886. [Google Scholar] [CrossRef] [PubMed]
- Rasche, M.; Zimmermann, M.; Borschel, L.; Bourquin, J.P.; Dworzak, M.; Klingebiel, T.; Lehrnbecher, T.; Creutzig, U.; Klusmann, J.H.; Reinhardt, D. Successes and challenges in the treatment of pediatric acute myeloid leukemia: A retrospective analysis of the AML-BFM trials from 1987 to 2012. Leukemia 2018, 32, 2167–2177. [Google Scholar] [CrossRef] [Green Version]
- Conneely, S.E.; Stevens, A.M. Acute Myeloid Leukemia in Children: Emerging Paradigms in Genetics and New Approaches to Therapy. Curr. Oncol. Rep. 2021, 23, 16. [Google Scholar] [CrossRef]
- Ravindranath, Y.; Chang, M.; Steuber, C.P.; Becton, D.; Dahl, G.; Civin, C.; Camitta, B.; Carroll, A.; Raimondi, S.C.; Weinstein, H.J.; et al. Pediatric Oncology Group (POG) studies of acute myeloid leukemia (AML): A review of four consecutive childhood AML trials conducted between 1981 and 2000. Leukemia 2005, 19, 2101–2116. [Google Scholar] [CrossRef] [Green Version]
- Zwaan, C.M.; Kolb, E.K.A.; Reinhardt, D.; Abrahamsson, J.; Adachi, S.; Aplenc, R.; De Bont, E.S.; De Moerloose, B.; Dworzak, M.; Gibson, B.E.; et al. Collaborative Efforts Driving Progress in Pediatric Acute Myeloid Leukemia. J. Clin. Oncol. 2015, 33, 2949–2962. [Google Scholar] [CrossRef] [Green Version]
- Kopp, L.M.; Gupta, P.; Pelayo-Katsanis, L.; Wittman, B.; Katsanis, E. Late effects in adult survivors of pediatric cancer: A guide for the primary care physician. Am. J. Med. 2012, 125, 636–641. [Google Scholar] [CrossRef]
- Daver, N.; Wei, A.H.; Pollyea, D.A.; Fathi, A.T.; Vyas, P.; DiNardo, C.D. New directions for emerging therapies in acute myeloid leukemia: The next chapter. Blood Cancer J. 2020, 10, 107. [Google Scholar] [CrossRef]
- Bolouri, H.; Farrar, J.E.; Triche, T., Jr.; Ries, R.E.; Lim, E.L.; Alonzo, T.A.; Ma, Y.; Moore, R.; Mungall, A.J.; Marra, M.A.; et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat. Med. 2018, 24, 103–112. [Google Scholar] [CrossRef]
- Gröbner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. The landscape of genomic alterations across childhood cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Parsons, D.W.; Janeway, K.A.; Patton, D.R.; Winter, C.L.; Coffey, B.; Williams, P.M.; Roy-Chowdhuri, S.; Tsongalis, G.J.; Routbort, M.; Ramirez, N.C.; et al. Actionable Tumor Alterations and Treatment Protocol Enrollment of Pediatric and Young Adult Patients with Refractory Cancers in the National Cancer Institute-Children’s Oncology Group Pediatric MATCH Trial. J Clin. Oncol. 2022, 40, 2224–2234. [Google Scholar] [CrossRef] [PubMed]
- Eckstein, O.S.; Allen, C.E.; Williams, P.M.; Roy-Chowdhuri, S.; Patton, D.R.; Coffey, B.; Reid, J.M.; Piao, J.; Saguilig, L.; Alonzo, T.A.; et al. Phase II Study of Selumetinib in Children and Young Adults with Tumors Harboring Activating Mitogen-Activated Protein Kinase Pathway Genetic Alterations: Arm E of the NCI-COG Pediatric MATCH Trial. J. Clin. Oncol. 2022, 40, 2235–2245. [Google Scholar] [CrossRef] [PubMed]
- Walter, R.B.; Michaelis, L.C.; Othus, M.; Uy, G.L.; Radich, J.P.; Little, R.F.; Hita, S.; Saini, L.; Foran, J.M.; Gerds, A.T.; et al. Intergroup LEAP trial (S1612): A randomized phase 2/3 platform trial to test novel therapeutics in medically less fit older adults with acute myeloid leukemia. Am. J. Hematol. 2018, 93, E49–E52. [Google Scholar] [CrossRef]
- Pikman, Y.; Tasian, S.K.; Sulis, M.L.; Stevenson, K.; Blonquist, T.M.; Apsel Winger, B.; Cooper, T.M.; Pauly, M.; Maloney, K.W.; Burke, M.J.; et al. Matched Targeted Therapy for Pediatric Patients with Relapsed, Refractory, or High-Risk Leukemias: A Report from the LEAP Consortium. Clin. Trial Cancer Discov. 2021, 11, 1424–1439. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.E.; Eide, C.A.; Kaempf, A.; Khanna, V.; Savage, S.L.; Rofelty, A.; English, I.; Ho, H.; Pandya, R.; Bolosky, W.J.; et al. Molecularly targeted drug combinations demonstrate selective effectiveness for myeloid- and lymphoid-derived hematologic malignancies. Proc. Natl. Acad. Sci. USA 2017, 114, E7554–E7563. [Google Scholar] [CrossRef] [Green Version]
- Tyner, J.W.; Yang, W.F.; Bankhead, A., 3rd; Fan, G.; Fletcher, L.B.; Bryant, J.; Glover, J.M.; Chang, B.H.; Spurgeon, S.E.; Fleming, W.H.; et al. Kinase pathway dependence in primary human leukemias determined by rapid inhibitor screening. Cancer Res. 2013, 73, 285–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spinner, M.A.; Aleshin, A.; Santaguida, M.T.; Schaffert, S.A.; Zehnder, J.L.; Patterson, A.S.; Gekas, C.; Heiser, D.; Greenberg, P.L. Ex vivo drug screening defines novel drug sensitivity patterns for informing personalized therapy in myeloid neoplasms. Blood Adv. 2020, 4, 2768–2778. [Google Scholar] [CrossRef]
- Malani, D.; Kumar, A.; Brück, O.; Kontro, M.; Yadav, B.; Hellesøy, M.; Kuusanmäki, H.; Dufva, O.; Kankainen, M.; Eldfors, S.; et al. Implementing a Functional Precision Medicine Tumor Board for Acute Myeloid Leukemia. Cancer Discov. 2022, 12, 388–401. [Google Scholar] [CrossRef]
- Kornauth, C.; Pemovska, T.; Vladimer, G.I.; Bayer, G.; Bergmann, M.; Eder, S.; Eichner, R.; Erl, M.; Esterbauer, H.; Exner, R.; et al. Functional Precision Medicine Provides Clinical Benefit in Advanced Aggressive Hematologic Cancers and Identifies Exceptional Responders. Cancer Discov. 2022, 12, 372–387. [Google Scholar] [CrossRef]
- Tyner, J.W.; Tognon, C.E.; Bottomly, D.; Wilmot, B.; Kurtz, S.E.; Savage, S.L.; Long, N.; Schultz, A.R.; Traer, E.; Abel, M.; et al. Functional genomic landscape of acute myeloid leukaemia. Nature 2018, 562, 526–531. [Google Scholar] [CrossRef]
- Eaton, A.; Wong, V.; Schiff, D.; Anderson, E.; Ding, H.; Capparelli, E.V.; Determan, D.; Kuo, D.J. Tumor Genomic Profiling and Ex Vivo Drug Sensitivity Testing for Pediatric Leukemia and Lymphoma Patients. J. Pediatr. Pharmacol. Ther. 2022, 27, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chan, K.; Cheng, C.K.; Ng, M.; Lee, P.Y.; Cheng, F.; Lam, G.; Chow, T.W.; Ha, S.Y.; Chiang, A.; et al. Pharmacogenomic profiling of pediatric acute myeloid leukemia to identify therapeutic vulnerabilities and inform functional precision medicine. Blood Cancer Discov. 2022, 3, 516–535. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ji, M.; Zhao, J.Y.; Wang, H.F.; Wang, C.W.; Li, W.; Ye, J.J.; Lu, F.; Lin, L.H.; Gao, Y.T.; et al. Ex Vivo Chemosensitivity Profiling of Acute Myeloid Leukemia and Its Correlation with Clinical Response and Outcome to Chemotherapy. Front. Oncol. 2021, 11, 793773. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Tong, Y.; Straube, J.; Zhao, J.; Gao, Y.; Bai, P.; Li, J.; Wang, J.; Wang, H.; Wang, X.; et al. Ex-vivo drug testing predicts chemosensitivity in acute myeloid leukemia. J. Leukoc. Biol. 2020, 107, 859–870. [Google Scholar] [CrossRef]
- Stevens, A.M.; Xiang, M.; Heppler, L.N.; Tošić, I.; Jiang, K.; Munoz, J.O.; Gaikwad, A.S.; Horton, T.M.; Long, X.; Narayanan, P.; et al. Atovaquone is active against AML by upregulating the integrated stress pathway and suppressing oxidative phosphorylation. Blood Adv. 2019, 3, 4215–4227. [Google Scholar] [CrossRef]
- Gamis, A.S.; Alonzo, T.A.; Meshinchi, S.; Sung, L.; Gerbing, R.B.; Raimondi, S.C.; Hirsch, B.A.; Kahwash, S.B.; Heerema-McKenney, A.; Winter, L.; et al. Gemtuzumab ozogamicin in children and adolescents with de novo acute myeloid leukemia improves event-free survival by reducing relapse risk: Results from the randomized phase III Children’s Oncology Group trial AAML0531. J. Clin. Oncol. 2014, 32, 3021–3032. [Google Scholar] [CrossRef] [Green Version]
- Karol, S.E.; Cooper, T.M.; Mead, P.E.; Crews, K.R.; Panetta, J.C.; Alexander, T.B.; Taub, J.W.; Lacayo, N.J.; Heym, K.M.; Kuo, D.J.; et al. Safety, pharmacokinetics, and pharmacodynamics of panobinostat in children, adolescents, and young adults with relapsed acute myeloid leukemia. Cancer 2020, 126, 4800–4805. [Google Scholar] [CrossRef]
- Winters, A.C.; Maloney, K.W.; Treece, A.L.; Gore, L.; Franklin, A.K. Single-center pediatric experience with venetoclax and azacitidine as treatment for myelodysplastic syndrome and acute myeloid leukemia. Pediatr. Blood Cancer 2020, 67, e28398. [Google Scholar] [CrossRef]
- Karol, S.E.; Alexander, T.B.; Budhraja, A.; Pounds, S.B.; Canavera, K.; Wang, L.; Wolf, J.; Klco, J.M.; Mead, P.E.; Das Gupta, S.; et al. Venetoclax in combination with cytarabine with or without idarubicin in children with relapsed or refractory acute myeloid leukaemia: A phase 1, dose-escalation study. Lancet Oncol. 2020, 21, 551–560. [Google Scholar] [CrossRef]
- Ivanov, V.; Yeh, S.P.; Mayer, J.; Saini, L.; Unal, A.; Boyiadzis, M.; Hoffman, D.M.; Kang, K.; Addo, S.N.; Mendes, W.L.; et al. Design of the VIALE-M phase III trial of venetoclax and oral azacitidine maintenance therapy in acute myeloid leukemia. Future Oncol. 2022, 18, 2879–2889. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UPN | Age, y; Sex | Disease Status | FAB | Final Risk | Cytogenetics | Mutations | Induction Chemotherapy | MRD, % | Relapse | Clinical Outcome |
|---|---|---|---|---|---|---|---|---|---|---|
| pAML1 | 15; M | De Novo | M2 | High Risk | t(8;21) | Negative | ADE + AQ | 9.7 | No | Alive |
| pAML2 | 16; F | De Novo | M2 | Low Risk | 11q23 dupl | Negative | ADE + AQ | 0 | No | Alive |
| pAML3 | 16; M | De Novo | M5a | High Risk | Complex | Negative | ADE + AQ | 4 | Yes | DD |
| pAML4 | 7; M | De Novo | M4 | High Risk | 8 | FLT3-ITD | ADE | 0 | No | Alive |
| pAML5 | 13; M | De Novo | M2 | Low Risk | t(8;21) | Negative | ADE + AQ | 0 | No | Alive |
| pAML6 | 11; M | De Novo | M1 | High Risk | t(10;11)+ complex | KMT2Ar | ADE + AQ | 50 | Yes | Alive |
| pAML7 | 19; F | De Novo | M1 | High Risk | Normal | FLT3-ITD; NPM1 | ADE + AQ | 0.7 | No | TRM |
| pAML8 | 2; M | De Novo | M7 | Low Risk | 10 | Negative | ADE + AQ | 0 | No | Alive |
| pAML9 | 16; F | De Novo | M1/M2 | Low Risk | Normal | CEBPa | ADE + AQ | 0 | No | Alive |
| pAML10 | 16; F | De Novo | M4/M5 | Low Risk | Normal | NPM1 | ADE | 0 | No | Alive |
| pAML11 | 11; M | De Novo | M1/M2 | Low Risk | Normal | NPM1 | ADE + AQ | 0 | No | Alive |
| pAML12 | 12; M | De Novo | M1/M2 | Low Risk | Normal | FLT3-ITD; CEBPa | ADE + AQ | 0 | No | Alive |
| pAML13 | 6; F | De Novo | M1/M2 | Low Risk | t(1;11) not KMT2A, del(11q) | CEBPa | ADE | 0 | No | Alive |
| pAML14 | 7; M | Secondary | M5 | High Risk | t(11;19p13.1) | FLT3 (2 PMs); KMT2Ar | CPX-351 | 0 | Yes | DD |
| pAML15 | 13; M | Refractory | M5 | High Risk | Normal | FLT3-ITD | ADE +AQ | 4.7 | Yes | Alive |
| pAML16 | unknown | Refractory | unknown | unknown | t(7;21), -17 | not done | unknown | unknown | Refractory | lost to f/u |
| pAML17 | 1; M | De Novo | M5 | Low Risk | t(10;11) cryptic | KMT2Ar | ADE | 0 | Yes | DD |
| pAML18 | 1; M | Refractory | M7 | High Risk | t(1;21) | Negative | ADE + AQ | 2 | Refractory | DD |
| pAML19 | 0.75; F | De Novo | M5 | Low Risk | inv(16) | Negative | ADE + AQ | 1.5 | No | Alive |
| pAML20 | 14; F | De Novo | M1/M2 | High Risk | Complex | Negative | CPX-351 | 4 | Refractory | DD |
| pAML21 | 11; F | De Novo | M5 | High Risk | Normal | FLT3-ITD | ADE + AQ | 0 | No | Alive |
| pAML22 | 0.83; M | De Novo | M7 | High Risk | CBFA1T3-GLIS2 | Negative | ADE + AQ | 0.02 | Yes | Alive |
| pAML23 | 15; F | De Novo | M2 | High Risk | 8 | FLT3-ITD; NPM1 | ADE + AQ | 0 | No | TRM |
| pAML24 | 6; F | Relapse | M2 | Low Risk | t(8;21), inv(8), del(9q) | Negative | ADE + AQ | 0 | Yes | Alive |
| pAML25 | 10; F | De Novo | M7 | High Risk | del(7q), +1, +8, der(1;7) | Negative | ADE + AQ | 0.23 | No | Alive |
| pAML26 | 14; F | De Novo | M5 | High Risk | t(10;11) | KMT2Ar | ADE + AQ | 2.5 | Yes | Alive |
| pAML27 | 1; F | Relapse | M4/M5 | High Risk | Complex | FLT3-ITD; KMT2Ar | ADEx2, AE, FLAG | 0 | Yes | DD |
| pAML28 | 1; M | De Novo | M5 | Low Risk | t(9;11) | KMT2Ar | ADE + AQ | 0 | Yes | Alive |
| pAML29 | 3; F | De Novo | M5 | High Risk | dup(Xq) | Negative | ADE | 0 | No | Alive |
| pAML30 | 17; F | Refractory | M7 | High Risk | Complex | Negative | ADE | 25 | Refractory | Alive |
| pAML31 | 0.58; F | De Novo | M5 | Low Risk | t(9;11) | KMT2Ar | DA+gemtuzumab + AQ | 0 | No | Alive |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strachan, D.C.; Gu, C.J.; Kita, R.; Anderson, E.K.; Richardson, M.A.; Yam, G.; Pimm, G.; Roselli, J.; Schweickert, A.; Terrell, M.; et al. Ex Vivo Drug Sensitivity Correlates with Clinical Response and Supports Personalized Therapy in Pediatric AML. Cancers 2022, 14, 6240. https://doi.org/10.3390/cancers14246240
Strachan DC, Gu CJ, Kita R, Anderson EK, Richardson MA, Yam G, Pimm G, Roselli J, Schweickert A, Terrell M, et al. Ex Vivo Drug Sensitivity Correlates with Clinical Response and Supports Personalized Therapy in Pediatric AML. Cancers. 2022; 14(24):6240. https://doi.org/10.3390/cancers14246240
Chicago/Turabian StyleStrachan, Debbie C., Christine J. Gu, Ryosuke Kita, Erica K. Anderson, Michelle A. Richardson, George Yam, Graham Pimm, Jordan Roselli, Alyssa Schweickert, Maci Terrell, and et al. 2022. "Ex Vivo Drug Sensitivity Correlates with Clinical Response and Supports Personalized Therapy in Pediatric AML" Cancers 14, no. 24: 6240. https://doi.org/10.3390/cancers14246240
APA StyleStrachan, D. C., Gu, C. J., Kita, R., Anderson, E. K., Richardson, M. A., Yam, G., Pimm, G., Roselli, J., Schweickert, A., Terrell, M., Rashid, R., Gonzalez, A. K., Oviedo, H. H., Alozie, M. C., Ilangovan, T., Marcogliese, A. N., Tada, H., Santaguida, M. T., & Stevens, A. M. (2022). Ex Vivo Drug Sensitivity Correlates with Clinical Response and Supports Personalized Therapy in Pediatric AML. Cancers, 14(24), 6240. https://doi.org/10.3390/cancers14246240