
AXL Receptor Tyrosine Kinase as a Promising Therapeutic Target Directing Multiple Aspects of Cancer Progression and Metastasis


{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. AXL Activation and Therapeutic Targeting
2.1. Ligand-Dependent and Ligand-Independent Mechanisms of Activation
2.2. AXL Therapeutic Targeting
3. Cancer Cell Intrinsic Implications of AXL Expression in the Metastatic Cascade and Therapy Resistance
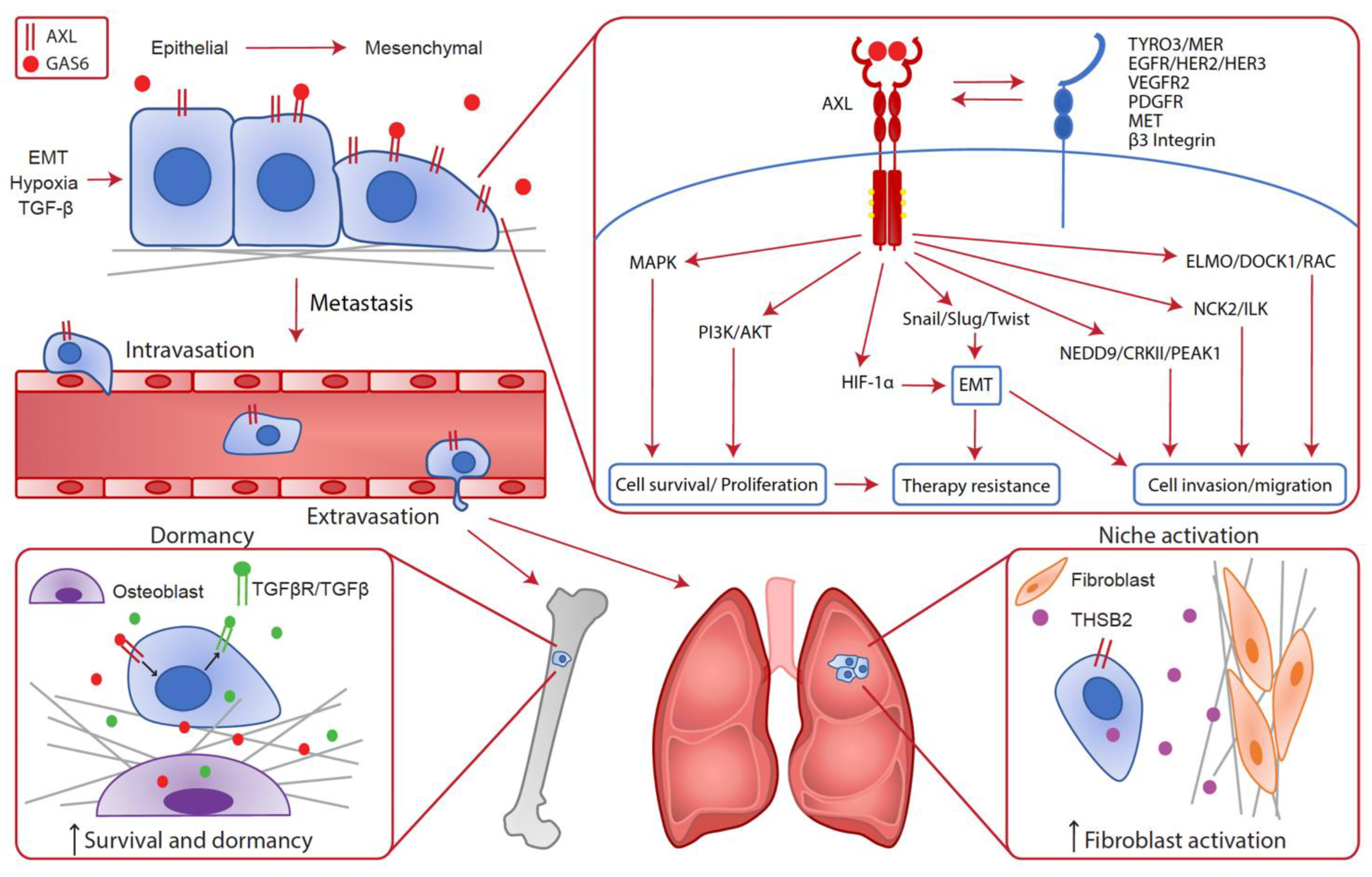
3.1. Epithelial-to-Mesenchymal Transition (EMT)
3.2. Cell Invasion and Migration
3.3. Modulation of the Metastatic Niche and Dormancy
3.4. Therapy Resistance
4. AXL as a Modulator of the Tumor Microenvironment
4.1. Angiogenesis
4.2. Fibrosis
4.3. Generation of an Immunosuppressive Microenvironment
4.4. Hypoxia
4.5. Therapeutic Implications
5. Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- O’Bryan, J.P.; Frye, R.A.; Cogswell, P.C.; Neubauer, A.; Kitch, B.; Prokop, C.; Espinosa, R., 3rd; Le Beau, M.M.; Earp, H.S.; Liu, E.T. axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol. Cell. Biol. 1991, 11, 5016–5031. [Google Scholar]
- Graham, D.K.; DeRyckere, D.; Davies, K.D.; Earp, H.S. The TAM family: Phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785. [Google Scholar] [CrossRef]
- Goyette, M.A.; Duhamel, S.; Aubert, L.; Pelletier, A.; Savage, P.; Thibault, M.P.; Johnson, R.M.; Carmeliet, P.; Basik, M.; Gaboury, L.; et al. The Receptor Tyrosine Kinase AXL Is Required at Multiple Steps of the Metastatic Cascade during HER2-Positive Breast Cancer Progression. Cell Rep. 2018, 23, 1476–1490. [Google Scholar] [CrossRef]
- Gjerdrum, C.; Tiron, C.; Hoiby, T.; Stefansson, I.; Haugen, H.; Sandal, T.; Collett, K.; Li, S.; McCormack, E.; Gjertsen, B.T.; et al. Axl is an essential epithelial-to-mesenchymal transition-induced regulator of breast cancer metastasis and patient survival. Proc. Natl. Acad. Sci. USA 2010, 107, 1124–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozneanu, L.; Pinciroli, P.; Ciobanu, D.A.; Carcangiu, M.L.; Canevari, S.; Tomassetti, A.; Caruntu, I.D. Computational and Immunohistochemical Analyses Highlight AXL as a Potential Prognostic Marker for Ovarian Cancer Patients. Anticancer Res. 2016, 36, 4155–4163. [Google Scholar]
- Rankin, E.B.; Fuh, K.C.; Taylor, T.E.; Krieg, A.J.; Musser, M.; Yuan, J.; Wei, K.; Kuo, C.J.; Longacre, T.A.; Giaccia, A.J. AXL is an essential factor and therapeutic target for metastatic ovarian cancer. Cancer Res. 2010, 70, 7570–7579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shieh, Y.S.; Lai, C.Y.; Kao, Y.R.; Shiah, S.G.; Chu, Y.W.; Lee, H.S.; Wu, C.W. Expression of axl in lung adenocarcinoma and correlation with tumor progression. Neoplasia 2005, 7, 1058–1064. [Google Scholar] [PubMed] [Green Version]
- Yu, H.; Liu, R.; Ma, B.; Li, X.; Yen, H.Y.; Zhou, Y.; Krasnoperov, V.; Xia, Z.; Zhang, X.; Bove, A.M.; et al. Axl receptor tyrosine kinase is a potential therapeutic target in renal cell carcinoma. Br. J. Cancer 2015, 113, 616–625. [Google Scholar] [CrossRef] [Green Version]
- Stitt, T.N.; Conn, G.; Gore, M.; Lai, C.; Bruno, J.; Radziejewski, C.; Mattsson, K.; Fisher, J.; Gies, D.R.; Jones, P.F.; et al. The anticoagulation factor protein S and its relative, Gas6, are ligands for the Tyro 3/Axl family of receptor tyrosine kinases. Cell 1995, 80, 661–670. [Google Scholar] [CrossRef] [Green Version]
- Manfioletti, G.; Brancolini, C.; Avanzi, G.; Schneider, C. The protein encoded by a growth arrest-specific gene (gas6) is a new member of the vitamin K-dependent proteins related to protein S, a negative coregulator in the blood coagulation cascade. Mol. Cell. Biol. 1993, 13, 4976–4985. [Google Scholar] [CrossRef] [Green Version]
- Mercer, J.; Helenius, A. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science 2008, 320, 531–535. [Google Scholar] [CrossRef]
- Chen, C.; Li, Q.; Darrow, A.L.; Wang, Y.; Derian, C.K.; Yang, J.; de Garavilla, L.; Andrade-Gordon, P.; Damiano, B.P. Mer receptor tyrosine kinase signaling participates in platelet function. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1118–1123. [Google Scholar] [CrossRef]
- Scott, R.S.; McMahon, E.J.; Pop, S.M.; Reap, E.A.; Caricchio, R.; Cohen, P.L.; Earp, H.S.; Matsushima, G.K. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 2001, 411, 207–211. [Google Scholar] [CrossRef]
- Meyer, A.S.; Zweemer, A.J.; Lauffenburger, D.A. The AXL Receptor is a Sensor of Ligand Spatial Heterogeneity. Cell Syst. 2015, 1, 25–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seitz, H.M.; Camenisch, T.D.; Lemke, G.; Earp, H.S.; Matsushima, G.K. Macrophages and dendritic cells use different Axl/Mertk/Tyro3 receptors in clearance of apoptotic cells. J. Immunol 2007, 178, 5635–5642. [Google Scholar] [CrossRef] [PubMed]
- Tsou, W.-I.; Nguyen, K.-Q.N.; Calarese, D.A.; Garforth, S.J.; Antes, A.L.; Smirnov, S.V.; Almo, S.C.; Birge, R.B.; Kotenko, S.V. Receptor tyrosine kinases, TYRO3, AXL, and MER, demonstrate distinct patterns and complex regulation of ligand-induced activation. J. Biol. Chem. 2014, 289, 25750–25763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemke, G. Biology of the TAM receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a009076. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.M.; Gray, Z.; Gomes, A.M.; Myers, L.; Behbod, F.; Machado, H.L. Gas6 expression is reduced in advanced breast cancers. NPJ Precis. Oncol. 2020, 4, 9. [Google Scholar] [CrossRef]
- Mc Cormack, O.; Chung, W.Y.; Fitzpatrick, P.; Cooke, F.; Flynn, B.; Harrison, M.; Fox, E.; Gallagher, E.; Goldrick, A.M.; Dervan, P.A.; et al. Growth arrest-specific gene 6 expression in human breast cancer. Br. J. Cancer 2008, 98, 1141–1146. [Google Scholar] [CrossRef]
- Bellosta, P.; Costa, M.; Lin, D.A.; Basilico, C. The receptor tyrosine kinase ARK mediates cell aggregation by homophilic binding. Mol. Cell. Biol. 1995, 15, 614–625. [Google Scholar] [CrossRef] [Green Version]
- Konishi, A.; Aizawa, T.; Mohan, A.; Korshunov, V.A.; Berk, B.C. Hydrogen peroxide activates the Gas6-Axl pathway in vascular smooth muscle cells. J. Biol. Chem. 2004, 279, 28766–28770. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.E.; Krodel, M.; Pazos, M.; Lai, C.; Prieto, A.L. Cross-phosphorylation, signaling and proliferative functions of the Tyro3 and Axl receptors in Rat2 cells. PLoS ONE 2012, 7, e36800. [Google Scholar] [CrossRef] [Green Version]
- Meyer, A.S.; Miller, M.A.; Gertler, F.B.; Lauffenburger, D.A. The receptor AXL diversifies EGFR signaling and limits the response to EGFR-targeted inhibitors in triple-negative breast cancer cells. Sci. Signal. 2013, 6, ra66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, G.X.; Kazlauskas, A. Axl is essential for VEGF-A-dependent activation of PI3K/Akt. EMBO J. 2012, 31, 1692–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salian-Mehta, S.; Xu, M.; Wierman, M.E. AXL and MET crosstalk to promote gonadotropin releasing hormone (GnRH) neuronal cell migration and survival. Mol. Cell Endocrinol. 2013, 374, 92–100. [Google Scholar] [CrossRef] [Green Version]
- Elkabets, M.; Pazarentzos, E.; Juric, D.; Sheng, Q.; Pelossof, R.A.; Brook, S.; Benzaken, A.O.; Rodon, J.; Morse, N.; Yan, J.J.; et al. AXL mediates resistance to PI3Kα inhibition by activating the EGFR/PKC/mTOR axis in head and neck and esophageal squamous cell carcinomas. Cancer Cell 2015, 27, 533–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, T.M.; Iida, M.; Corrigan, K.L.; Braverman, C.M.; Coan, J.P.; Flanigan, B.G.; Stein, A.P.; Salgia, R.; Rolff, J.; Kimple, R.J.; et al. The receptor tyrosine kinase AXL mediates nuclear translocation of the epidermal growth factor receptor. Sci. Signal 2017, 10. [Google Scholar] [CrossRef]
- Vouri, M.; Croucher, D.R.; Kennedy, S.P.; An, Q.; Pilkington, G.J.; Hafizi, S. Axl-EGFR receptor tyrosine kinase hetero-interaction provides EGFR with access to pro-invasive signalling in cancer cells. Oncogenesis 2016, 5, e266. [Google Scholar] [CrossRef] [Green Version]
- Revach, O.Y.; Sandler, O.; Samuels, Y.; Geiger, B. Cross-Talk between Receptor Tyrosine Kinases AXL and ERBB3 Regulates Invadopodia Formation in Melanoma Cells. Cancer Res. 2019, 79, 2634–2648. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, H.; Yamada, T.; Wang, R.; Tanimura, K.; Adachi, Y.; Nishiyama, A.; Tanimoto, A.; Takeuchi, S.; Araujo, L.H.; Boroni, M.; et al. AXL confers intrinsic resistance to osimertinib and advances the emergence of tolerant cells. Nat. Commun. 2019, 10, 259. [Google Scholar] [CrossRef]
- Park, I.K.; Mundy-Bosse, B.; Whitman, S.P.; Zhang, X.; Warner, S.L.; Bearss, D.J.; Blum, W.; Marcucci, G.; Caligiuri, M.A. Receptor tyrosine kinase Axl is required for resistance of leukemic cells to FLT3-targeted therapy in acute myeloid leukemia. Leukemia 2015, 29, 2382–2389. [Google Scholar] [CrossRef]
- Niu, X.; Rothe, K.; Chen, M.; Grasedieck, S.; Li, R.; Nam, S.E.; Zhang, X.; Novakovskiy, G.E.; Ahn, Y.H.; Maksakova, I.; et al. Targeting AXL kinase sensitizes leukemic stem and progenitor cells to venetoclax treatment in acute myeloid leukemia. Blood 2021, 137, 3641–3655. [Google Scholar] [CrossRef]
- Mahadevan, D.; Cooke, L.; Riley, C.; Swart, R.; Simons, B.; Della Croce, K.; Wisner, L.; Iorio, M.; Shakalya, K.; Garewal, H.; et al. A novel tyrosine kinase switch is a mechanism of imatinib resistance in gastrointestinal stromal tumors. Oncogene 2007, 26, 3909–3919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Greger, J.; Shi, H.; Liu, Y.; Greshock, J.; Annan, R.; Halsey, W.; Sathe, G.M.; Martin, A.M.; Gilmer, T.M. Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: Activation of AXL. Cancer Res. 2009, 69, 6871–6878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.; Wei, Y.; Wei, X. AXL receptor tyrosine kinase as a promising anti-cancer approach: Functions, molecular mechanisms and clinical applications. Mol. Cancer 2019, 18, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Chen, X.; He, J.; Liao, D.; Zu, X. Axl inhibitors as novel cancer therapeutic agents. Life Sci. 2018, 198, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Li, Y.; Stawicki, S.; Couto, S.; Eastham-Anderson, J.; Kallop, D.; Weimer, R.; Wu, Y.; Pei, L. An anti-Axl monoclonal antibody attenuates xenograft tumor growth and enhances the effect of multiple anticancer therapies. Oncogene 2010, 29, 5254–5264. [Google Scholar] [CrossRef] [Green Version]
- Leconet, W.; Chentouf, M.; du Manoir, S.; Chevalier, C.; Sirvent, A.; Ait-Arsa, I.; Busson, M.; Jarlier, M.; Radosevic-Robin, N.; Theillet, C.; et al. Therapeutic Activity of Anti-AXL Antibody against Triple-Negative Breast Cancer Patient-Derived Xenografts and Metastasis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 2806–2816. [Google Scholar] [CrossRef] [Green Version]
- Holland, S.J.; Pan, A.; Franci, C.; Hu, Y.; Chang, B.; Li, W.; Duan, M.; Torneros, A.; Yu, J.; Heckrodt, T.J.; et al. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 2010, 70, 1544–1554. [Google Scholar] [CrossRef] [Green Version]
- Myers, S.H.; Brunton, V.G.; Unciti-Broceta, A. AXL Inhibitors in Cancer: A Medicinal Chemistry Perspective. J. Med. Chem. 2016, 59, 3593–3608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirane, A.; Ludwig, K.F.; Sorrelle, N.; Haaland, G.; Sandal, T.; Ranaweera, R.; Toombs, J.E.; Wang, M.; Dineen, S.P.; Micklem, D.; et al. Warfarin Blocks Gas6-Mediated Axl Activation Required for Pancreatic Cancer Epithelial Plasticity and Metastasis. Cancer Res. 2015, 75, 3699–3705. [Google Scholar] [CrossRef] [Green Version]
- Paolino, M.; Choidas, A.; Wallner, S.; Pranjic, B.; Uribesalgo, I.; Loeser, S.; Jamieson, A.M.; Langdon, W.Y.; Ikeda, F.; Fededa, J.P.; et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature 2014, 507, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Kariolis, M.S.; Miao, Y.R.; Jones, D.S., 2nd; Kapur, S.; Mathews, I.I.; Giaccia, A.J.; Cochran, J.R. An engineered Axl ‘decoy receptor’ effectively silences the Gas6-Axl signaling axis. Nat. Chem. Biol. 2014, 10, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Sun, H.; Zhang, A.; Wu, X.; Li, Y.; Liu, J.; Duan, Y.; Xiao, F.; Wang, H.; Lv, M.; et al. A novel AXL chimeric antigen receptor endows T cells with anti-tumor effects against triple negative breast cancers. Cell. Immunol. 2018, 331, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Li, Y.; Liu, W.; Li, X. Engineered IL-7 Receptor Enhances the Therapeutic Effect of AXL-CAR-T Cells on Triple-Negative Breast Cancer. BioMed Res. Int. 2020, 2020, 4795171. [Google Scholar] [CrossRef]
- Vuoriluoto, K.; Haugen, H.; Kiviluoto, S.; Mpindi, J.P.; Nevo, J.; Gjerdrum, C.; Tiron, C.; Lorens, J.B.; Ivaska, J. Vimentin regulates EMT induction by Slug and oncogenic H-Ras and migration by governing Axl expression in breast cancer. Oncogene 2011, 30, 1436–1448. [Google Scholar] [CrossRef] [Green Version]
- Rankin, E.B.; Fuh, K.C.; Castellini, L.; Viswanathan, K.; Finger, E.C.; Diep, A.N.; LaGory, E.L.; Kariolis, M.S.; Chan, A.; Lindgren, D.; et al. Direct regulation of GAS6/AXL signaling by HIF promotes renal metastasis through SRC and MET. Proc. Natl. Acad. Sci. USA 2014, 111, 13373–13378. [Google Scholar] [CrossRef] [Green Version]
- Goyette, M.A.; Elkholi, I.E.; Apcher, C.; Kuasne, H.; Rothlin, C.V.; Muller, W.J.; Richard, D.E.; Park, M.; Gratton, J.P.; Côté, J.F. Targeting Axl favors an antitumorigenic microenvironment that enhances immunotherapy responses by decreasing Hif-1α levels. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Mak, M.P.; Tong, P.; Diao, L.; Cardnell, R.J.; Gibbons, D.L.; William, W.N.; Skoulidis, F.; Parra, E.R.; Rodriguez-Canales, J.; Wistuba, I.I.; et al. A Patient-Derived, Pan-Cancer EMT Signature Identifies Global Molecular Alterations and Immune Target Enrichment Following Epithelial-to-Mesenchymal Transition. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 609–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asiedu, M.K.; Beauchamp-Perez, F.D.; Ingle, J.N.; Behrens, M.D.; Radisky, D.C.; Knutson, K.L. AXL induces epithelial-to-mesenchymal transition and regulates the function of breast cancer stem cells. Oncogene 2014, 33, 1316–1324. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Jeng, Y.M.; Chen, Y.L.; Chung, L.; Yuan, R.H. Gas6/Axl pathway promotes tumor invasion through the transcriptional activation of Slug in hepatocellular carcinoma. Carcinogenesis 2014, 35, 769–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halmos, B.; Haura, E.B. New twists in the AXL(e) of tumor progression. Sci. Signal. 2016, 9, fs14. [Google Scholar] [CrossRef]
- Hafizi, S.; Dahlback, B. Signalling and functional diversity within the Axl subfamily of receptor tyrosine kinases. Cytokine Growth Factor Rev. 2006, 17, 295–304. [Google Scholar] [CrossRef] [Green Version]
- Allen, M.P.; Xu, M.; Linseman, D.A.; Pawlowski, J.E.; Bokoch, G.M.; Heidenreich, K.A.; Wierman, M.E. Adhesion-related kinase repression of gonadotropin-releasing hormone gene expression requires Rac activation of the extracellular signal-regulated kinase pathway. J. Biol. Chem. 2002, 277, 38133–38140. [Google Scholar] [CrossRef] [Green Version]
- Dunne, P.D.; McArt, D.G.; Blayney, J.K.; Kalimutho, M.; Greer, S.; Wang, T.; Srivastava, S.; Ong, C.W.; Arthur, K.; Loughrey, M.; et al. AXL is a key regulator of inherent and chemotherapy-induced invasion and predicts a poor clinical outcome in early-stage colon cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 164–175. [Google Scholar] [CrossRef] [Green Version]
- Giles, K.M.; Kalinowski, F.C.; Candy, P.A.; Epis, M.R.; Zhang, P.M.; Redfern, A.D.; Stuart, L.M.; Goodall, G.J.; Leedman, P.J. Axl mediates acquired resistance of head and neck cancer cells to the epidermal growth factor receptor inhibitor erlotinib. Mol. Cancer Ther. 2013, 12, 2541–2558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linger, R.M.; Keating, A.K.; Earp, H.S.; Graham, D.K. TAM receptor tyrosine kinases: Biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv. Cancer Res. 2008, 100, 35–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Lieu, Z.Z.; Wolfenson, H.; Hameed, F.M.; Bershadsky, A.D.; Sheetz, M.P. Mechanosensing Controlled Directly by Tyrosine Kinases. Nano Lett. 2016, 16, 5951–5961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rea, K.; Pinciroli, P.; Sensi, M.; Alciato, F.; Bisaro, B.; Lozneanu, L.; Raspagliesi, F.; Centritto, F.; Cabodi, S.; Defilippi, P.; et al. Novel Axl-driven signaling pathway and molecular signature characterize high-grade ovarian cancer patients with poor clinical outcome. Oncotarget 2015, 6, 30859–30875. [Google Scholar] [CrossRef] [Green Version]
- Abu-Thuraia, A.; Gauthier, R.; Chidiac, R.; Fukui, Y.; Screaton, R.A.; Gratton, J.P.; Cote, J.F. Axl phosphorylates Elmo scaffold proteins to promote Rac activation and cell invasion. Mol. Cell. Biol. 2015, 35, 76–87. [Google Scholar] [CrossRef] [Green Version]
- Abu-Thuraia, A.; Goyette, M.A.; Boulais, J.; Delliaux, C.; Apcher, C.; Schott, C.; Chidiac, R.; Bagci, H.; Thibault, M.P.; Davidson, D.; et al. AXL confers cell migration and invasion by hijacking a PEAK1-regulated focal adhesion protein network. Nat. Commun. 2020, 11, 3586. [Google Scholar] [CrossRef] [PubMed]
- D'Alfonso, T.M.; Hannah, J.; Chen, Z.; Liu, Y.; Zhou, P.; Shin, S.J. Axl receptor tyrosine kinase expression in breast cancer. J. Clin. Pathol. 2014, 67, 690–696. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Sarkissyan, M.; Vadgama, J.V. Epithelial-Mesenchymal Transition and Breast Cancer. J. Clin. Med. 2016, 5, 13. [Google Scholar] [CrossRef] [Green Version]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef] [Green Version]
- Giampieri, S.; Manning, C.; Hooper, S.; Jones, L.; Hill, C.S.; Sahai, E. Localized and reversible TGFbeta signalling switches breast cancer cells from cohesive to single cell motility. Nat. Cell Biol. 2009, 11, 1287–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshimori, N.; Oristian, D.; Fuchs, E. TGF-beta promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell 2015, 160, 963–976. [Google Scholar] [CrossRef] [Green Version]
- Wilson, C.; Ye, X.; Pham, T.; Lin, E.; Chan, S.; McNamara, E.; Neve, R.M.; Belmont, L.; Koeppen, H.; Yauch, R.L.; et al. AXL inhibition sensitizes mesenchymal cancer cells to antimitotic drugs. Cancer Res. 2014, 74, 5878–5890. [Google Scholar] [CrossRef] [Green Version]
- Del Pozo Martin, Y.; Park, D.; Ramachandran, A.; Ombrato, L.; Calvo, F.; Chakravarty, P.; Spencer-Dene, B.; Derzsi, S.; Hill, C.S.; Sahai, E.; et al. Mesenchymal Cancer Cell-Stroma Crosstalk Promotes Niche Activation, Epithelial Reversion, and Metastatic Colonization. Cell Rep. 2015, 13, 2456–2469. [Google Scholar] [CrossRef] [Green Version]
- Sosa, M.S.; Bragado, P.; Aguirre-Ghiso, J.A. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat. Rev. Cancer 2014, 14, 611–622. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Pedersen, E.A.; Patel, L.R.; Ziegler, A.M.; Havens, A.M.; Jung, Y.; Wang, J.; Zalucha, S.; Loberg, R.D.; Pienta, K.J.; et al. GAS6/AXL axis regulates prostate cancer invasion, proliferation, and survival in the bone marrow niche. Neoplasia 2010, 12, 116–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yumoto, K.; Eber, M.R.; Wang, J.; Cackowski, F.C.; Decker, A.M.; Lee, E.; Nobre, A.R.; Aguirre-Ghiso, J.A.; Jung, Y.; Taichman, R.S. Axl is required for TGF-beta2-induced dormancy of prostate cancer cells in the bone marrow. Sci. Rep. 2016, 6, 36520. [Google Scholar] [CrossRef] [PubMed]
- Khoo, W.H.; Ledergor, G.; Weiner, A.; Roden, D.L.; Terry, R.L.; McDonald, M.M.; Chai, R.C.; De Veirman, K.; Owen, K.L.; Opperman, K.S.; et al. A niche-dependent myeloid transcriptome signature defines dormant myeloma cells. Blood 2019, 134, 30–43. [Google Scholar] [CrossRef]
- Li, Y.; Wang, X.; Bi, S.; Zhao, K.; Yu, C. Inhibition of Mer and Axl receptor tyrosine kinases leads to increased apoptosis and improved chemosensitivity in human neuroblastoma. Biochem. Biophys. Res. Commun. 2015, 457, 461–466. [Google Scholar] [CrossRef]
- Lin, J.Z.; Wang, Z.J.; De, W.; Zheng, M.; Xu, W.Z.; Wu, H.F.; Armstrong, A.; Zhu, J.G. Targeting AXL overcomes resistance to docetaxel therapy in advanced prostate cancer. Oncotarget 2017, 8, 41064–41077. [Google Scholar] [CrossRef]
- Oien, D.B.; Garay, T.; Eckstein, S.; Chien, J. Cisplatin and Pemetrexed Activate AXL and AXL Inhibitor BGB324 Enhances Mesothelioma Cell Death from Chemotherapy. Front. Pharm. 2017, 8, 970. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, K.F.; Du, W.; Sorrelle, N.B.; Wnuk-Lipinska, K.; Topalovski, M.; Toombs, J.E.; Cruz, V.H.; Yabuuchi, S.; Rajeshkumar, N.V.; Maitra, A.; et al. Small-Molecule Inhibition of Axl Targets Tumor Immune Suppression and Enhances Chemotherapy in Pancreatic Cancer. Cancer Res. 2018, 78, 246–255. [Google Scholar] [CrossRef] [Green Version]
- Linger, R.M.; Cohen, R.A.; Cummings, C.T.; Sather, S.; Migdall-Wilson, J.; Middleton, D.H.; Lu, X.; Baron, A.E.; Franklin, W.A.; Merrick, D.T.; et al. Mer or Axl receptor tyrosine kinase inhibition promotes apoptosis, blocks growth and enhances chemosensitivity of human non-small cell lung cancer. Oncogene 2013, 32, 3420–3431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef]
- Debruyne, D.N.; Bhatnagar, N.; Sharma, B.; Luther, W.; Moore, N.F.; Cheung, N.K.; Gray, N.S.; George, R.E. ALK inhibitor resistance in ALK(F1174L)-driven neuroblastoma is associated with AXL activation and induction of EMT. Oncogene 2016, 35, 3681–3691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, M.; Siemann, D.W. Axl signaling is an important mediator of tumor angiogenesis. Oncotarget 2019, 10, 2887–2898. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Zhao, H.; Tian, L.; Nolley, R.; Diep, A.N.; Ernst, A.; Fuh, K.C.; Miao, Y.R.; von Eyben, R.; Leppert, J.T.; et al. S100A10 Is a Critical Mediator of GAS6/AXL-Induced Angiogenesis in Renal Cell Carcinoma. Cancer Res. 2019, 79, 5758–5768. [Google Scholar] [CrossRef] [Green Version]
- Melaragno, M.G.; Fridell, Y.W.; Berk, B.C. The Gas6/Axl system: A novel regulator of vascular cell function. Trends Cardiovasc. Med. 1999, 9, 250–253. [Google Scholar] [CrossRef]
- Holland, S.J.; Powell, M.J.; Franci, C.; Chan, E.W.; Friera, A.M.; Atchison, R.E.; McLaughlin, J.; Swift, S.E.; Pali, E.S.; Yam, G.; et al. Multiple roles for the receptor tyrosine kinase axl in tumor formation. Cancer Res. 2005, 65, 9294–9303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Ye, X.; Tan, C.; Hongo, J.A.; Zha, J.; Liu, J.; Kallop, D.; Ludlam, M.J.; Pei, L. Axl as a potential therapeutic target in cancer: Role of Axl in tumor growth, metastasis and angiogenesis. Oncogene 2009, 28, 3442–3455. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Xia, X.; Tang, L.; Yao, F.; Xu, H.; Lei, H. Normal vitreous promotes angiogenesis via activation of Axl. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2021, 35, e21152. [Google Scholar] [CrossRef]
- Lechertier, T.; Reynolds, L.E.; Kim, H.; Pedrosa, A.R.; Gomez-Escudero, J.; Munoz-Felix, J.M.; Batista, S.; Dukinfield, M.; Demircioglu, F.; Wong, P.P.; et al. Pericyte FAK negatively regulates Gas6/Axl signalling to suppress tumour angiogenesis and tumour growth. Nat. Commun. 2020, 11, 2810. [Google Scholar] [CrossRef]
- Staufer, K.; Dengler, M.; Huber, H.; Marculescu, R.; Stauber, R.; Lackner, C.; Dienes, H.-P.; Kivaranovic, D.; Schachner, C.; Zeitlinger, M.; et al. The non-invasive serum biomarker soluble Axl accurately detects advanced liver fibrosis and cirrhosis. Cell Death Dis. 2017, 8, e3135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tutusaus, A.; de Gregorio, E.; Cucarull, B.; Cristóbal, H.; Aresté, C.; Graupera, I.; Coll, M.; Colell, A.; Gausdal, G.; Lorens, J.B.; et al. A Functional Role of GAS6/TAM in Nonalcoholic Steatohepatitis Progression Implicates AXL as Therapeutic Target. Cell Mol. Gastroenterol. Hepatol. 2020, 9, 349–368. [Google Scholar] [CrossRef]
- Bárcena, C.; Stefanovic, M.; Tutusaus, A.; Joannas, L.; Menéndez, A.; García-Ruiz, C.; Sancho-Bru, P.; Marí, M.; Caballeria, J.; Rothlin, C.V.; et al. Gas6/Axl pathway is activated in chronic liver disease and its targeting reduces fibrosis via hepatic stellate cell inactivation. J. Hepatol. 2015, 63, 670–678. [Google Scholar] [CrossRef] [Green Version]
- El-Serag, H.B. Hepatocellular carcinoma. N. Engl. J. Med. 2011, 365, 1118–1127. [Google Scholar] [CrossRef]
- Aguilera, T.A.; Rafat, M.; Castellini, L.; Shehade, H.; Kariolis, M.S.; Hui, A.B.; Stehr, H.; von Eyben, R.; Jiang, D.; Ellies, L.G.; et al. Reprogramming the immunological microenvironment through radiation and targeting Axl. Nat. Commun. 2016, 7, 13898. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Li, Y.; Zhang, D.; Ma, J. Axl inhibition induces the antitumor immune response which can be further potentiated by PD-1 blockade in the mouse cancer models. Oncotarget 2017, 8, 89761–89774. [Google Scholar] [CrossRef] [Green Version]
- Chiu, K.C.; Lee, C.H.; Liu, S.Y.; Chou, Y.T.; Huang, R.Y.; Huang, S.M.; Shieh, Y.S. Polarization of tumor-associated macrophages and Gas6/Axl signaling in oral squamous cell carcinoma. Oral Oncol. 2015, 51, 683–689. [Google Scholar] [CrossRef]
- Bottai, G.; Raschioni, C.; Székely, B.; Di Tommaso, L.; Szász, A.M.; Losurdo, A.; Győrffy, B.; Ács, B.; Torrisi, R.; Karachaliou, N.; et al. AXL-associated tumor inflammation as a poor prognostic signature in chemotherapy-treated triple-negative breast cancer patients. NPJ Breast Cancer 2016, 2, 16033. [Google Scholar] [CrossRef]
- Skinner, H.D.; Giri, U.; Yang, L.P.; Kumar, M.; Liu, Y.; Story, M.D.; Pickering, C.R.; Byers, L.A.; Williams, M.D.; Wang, J.; et al. Integrative Analysis Identifies a Novel AXL-PI3 Kinase-PD-L1 Signaling Axis Associated with Radiation Resistance in Head and Neck Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 2713–2722. [Google Scholar] [CrossRef] [Green Version]
- Kasikara, C.; Kumar, S.; Kimani, S.; Tsou, W.I.; Geng, K.; Davra, V.; Sriram, G.; Devoe, C.; Nguyen, K.N.; Antes, A.; et al. Phosphatidylserine Sensing by TAM Receptors Regulates AKT-Dependent Chemoresistance and PD-L1 Expression. Mol. Cancer Res. 2017, 15, 753–764. [Google Scholar] [CrossRef] [Green Version]
- Najafov, A.; Mookhtiar, A.K.; Luu, H.S.; Ordureau, A.; Pan, H.; Amin, P.P.; Li, Y.; Lu, Q.; Yuan, J. TAM Kinases Promote Necroptosis by Regulating Oligomerization of MLKL. Mol. Cell 2019, 75, 457–468.e4. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Lemke, G. Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science 2001, 293, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Gore, M.; Zhang, Q.; Camenisch, T.; Boast, S.; Casagranda, F.; Lai, C.; Skinner, M.K.; Klein, R.; Matsushima, G.K.; et al. Tyro-3 family receptors are essential regulators of mammalian spermatogenesis. Nature 1999, 398, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.L.; LaVail, M.M.; Yasumura, D.; Matthes, M.T.; Yang, H.; Trautmann, N.; Chappelow, A.V.; Feng, W.; Earp, H.S.; Matsushima, G.K.; et al. An RCS-like retinal dystrophy phenotype in mer knockout mice. Investig. Ophthalmol. Vis. Sci. 2003, 44, 826–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothlin, C.V.; Ghosh, S.; Zuniga, E.I.; Oldstone, M.B.; Lemke, G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 2007, 131, 1124–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, E.T.; Pang, I.K.; Carrera Silva, E.A.; Bosurgi, L.; Miner, J.J.; Diamond, M.S.; Iwasaki, A.; Rothlin, C.V. AXL receptor tyrosine kinase is required for T cell priming and antiviral immunity. Elife 2016, 5. [Google Scholar] [CrossRef]
- Carrera Silva, E.A.; Chan, P.Y.; Joannas, L.; Errasti, A.E.; Gagliani, N.; Bosurgi, L.; Jabbour, M.; Perry, A.; Smith-Chakmakova, F.; Mucida, D.; et al. T cell-derived protein S engages TAM receptor signaling in dendritic cells to control the magnitude of the immune response. Immunity 2013, 39, 160–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, G.J.; Zheng, J.Y.; Bian, J.L.; Chen, L.W.; Dong, N.; Yu, Y.; Hong, G.L.; Chandoo, A.; Yao, Y.M.; Lu, Z.Q. Growth Arrest-Specific 6 Enhances the Suppressive Function of CD4+CD25+ Regulatory T Cells Mainly through Axl Receptor. Mediat. Inflamm. 2017, 2017, 6848430. [Google Scholar] [CrossRef] [Green Version]
- Holtzhausen, A.; Harris, W.; Ubil, E.; Hunter, D.M.; Zhao, J.; Zhang, Y.; Zhang, D.; Liu, Q.; Wang, X.; Graham, D.K.; et al. TAM Family Receptor Kinase Inhibition Reverses MDSC-Mediated Suppression and Augments Anti-PD-1 Therapy in Melanoma. Cancer Immunol. Res. 2019, 7, 1672–1686. [Google Scholar] [CrossRef]
- Maier, B.; Leader, A.M.; Chen, S.T.; Tung, N.; Chang, C.; LeBerichel, J.; Chudnovskiy, A.; Maskey, S.; Walker, L.; Finnigan, J.P.; et al. A conserved dendritic-cell regulatory program limits antitumour immunity. Nature 2020, 580, 257–262. [Google Scholar] [CrossRef]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Liu, X.D.; Sun, M.; Zhang, X.; German, P.; Bai, S.; Ding, Z.; Tannir, N.; Wood, C.G.; Matin, S.F.; et al. Targeting MET and AXL overcomes resistance to sunitinib therapy in renal cell carcinoma. Oncogene 2016, 35, 2687–2697. [Google Scholar] [CrossRef]
- Gustafsson, A.; Fritz, H.K.M.; Dahlback, B. Gas6-Axl signaling in presence of Sunitinib is enhanced, diversified and sustained in renal tumor cells, resulting in tumor-progressive advantages. Exp. Cell Res. 2017, 355, 47–56. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [Green Version]
- Davra, V.; Kumar, S.; Geng, K.; Calianese, D.; Mehta, D.; Gadiyar, V.; Kasikara, C.; Lahey, K.C.; Chang, Y.-j.; Wichroski, M.; et al. Axl and Mertk receptors cooperate to promote breast cancer progression by combined oncogenic signaling and evasion of host anti-tumor immunity. Cancer Res. 2021, 81, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Kasikara, C.; Davra, V.; Calianese, D.; Geng, K.; Spires, T.E.; Quigley, M.; Wichroski, M.; Sriram, G.; Suarez-Lopez, L.; Yaffe, M.B.; et al. Pan-TAM Tyrosine Kinase Inhibitor BMS-777607 Enhances Anti-PD-1 mAb Efficacy in a Murine Model of Triple-Negative Breast Cancer. Cancer Res. 2019, 79, 2669–2683. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Ma, W.; Zhou, Q.; Yan, H.; Lim, Z.F.; Huang, M.; Deng, C.; Yu, X.; Su, H.; Komo, S.; et al. AXL-GAS6 expression can predict for adverse prognosis in non-small cell lung cancer with brain metastases. J. Cancer Res. Clin. Oncol 2017, 143, 1947–1957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Xu, X.S.; Yang, J.X.; Guo, J.H.; Chao, T.F.; Tong, Y. The prognostic role of Gas6/Axl axis in solid malignancies: A meta-analysis and literature review. Oncol. Targets 2018, 11, 509–519. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Wang, Z.; Tang, Y.; Lu, X.; Chen, J.; Dong, Y.; Wu, B.; Wang, C.; Yang, L.; Guo, Z.; et al. Liquid biopsy-based single-cell metabolic phenotyping of lung cancer patients for informative diagnostics. Nat. Commun. 2019, 10, 3856. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goyette, M.-A.; Côté, J.-F. AXL Receptor Tyrosine Kinase as a Promising Therapeutic Target Directing Multiple Aspects of Cancer Progression and Metastasis. Cancers 2022, 14, 466. https://doi.org/10.3390/cancers14030466
Goyette M-A, Côté J-F. AXL Receptor Tyrosine Kinase as a Promising Therapeutic Target Directing Multiple Aspects of Cancer Progression and Metastasis. Cancers. 2022; 14(3):466. https://doi.org/10.3390/cancers14030466
Chicago/Turabian StyleGoyette, Marie-Anne, and Jean-François Côté. 2022. "AXL Receptor Tyrosine Kinase as a Promising Therapeutic Target Directing Multiple Aspects of Cancer Progression and Metastasis" Cancers 14, no. 3: 466. https://doi.org/10.3390/cancers14030466
APA StyleGoyette, M.-A., & Côté, J.-F. (2022). AXL Receptor Tyrosine Kinase as a Promising Therapeutic Target Directing Multiple Aspects of Cancer Progression and Metastasis. Cancers, 14(3), 466. https://doi.org/10.3390/cancers14030466