
Evolution and Targeting of Myeloid Suppressor Cells in Cancer: A Translational Perspective

 , ,
, ,  ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
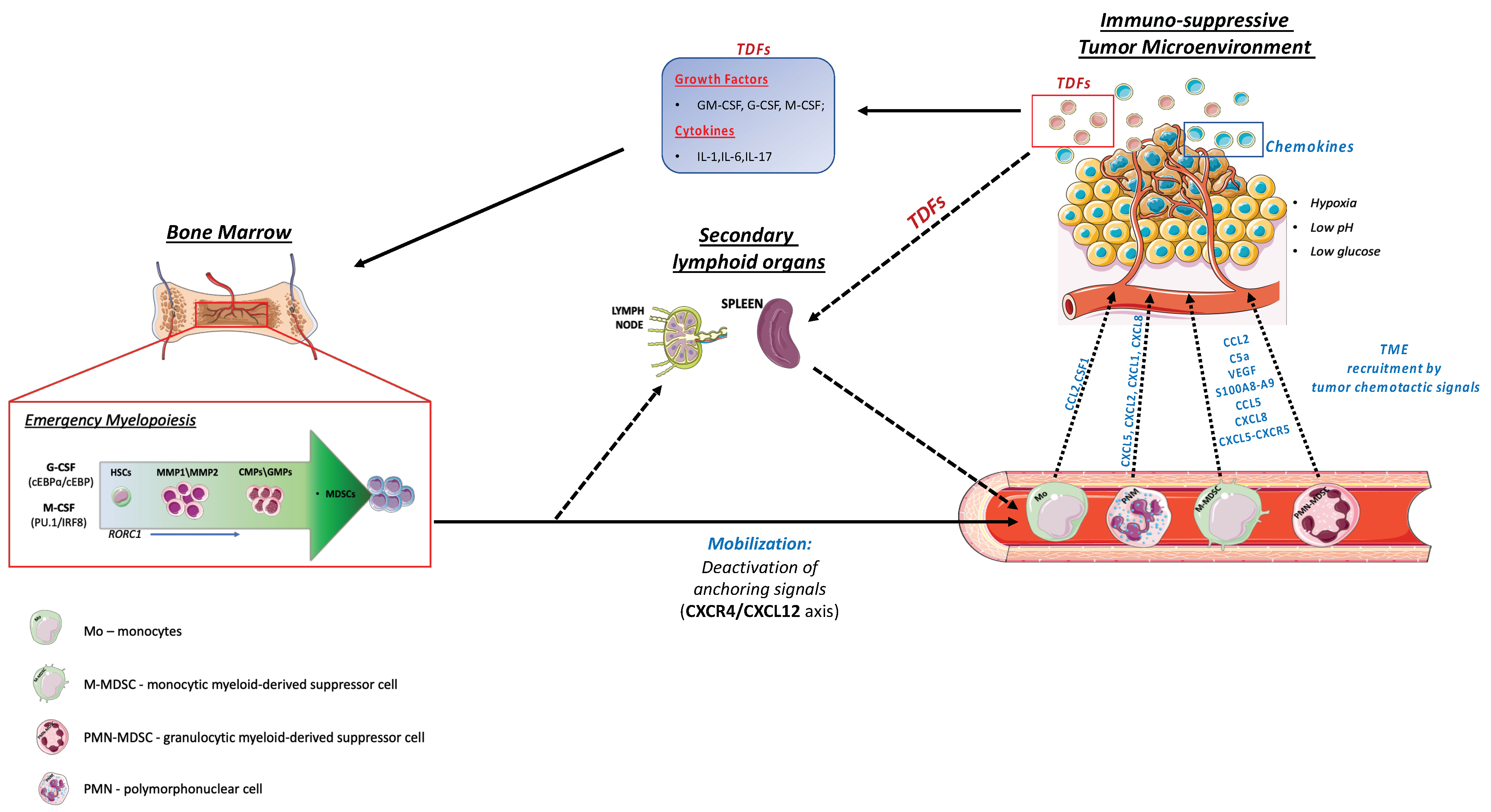
2. Emergency Myelopoiesis
3. Myeloid Cells Mobilization
4. Functional Heterogeneity of Tumor-Associated Myeloid Cells
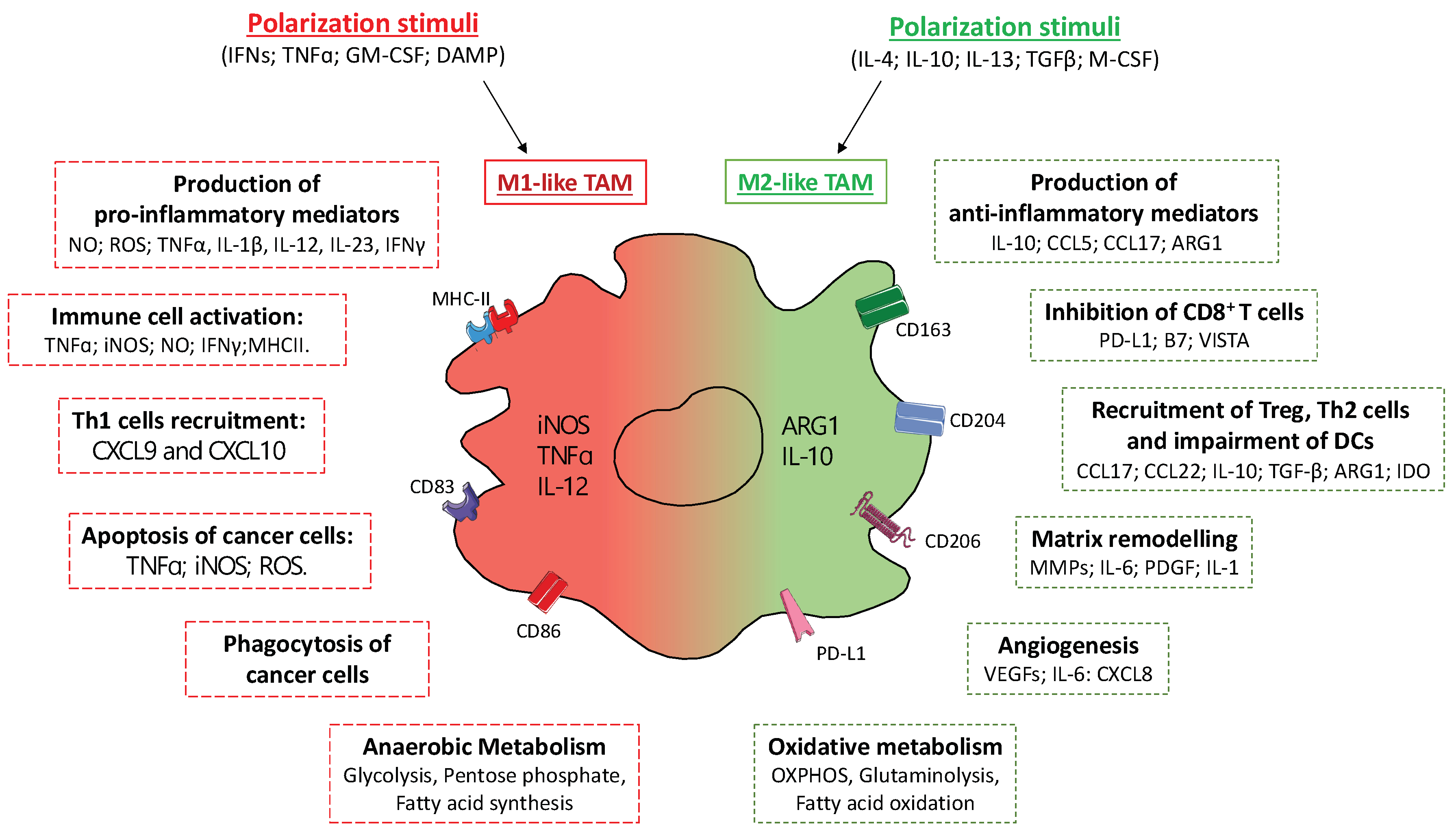
4.1. Tumor Associated Macrophages (TAMs)
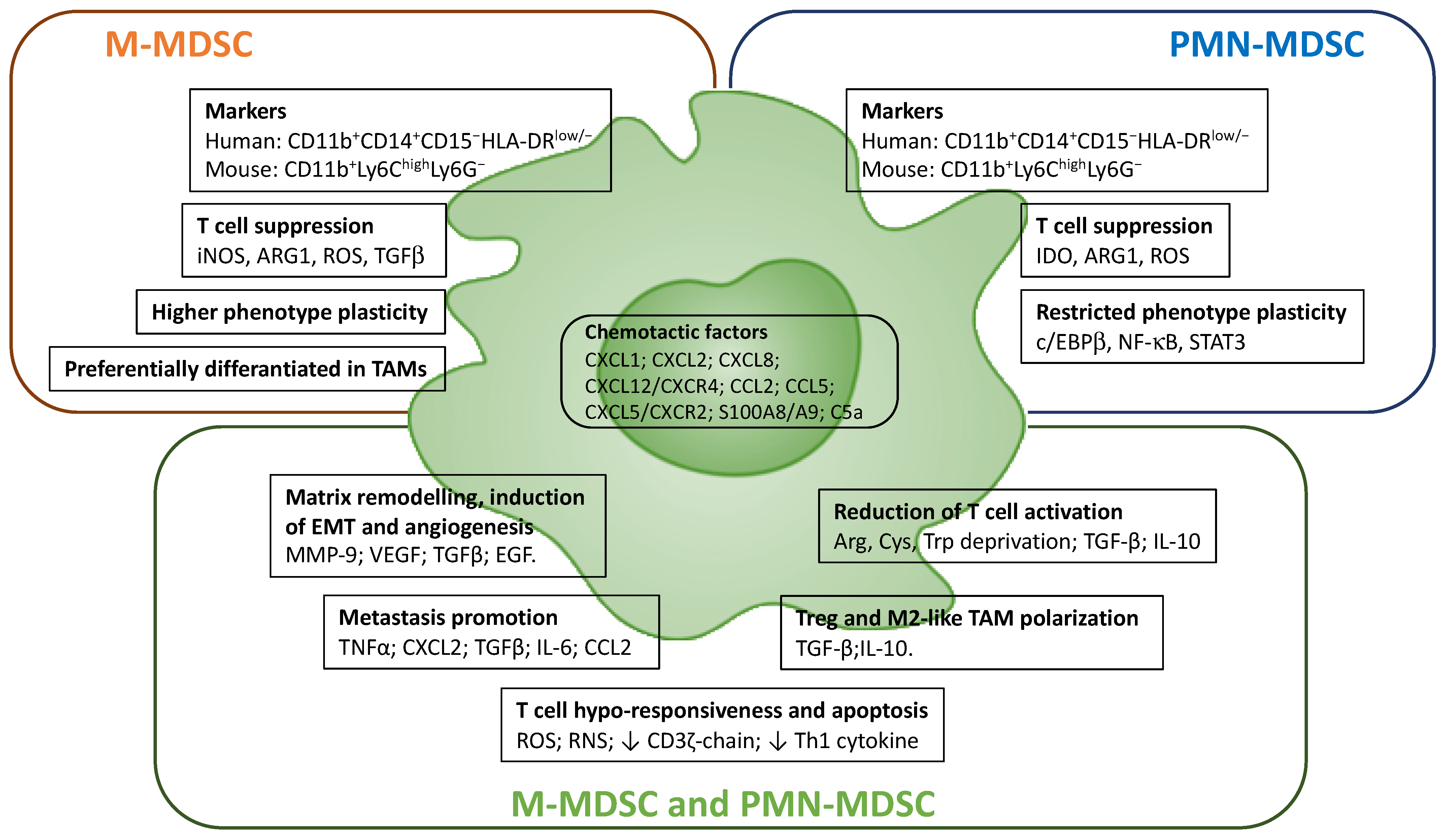
4.2. Myeloid-Derived Suppressor Cells (MDSCs)
4.3. Tumor-Associated Neutrophils (TANs)
5. Pre-Clinical Targeting of Myeloid Cells in Cancer
5.1. TAMs Targeting Approaches
5.2. MDSCs Targeting Approaches
6. Clinical Advances in Targeting Tumor-Associated Myeloid Cells
6.1. Clinical Trials Targeting TAMs
6.1.1. Abrogating TAM Enrichment
- Depleting TAMs
- Inhibition of TAMs Recruitment
6.1.2. Re-Education of TAMs
- Targeting TAM Polarization
- Re-Activation of Phagocytosis
- Macrophage Engineering
6.2. Clinical Trials Targeting MDSCs
6.2.1. Abrogating MDSCs Enrichment
- Inhibition of MDSCs Recruitment
- Depletion of MDSCs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strategy | Target | Drug Name | Combined Therapy | Disease | Clinical Trial |
|---|---|---|---|---|---|
| Inhibition of recruitment, mobilization, expansion | CXCR1/2-CXCL8 | SX-682 | Nivolumab | Metastatic Colorectal Cancer | NCT04599140 |
| Nivolumab | Pancreatic Cancer | NCT04477343 | |||
| Pembrolizumab | Metastatic Melanoma | NCT03161431 | |||
| BinTrafusp Alfa, CV301 | Advanced Solid Cancer | NCT04574583 | |||
| Navarixin | Pembrolizumab | Advanced Solid Cancer | NCT03473925 | ||
| Reparixin | Paclitaxel | Metastatic Breast Cancer | NCT02370238 | ||
| Paclitaxel | HER2-neg Metastatic Breast Cancer | NCT02001974 | |||
| Single Agent | Early Breast Cancer | NCT01861054 | |||
| CXCR2 | AZD5069 | Enzalutamide | Metastatic Prostate Cancer | NCT03177187 | |
| Nab-paclitaxel, Gemcitabine, MEDI4736 | Metastatic Pancreatic Ductal Carcinoma | NCT02583477 | |||
| AZD9150, MEDI4736, Tremelimumab | Head and Neck Carcinoma | NCT02499328 | |||
| CXCR4 | Plerixafor | Pembrolizumab | Head and Neck Cancer | NCT04058145 | |
| Motixafortide | Atezolizumab | Metastatic Pancreatic Adenocarcinoma | NCT03193190 | ||
| VEGF/VEGFR | Bevacizumab | Capecitabine | Glioblastoma | NCT02669173 | |
| Pazopanib Hydrochloride | Renal Cell Cancer | NCT01684397 | |||
| Anakinra | Metastatic Colorectal Cancer | NCT02090101 | |||
| Cabozantinib | Single agent | Prostate Cancer | NCT03964337 | ||
| Depleting MDSCs | Whole cell | Gemcitabine | Nivolumab | Non-small Cell Lung Cancer | NCT04331626 |
| Modified vaccine expressing p53 | Gynecological Cancers | NCT02275039 | |||
| DC vaccine | Breast Cancer | NCT02479230 | |||
| DC vaccine, imiquimod | Sarcomas | NCT01803152 | |||
| Fluorouracil | Avelumab, Cisplatin, Mitomycin | Bladder Cancer | NCT03617913 | ||
| Aldesleukin, Chemotherapies | Pancreatic Cancer | NCT02620865 | |||
| Capecitabine | Avelumab | Colorectal Cancer | NCT03854799 | ||
| Cisplatin, Rituximab | Head and Neck Squamous Cell Cancer | NCT04361409 | |||
| Cyclophosphamide | iNKT cells, hrIL-2 | Hepatocellular Carcinoma | NCT04011033 | ||
| Pembrolizumab, Vit D, Aspirin | Gynecological Cancer | NCT03192059 | |||
| Promoting MDSC differentiation | TLRs | Poly ICLC | IMA 950 | CNS Tumor | NCT01920191 |
| Imiquimod | DC vaccine | Glioblastoma | NCT01808820 | ||
| Motolimod | Cetuximab, Nivolumab | Head and Neck Squamous Cell Cancer | NCT02124850 | ||
| CpG | Nivolumab | Pancreatic Cancer | NCT04612530 | ||
| RAR/RXR | ATRA | Ipilimumab | Melanoma | NCT02403778 | |
| Pembrolizumab | Melanoma | NCT03200847 | |||
| Vaccine, Cyclophosphamide | Lung Cancer | NCT00601796 | |||
| Paclitaxel, p53-DC vaccines | Small Cell Lung Cancer | NCT00617409 | |||
| STAT3 | Danvatirsen | Durvalumab | Pancreatic, Colorectal, Lung Cancer | NCT02983578 | |
| Durvalumab | Non-Small Cell Lung Cancer | NCT03794544 | |||
| Inhibiting suppressive functions | TGFβ | ABBV-151 | Budigalimab | Advanced Solid Cancer | NCT03821935 |
| Pirfenidone | Atezolizumab | Advanced Non-Small Cell Lung Cancer | NCT04467723 | ||
| NIS793 | PDR001 | Advanced Solid Cancer | NCT02947165 | ||
| SAR439459 | Cemiplimab | Advanced Solid Cancer | NCT04729725 | ||
| Bintrafusp alfa | Single agent | Advanced Solid Cancer | NCT02517398 | ||
| Single agent | Advanced Solid Cancer | NCT02699515 | |||
| Single agent | HPV-associated malignancies | NCT03427411 | |||
| Cheotherapy | Non-Small Cell Lung Cancer | NCT03840915 | |||
| COX2 | Acetylsalicylic acid | Pembrolizumab, Clopidogrel | Head and Neck Cancer | NCT03245489 | |
| Celecoxib | DC vaccine, cisplatin | Ovarian Cancer | NCT02432378 | ||
| Nivolumab, Ipilimumab | Colorectal Cancer | NCT03026140 | |||
| Glucoferon, Rintatolimod | Metastatic Breast Cancer | NCT03599453 | |||
| PDE5 | Tadalafil | Single agent | Head and Neck Cancer | NCT01697800 | |
| Anti-Tumor Mucin-1 Vaccine | Head and Neck Squamous Cell Cancer | NCT02544880 | |||
| HDACs | Entinostat | Ipilimumab, Nivolumab | Breast Cancer | NCT02453620 | |
| Nivolumab | Pancreatic Cancer | NCT03250273 | |||
| Azacitidine, Nivolumab | Non-Small Cell Lung Cancer | NCT01928576 | |||
| NRF2 | Omaveloxolone | Ipilimumab, Nivolumab | Melanoma | NCT02259231 | |
| Single Agent | NSC Lung Cancer, Melanoma | NCT02029729 | |||
| Modulation of MDSC metabolism | CD39/CD73 | TTX-030 | Pembrolizumab, Chemotherapies | Advanced Solid Cancer | NCT04306900 |
| SRF617 | Chemotherapies, Pembrolizumab | Advanced Solid Cancer | NCT04336098 | ||
| Oleclumab | Durvalumab | Muscle Invasive Bladder Cancer | NCT03773666 | ||
| Durvalumab | Lung and Renal Cancer | NCT04262375 | |||
| Durvalumab | Head and Neck, Lung, Pancreatic Cancer | NCT04262388 | |||
| Paclitaxel, Carboplatin, MEDI4736 | Triple Negative Breast Cancer | NCT03616886 | |||
| Durvalumab | Sarcomas | NCT04668300 | |||
| IDO | Indoximod | Docetaxel, Paclitaxel | Metastatic Breast Cancer | NCT01792050 | |
| Epacadostat | Pembrolizumab | Melanoma | NCT02752074 | ||
| BMS-986205 | Nivolumab, Radiation, Temozolomide | Glioblastoma | NCT04047706 | ||
| ARG1 | INCB001158 | Retifanlimab | Advanced Solid Cancer | NCT03910530 | |
| Epacadostat, Pembrolizumab | Advanced Solid Cancer | NCT03361228 | |||
| Pembrolizumab | Advanced Solid Cancer | NCT02903914 | |||
| Chemotherapies | Advanced Solid Cancer | NCT03314935 | |||
| LXRs | RGX-104 | ICIs, Chemotherapies | Advanced Solid Cancer, Lymphoma | NCT02922764 |
6.2.2. Re-Education of MDSCs
- Promoting MDSCs Maturation
- Inhibition of MDSCs Immunosuppressive Functions
- Modulation of MDSCs Metabolism
7. Conclusions and Future Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Sica, A.; Guarneri, V.; Gennari, A. Myelopoiesis, metabolism and therapy: A crucial crossroads in cancer progression. Cell Stress 2019, 3, 284–294. [Google Scholar] [CrossRef] [Green Version]
- Chavakis, T.; Mitroulis, I.; Hajishengallis, G. Hematopoietic progenitor cells as integrative hubs for adaptation to and fine-tuning of inflammation. Nat. Immunol. 2019, 20, 802–811. [Google Scholar] [CrossRef]
- Escamilla-Tilch, M.; Filio-Rodríguez, G.; García-Rocha, R.; Mancilla-Herrera, I.; Mitchison, N.A.; Ruiz-Pacheco, J.A.; Sánchez-García, F.J.; Sandoval-Borrego, D.; Vázquez-Sánchez, E.A. The interplay between pathogen-associated and danger-associated molecular patterns: An inflammatory code in cancer. Immunol. Cell Biol. 2013, 91, 601–610. [Google Scholar] [CrossRef]
- Janeway, C.A. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb. Symp. Quant. Biol. 1989, 54, 1–13. [Google Scholar] [CrossRef]
- Strauss, L.; Guarneri, V.; Gennari, A.; Sica, A. Implications of metabolism-driven myeloid dysfunctions in cancer therapy. Cell. Mol. Immunol. 2021, 18, 829–841. [Google Scholar] [CrossRef]
- Travelli, C.; Consonni, F.M.; Sangaletti, S.; Storto, M.; Morlacchi, S.; Grolla, A.A.; Galli, U.; Tron, G.C.; Portararo, P.; Rimassa, L.; et al. Nicotinamide phosphoribosyltransferase acts as a metabolic gate for mobilization of myeloid-derived suppressor cells. Cancer Res. 2019, 79, 1938–1951. [Google Scholar] [CrossRef] [Green Version]
- Porta, C.; Consonni, F.M.; Morlacchi, S.; Sangaletti, S.; Bleve, A.; Totaro, M.G.; Larghi, P.; Rimoldi, M.; Tripodo, C.; Strauss, L.; et al. Tumor-derived prostaglandin E2 promotes p50 NF-kB-dependent differentiation of monocytic MDSCs. Cancer Res. 2020, 80, 2874–2888. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Weiss, J.M. The promise and peril of targeting cell metabolism for cancer therapy. Cancer Immunol. Immunother. 2020, 69, 255–261. [Google Scholar] [CrossRef]
- Iwasaki, H.; Akashi, K. Myeloid Lineage Commitment from the Hematopoietic Stem Cell. Immunity 2007, 26, 726–740. [Google Scholar] [CrossRef] [Green Version]
- Trumpp, A.; Essers, M.; Wilson, A. Awakening dormant haematopoietic stem cells. Nat. Rev. Immunol. 2010, 10, 201–209. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Hedrick, C.C.; Malanchi, I. Neutrophils in cancer: Heterogeneous and multifaceted. Nat. Rev. Immunol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, D.C.; Shevde, L.A. The tumor microenvironment innately modulates cancer progression. Cancer Res. 2019, 79, 4557–4567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lichterman, J.N.; Reddy, S.M. Mast cells: A new frontier for cancer immunotherapy. Cells 2021, 10, 1270. [Google Scholar] [CrossRef] [PubMed]
- Manz, M.G.; Boettcher, S. Emergency granulopoiesis. Nat. Rev. Immunol. 2014, 14, 302–314. [Google Scholar] [CrossRef]
- Pietras, E.M.; Mirantes-Barbeito, C.; Fong, S.; Loeffler, D.; Kovtonyuk, L.V.; Zhang, S.; Lakshminarasimhan, R.; Chin, C.P.; Techner, J.M.; Will, B.; et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat. Cell Biol. 2016, 18, 607–618. [Google Scholar] [CrossRef]
- Montfort, A.; Colacios, C.; Levade, T.; Andrieu-Abadie, N.; Meyer, N.; Ségui, B. The TNF paradox in cancer progression and immunotherapy. Front. Immunol. 2019, 10, 1818. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Rong, L.; Zhao, X.; Li, X.; Liu, X.; Deng, J.; Wu, H.; Xu, X.; Erben, U.; Wu, P.; et al. TNF signaling drives myeloid-derived suppressor cell accumulation. J. Clin. Investig. 2012, 122, 4094–4104. [Google Scholar] [CrossRef]
- Condamine, T.; Mastio, J.; Gabrilovich, D.I. Transcriptional regulation of myeloid-derived suppressor cells. J. Leukoc. Biol. 2015, 98, 913–922. [Google Scholar] [CrossRef]
- Hirai, H.; Zhang, P.; Dayaram, T.; Hetherington, C.J.; Mizuno, S.I.; Imanishi, J.; Akashi, K.; Tenen, D.G. C/EBPβ is required for “emergency” granulopoiesis. Nat. Immunol. 2006, 7, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Strauss, L.; Sangaletti, S.; Consonni, F.M.; Szebeni, G.; Morlacchi, S.; Totaro, M.G.; Porta, C.; Anselmo, A.; Tartari, S.; Doni, A.; et al. RORC1 Regulates Tumor-Promoting “Emergency” Granulo-Monocytopoiesis. Cancer Cell 2015, 28, 253–269. [Google Scholar] [CrossRef] [Green Version]
- Consonni, F.M.; Bleve, A.; Totaro, M.G.; Storto, M.; Kunderfranco, P.; Termanini, A.; Pasqualini, F.; Alì, C.; Pandolfo, C.; Sgambelluri, F.; et al. Heme catabolism by tumor-associated macrophages controls metastasis formation. Nat. Immunol. 2021, 22, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Richter, R.; Forssmann, W.; Henschler, R. Current Developments in Mobilization of Hematopoietic Stem and Progenitor Cells and Their Interaction with Niches in Bone Marrow. Transfus. Med. Hemotherapy 2017, 44, 151–164. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.W.; Wu, W.C.; Chen, H.T.; Xu, Y.T.; Yang, Y.Y.; Chen, J.; Yu, X.J.; Wang, Z.; Shuang, Z.Y.; Zheng, L. Glutamine Deprivation Promotes the Generation and Mobilization of MDSCs by Enhancing Expression of G-CSF and GM-CSF. Front. Immunol. 2021, 11, 616367. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.L.; Carvalho, T.; Serpa, J.; Torre, C.; Dias, S. Hypercholesterolemia promotes bone marrow cell mobilization by perturbing the SDF-1:CXCR4 axis. Blood 2010, 115, 3886–3894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamiak, M.; Poniewierska-Baran, A.; Borkowska, S.; Schneider, G.; Abdelbaset-Ismail, A.; Suszynska, M.; Abdel-Latif, A.; Kucia, M.; Ratajczak, J.; Ratajczak, M.Z. Evidence that a lipolytic enzyme-hematopoietic-specific phospholipase C-β2-promotes mobilization of hematopoietic stem cells by decreasing their lipid raft-mediated bone marrow retention and increasing the promobilizing effects of granulocytes. Leukemia 2016, 30, 919–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, F.H.; Jogia, T.; Gillespie, E.R.; Blomster, L.V.; Li, X.X.; Nowlan, B.; Williams, G.M.; Jacobson, E.; Osborne, G.W.; Meunier, F.A.; et al. Complement receptor C3aR1 controls neutrophil mobilization following spinal cord injury through physiological antagonism of CXCR2. JCI Insight 2019, 4, e98254. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.; Mithal, D.S.; Park, J.E.; Miller, R.J. Localized CCR2 activation in the bone marrow niche mobilizes monocytes by desensitizing CXCR4. PLoS ONE 2015, 10, e0128387. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sozzani, S.; Vecchi, A.; Locati, M.; Sica, A. Chemokines in the recruitment and shaping of the leukocyte infiltrate of tumors. Semin. Cancer Biol. 2004, 14, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Korbecki, J.; Grochans, S.; Gutowska, I.; Barczak, K.; Baranowska-Bosiacka, I. Cc chemokines in a tumor: A review of pro-cancer and anti-cancer properties of receptors ccr5, ccr6, ccr7, ccr8, ccr9, and ccr10 ligands. Int. J. Mol. Sci. 2020, 21, 7619. [Google Scholar] [CrossRef]
- Li, B.H.; Garstka, M.A.; Li, Z.F. Chemokines and their receptors promoting the recruitment of myeloid-derived suppressor cells into the tumor. Mol. Immunol. 2020, 117, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Engblom, C.; Pfirschke, C.; Pittet, M.J. The role of myeloid cells in cancer therapies. Nat. Rev. Cancer 2016, 16, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Bruni, D.; Angell, H.K.; Galon, J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer 2020, 20, 662–680. [Google Scholar] [CrossRef] [PubMed]
- Kiss, M.; Van Gassen, S.; Movahedi, K.; Saeys, Y.; Laoui, D. Myeloid cell heterogeneity in cancer: Not a single cell alike. Cell. Immunol. 2018, 330, 188–201. [Google Scholar] [CrossRef]
- Geissmann, F.; Gordon, S.; Hume, D.A.; Mowat, A.M.; Randolph, G.J. Unravelling mononuclear phagocyte heterogeneity. Nat. Rev. Immunol. 2010, 10, 453–460. [Google Scholar] [CrossRef] [Green Version]
- Casanova-Acebes, M.; Dalla, E.; Leader, A.M.; LeBerichel, J.; Nikolic, J.; Morales, B.M.; Brown, M.; Chang, C.; Troncoso, L.; Chen, S.T.; et al. Tissue-resident macrophages provide a pro-tumorigenic niche to early NSCLC cells. Nature 2021, 595, 578–584. [Google Scholar] [CrossRef]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Chevrier, S.; Levine, J.H.; Zanotelli, V.R.T.; Silina, K.; Schulz, D.; Bacac, M.; Ries, C.H.; Ailles, L.; Jewett, M.A.S.; Moch, H.; et al. An Immune Atlas of Clear Cell Renal Cell Carcinoma. Cell 2017, 169, 736–749.e18. [Google Scholar] [CrossRef] [Green Version]
- Azizi, E.; Carr, A.J.; Plitas, G.; Cornish, A.E.; Konopacki, C.; Prabhakaran, S.; Nainys, J.; Wu, K.; Kiseliovas, V.; Setty, M.; et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 2018, 174, 1293–1308.e36. [Google Scholar] [CrossRef] [Green Version]
- Donadon, M.; Torzilli, G.; Cortese, N.; Soldani, C.; Di Tommaso, L.; Franceschini, B.; Carriero, R.; Barbagallo, M.; Rigamonti, A.; Anselmo, A.; et al. Macrophage morphology correlates with single-cell diversity and prognosis in colorectal liver metastasis. J. Exp. Med. 2020, 217, e20191847. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Z.; Skrzypczynska, K.M.; Fang, Q.; Zhang, W.; O’Brien, S.A.; He, Y.; Wang, L.; Zhang, Q.; Kim, A.; et al. Single-Cell Analyses Inform Mechanisms of Myeloid-Targeted Therapies in Colon Cancer. Cell 2020, 181, 442–459.e29. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Tang, R.; Xu, J.; Wang, W.; Zhang, B.; Liu, J.; Yu, X.; Shi, S. Applications of single-cell sequencing in cancer research: Progress and perspectives. J. Hematol. Oncol. 2021, 14, 91. [Google Scholar] [CrossRef]
- Maynard, A.; McCoach, C.E.; Rotow, J.K.; Harris, L.; Haderk, F.; Kerr, D.L.; Yu, E.A.; Schenk, E.L.; Tan, W.; Zee, A.; et al. Therapy-Induced Evolution of Human Lung Cancer Revealed by Single-Cell RNA Sequencing. Cell 2020, 182, 1232–1251.e22. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; McKay, D.; Pollard, J.W.; Lewis, C.E. Diverse functions of macrophages in different tumor microenvironments. Cancer Res. 2018, 78, 5492–5503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, L.Q.; Du, W.L.; Cai, M.H.; Yao, J.Y.; Zhao, Y.Y.; Mou, X.Z. The roles of tumor-associated macrophages in tumor angiogenesis and metastasis. Cell. Immunol. 2020, 353, 104119. [Google Scholar] [CrossRef]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef]
- De Palma, M.; Naldini, L. Angiopoietin-2 TIEs up macrophages in tumor angiogenesis. Clin. Cancer Res. 2011, 17, 5226–5232. [Google Scholar] [CrossRef] [Green Version]
- Kato, S.; Okamura, R.; Kumaki, Y.; Ikeda, S.; Nikanjam, M.; Eskander, R.; Goodman, A.; Lee, S.; Glenn, S.T.; Dressman, D.; et al. Expression of TIM3/VISTA checkpoints and the CD68 macrophage-associated marker correlates with anti-PD1/PDL1 resistance: Implications of immunogram heterogeneity. Oncoimmunology 2020, 9, 1708065. [Google Scholar] [CrossRef] [Green Version]
- Ruffell, B.; Affara, N.I.; Coussens, L.M. Differential macrophage programming in the tumor microenvironment. Trends Immunol. 2012, 33, 119–126. [Google Scholar] [CrossRef] [Green Version]
- Bosurgi, L.; Cao, Y.G.; Cabeza-Cabrerizo, M.; Tucci, A.; Hughes, L.D.; Kong, Y.; Weinstein, J.S.; Licona-Limon, P.; Schmid, E.T.; Pelorosso, F.; et al. Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science 2017, 356, 1072–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastò, A.; Consonni, F.M.; Sica, A. Influence of innate immunity on cancer cell stemness. Int. J. Mol. Sci. 2020, 21, 3352. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Forssell, J.; Öberg, Å.; Henriksson, M.L.; Stenling, R.; Jung, A.; Palmqvist, R. High macrophage infiltration along the tumor front correlates with improved survival in colon cancer. Clin. Cancer Res. 2007, 3, 1472–1479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, J.; Rapsomaniki, M.A.; Chevrier, S.; Anzeneder, T.; Langwieder, C.; Dykgers, A.; Rees, M.; Ramaswamy, A.; Muenst, S.; Soysal, S.D.; et al. A Single-Cell Atlas of the Tumor and Immune Ecosystem of Human Breast Cancer. Cell 2019, 177, 1330–1345.e18. [Google Scholar] [CrossRef] [Green Version]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; He, Y.; Luo, N.; Patel, S.J.; Han, Y.; Gao, R.; Modak, M.; Carotta, S.; Haslinger, C.; Kind, D.; et al. Landscape and Dynamics of Single Immune Cells in Hepatocellular Carcinoma. Cell 2019, 179, 829–845.e20. [Google Scholar] [CrossRef]
- Cassetta, L.; Fragkogianni, S.; Sims, A.H.; Swierczak, A.; Forrester, L.M.; Zhang, H.; Soong, D.Y.H.; Cotechini, T.; Anur, P.; Lin, E.Y.; et al. Human Tumor-Associated Macrophage and Monocyte Transcriptional Landscapes Reveal Cancer-Specific Reprogramming, Biomarkers, and Therapeutic Targets. Cancer Cell 2019, 35, 588–602.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, T.; Wang, F.; Deng, H.; Condamine, T.; Kumar, V.; Perego, M.; Kossenkov, A.; Montaner, L.J.; Xu, X.; Xu, W.; et al. Distinct Populations of Immune-Suppressive Macrophages Differentiate from Monocytic Myeloid-Derived Suppressor Cells in Cancer. Cell Rep. 2020, 33, 108571. [Google Scholar] [CrossRef] [PubMed]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.A.; Liao, W.; Sarkar, A.; Kim, M.V.; Bivona, M.R.; Liu, K.; Pamer, E.G.; Li, M.O. The cellular and molecular origin of tumor-associated macrophages. Science 2014, 344, 921–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, R.L.; Klemm, F.; Akkari, L.; Pyonteck, S.M.; Sevenich, L.; Quail, D.F.; Dhara, S.; Simpson, K.; Gardner, E.E.; Iacobuzio-Donahue, C.A.; et al. Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep. 2016, 17, 2445–2459. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Herndon, J.M.; Sojka, D.K.; Kim, K.W.; Knolhoff, B.L.; Zuo, C.; Cullinan, D.R.; Luo, J.; Bearden, A.R.; Lavine, K.J.; et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 2017, 47, 323–338.e6. [Google Scholar] [CrossRef]
- Loyher, P.L.; Hamon, P.; Laviron, M.; Meghraoui-Kheddar, A.; Goncalves, E.; Deng, Z.; Torstensson, S.; Bercovici, N.; De Chanville, C.B.; Combadière, B.; et al. Macrophages of distinct origins contribute to tumor development in the lung. J. Exp. Med. 2018, 215, 2536–2553. [Google Scholar] [CrossRef] [PubMed]
- Etzerodt, A.; Moulin, M.; Doktor, T.K.; Delfini, M.; Mossadegh-Keller, N.; Bajenoff, M.; Sieweke, M.H.; Moestrup, S.K.; Auphan-Anezin, N.; Lawrence, T. Tissue-resident macrophages in omentum promote metastatic spread of ovarian cancer. J. Exp. Med. 2020, 217, e20191869. [Google Scholar] [CrossRef]
- Grover, A.; Sanseviero, E.; Timosenko, E.; Gabrilovich, D.I. Myeloid-Derived Suppressor Cells: A Propitious Road to Clinic. Cancer Discov. 2021, 11, 2693–2706. [Google Scholar] [CrossRef]
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [Green Version]
- Hegde, S.; Leader, A.M.; Merad, M. MDSC: Markers, development, states, and unaddressed complexity. Immunity 2021, 54, 875–884. [Google Scholar] [CrossRef]
- Koh, J.; Kim, Y.; Lee, K.Y.; Hur, J.Y.; Kim, M.S.; Kim, B.; Cho, H.J.; Lee, Y.C.; Bae, Y.H.; Ku, B.M.; et al. MDSC subtypes and CD39 expression on CD8+ T cells predict the efficacy of anti-PD-1 immunotherapy in patients with advanced NSCLC. Eur. J. Immunol. 2020, 50, 1810–1819. [Google Scholar] [CrossRef] [PubMed]
- Joseph, E.L.M.; Laheurte, C.; Jary, M.; Boullerot, L.; Asgarov, K.; Gravelin, E.; Bouard, A.; Rangan, L.; Dosset, M.; Borg, C.; et al. Immunoregulation and clinical implications of ANGPT2/Tie2+ m-MDSC signature in non–small cell lung cancer. Cancer Immunol. Res. 2020, 8, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Sade-Feldman, M.; Kanterman, J.; Klieger, Y.; Ish-Shalom, E.; Olga, M.; Saragovi, A.; Shtainberg, H.; Lotem, M.; Baniyash, M. Clinical significance of circulating CD33+ CD11bHLA-DR myeloid cells in patients with stage IV melanoma treated with ipilimumab. Clin. Cancer Res. 2016, 22, 5661–5672. [Google Scholar] [CrossRef] [Green Version]
- Porta, C.; Marino, A.; Consonni, F.M.; Bleve, A.; Mola, S.; Storto, M.; Riboldi, E.; Sica, A. Metabolic influence on the differentiation of suppressive myeloid cells in cancer. Carcinogenesis 2018, 39, 1095–1104. [Google Scholar] [CrossRef] [Green Version]
- Condamine, T.; Ramachandran, I.; Youn, J.-I.; Gabrilovich, D.I. Regulation of Tumor Metastasis by Myeloid-Derived Suppressor Cells. Annu. Rev. Med. 2015, 66, 97–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consonni, F.M.; Porta, C.; Marino, A.; Pandolfo, C.; Mola, S.; Bleve, A.; Sica, A. Myeloid-derived suppressor cells: Ductile targets in disease. Front. Immunol. 2019, 10, 949. [Google Scholar] [CrossRef]
- Yang, Y.; Li, C.; Liu, T.; Dai, X.; Bazhin, A.V. Myeloid-Derived Suppressor Cells in Tumors: From Mechanisms to Antigen Specificity and Microenvironmental Regulation. Front. Immunol. 2020, 11, 1371. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Redd, P.S.; Lee, J.R.; Savage, N.; Liu, K. The expression profiles and regulation of PD-L1 in tumor-induced myeloid-derived suppressor cells. Oncoimmunology 2016, 5, e1247135. [Google Scholar] [CrossRef] [Green Version]
- Trovato, R.; Fiore, A.; Sartori, S.; Canè, S.; Giugno, R.; Cascione, L.; Paiella, S.; Salvia, R.; De Sanctis, F.; Poffe, O.; et al. Immunosuppression by monocytic myeloid-derived suppressor cells in patients with pancreatic ductal carcinoma is orchestrated by STAT3. J. Immunother. Cancer 2019, 7, 255. [Google Scholar] [CrossRef] [PubMed]
- Ugel, S.; De Sanctis, F.; Mandruzzato, S.; Bronte, V. Tumor-induced myeloid deviation: When myeloid-derived suppressor cells meet tumor-Associated macrophages. J. Clin. Investig. 2015, 125, 3365–3376. [Google Scholar] [CrossRef] [Green Version]
- Marvel, D.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the tumor microenvironment: Expect the unexpected. J. Clin. Investig. 2015, 125, 3356–3364. [Google Scholar] [CrossRef] [PubMed]
- Condamine, T.; Dominguez, G.A.; Youn, J.I.; Kossenkov, A.V.; Mony, S.; Alicea-Torres, K.; Tcyganov, E.; Hashimoto, A.; Nefedova, Y.; Lin, C.; et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci. Immunol. 2016, 1, aaf8943. [Google Scholar] [CrossRef] [Green Version]
- Alshetaiwi, H.; Pervolarakis, N.; McIntyre, L.L.; Ma, D.; Nguyen, Q.; Rath, J.A.; Nee, K.; Hernandez, G.; Evans, K.; Torosian, L.; et al. Defining the emergence of myeloid-derived suppressor cells in breast cancer using single-cell transcriptomics. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef]
- Song, Q.; Hawkins, G.A.; Wudel, L.; Chou, P.C.; Forbes, E.; Pullikuth, A.K.; Liu, L.; Jin, G.; Craddock, L.; Topaloglu, U.; et al. Dissecting intratumoral myeloid cell plasticity by single cell RNA-seq. Cancer Med. 2019, 8, 3072–3085. [Google Scholar] [CrossRef] [Green Version]
- Masucci, M.T.; Minopoli, M.; Carriero, M.V. Tumor Associated Neutrophils. Their Role in Tumorigenesis, Metastasis, Prognosis and Therapy. Front. Oncol. 2019, 9, 1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaillon, S.; Ponzetta, A.; Di Mitri, D.; Santoni, A.; Bonecchi, R.; Mantovani, A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat. Rev. Cancer 2020, 20, 485–503. [Google Scholar] [CrossRef] [PubMed]
- Coffelt, S.B.; Wellenstein, M.D.; De Visser, K.E. Neutrophils in cancer: Neutral no more. Nat. Rev. Cancer 2016, 16, 431–446. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.P.; Eggert, T.; Araujo, D.J.; Vijayanand, P.; Ottensmeier, C.H.; Hedrick, C.C. CyTOF mass cytometry reveals phenotypically distinct human blood neutrophil populations differentially correlated with melanoma stage. J. Immunother. Cancer 2020, 8, e000473. [Google Scholar] [CrossRef]
- Engblom, C.; Pfirschke, C.; Zilionis, R.; Da Silva Martins, J.; Bos, S.A.; Courties, G.; Rickelt, S.; Severe, N.; Baryawno, N.; Faget, J.; et al. Osteoblasts remotely supply lung tumors with cancer-promoting SiglecFhigh neutrophils. Science 2017, 358, eaal5081. [Google Scholar] [CrossRef] [Green Version]
- Zilionis, R.; Engblom, C.; Pfirschke, C.; Savova, V.; Zemmour, D.; Saatcioglu, H.D.; Krishnan, I.; Maroni, G.; Meyerovitz, C.V.; Kerwin, C.M.; et al. Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity 2019, 50, 1317–1334.e10. [Google Scholar] [CrossRef] [PubMed]
- Lavin, Y.; Kobayashi, S.; Leader, A.; Amir, E.a.D.; Elefant, N.; Bigenwald, C.; Remark, R.; Sweeney, R.; Becker, C.D.; Levine, J.H.; et al. Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses. Cell 2017, 169, 750–765.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of Tumor-Associated Neutrophil Phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reis, E.S.; Mastellos, D.C.; Ricklin, D.; Mantovani, A.; Lambris, J.D. Complement in cancer: Untangling an intricate relationship. Nat. Rev. Immunol. 2018, 18, 5–18. [Google Scholar] [CrossRef]
- Singhal, S.; Bhojnagarwala, P.S.; O’Brien, S.; Moon, E.K.; Garfall, A.L.; Rao, A.S.; Quatromoni, J.G.; Stephen, T.L.; Litzky, L.; Deshpande, C.; et al. Origin and Role of a Subset of Tumor-Associated Neutrophils with Antigen-Presenting Cell Features in Early Stage Human Lung Cancer. Cancer Cell 2016, 30, 120–135. [Google Scholar] [CrossRef] [Green Version]
- Wculek, S.K.; Malanchi, I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature 2015, 528, 413–417. [Google Scholar] [CrossRef] [Green Version]
- He, G.; Zhang, H.; Zhou, J.; Wang, B.; Chen, Y.; Kong, Y.; Xie, X.; Wang, X.; Fei, R.; Wei, L.; et al. Peritumoural neutrophils negatively regulate adaptive immunity via the PD-L1/PD-1 signalling pathway in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2015, 34, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldmeyer, N.; Wabnitz, G.; Leicht, S.; Luckner-minden, C.; Schiller, M.; Franz, T.; Conradi, R.; Kropf, P.; Müller, I.; Ho, A.D.; et al. Arginine deficiency leads to impaired cofilin dephosphorylation in activated human T lymphocytes. Int. Immunol. 2012, 24, 303–313. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Wysocki, R.W.; Amoozgar, Z.; Maiorino, L.; Fein, M.R.; Jorns, J.; Schott, A.F.; Kinugasa-Katayama, Y.; Lee, Y.; Won, N.H.; et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci. Transl. Med. 2016, 8, 361ra138. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.J.; Ko, S.Y.; Mohamed, M.S.; Kenny, H.A.; Lengyel, E.; Naora, H. Neutrophils facilitate ovarian cancer premetastatic niche formation in the omentum. J. Exp. Med. 2019, 216, 176–194. [Google Scholar] [CrossRef]
- Tohme, S.; Yazdani, H.O.; Al-Khafaji, A.B.; Chidi, A.P.; Loughran, P.; Mowen, K.; Wang, Y.; Simmons, R.L.; Huang, H.; Tsung, A. Neutrophil extracellular traps promote the development and progression of liver metastases after surgical stress. Cancer Res. 2016, 76, 1367–1380. [Google Scholar] [CrossRef] [Green Version]
- Albrengues, J.; Shields, M.A.; Ng, D.; Park, C.G.; Ambrico, A.; Poindexter, M.E.; Upadhyay, P.; Uyeminami, D.L.; Pommier, A.; Küttner, V.; et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018, 361, eaao4227. [Google Scholar] [CrossRef] [Green Version]
- Teijeira, Á.; Garasa, S.; Gato, M.; Alfaro, C.; Migueliz, I.; Cirella, A.; de Andrea, C.; Ochoa, M.C.; Otano, I.; Etxeberria, I.; et al. CXCR1 and CXCR2 Chemokine Receptor Agonists Produced by Tumors Induce Neutrophil Extracellular Traps that Interfere with Immune Cytotoxicity. Immunity 2020, 52, 856–871.e8. [Google Scholar] [CrossRef] [PubMed]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014, 25, 846–859. [Google Scholar] [CrossRef] [Green Version]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [Green Version]
- Lesokhin, A.M.; Hohl, T.M.; Kitano, S.; Cortez, C.; Hirschhorn-Cymerman, D.; Avogadri, F.; Rizzuto, G.A.; Lazarus, J.J.; Pamer, E.G.; Houghton, A.N.; et al. Monocytic CCR2 + myeloid-derived suppressor cells promote immune escape by limiting activated CD8 T-cell infiltration into the tumor microenvironment. Cancer Res. 2012, 72, 876–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonapace, L.; Coissieux, M.M.; Wyckoff, J.; Mertz, K.D.; Varga, Z.; Junt, T.; Bentires-Alj, M. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature 2014, 515, 130–133. [Google Scholar] [CrossRef]
- Litmanovich, A.; Khazim, K.; Cohen, I. The Role of Interleukin-1 in the Pathogenesis of Cancer and its Potential as a Therapeutic Target in Clinical Practice. Oncol. Ther. 2018, 6, 109–127. [Google Scholar] [CrossRef] [Green Version]
- Germano, G.; Frapolli, R.; Belgiovine, C.; Anselmo, A.; Pesce, S.; Liguori, M.; Erba, E.; Uboldi, S.; Zucchetti, M.; Pasqualini, F.; et al. Role of Macrophage Targeting in the Antitumor Activity of Trabectedin. Cancer Cell 2013, 23, 249–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etzerodt, A.; Tsalkitzi, K.; Maniecki, M.; Damsky, W.; Delfini, M.; Baudoin, E.; Moulin, M.; Bosenberg, M.; Graversen, J.H.; Auphan-Anezin, N.; et al. Specific targeting of CD163+ TAMs mobilizes inflammatory monocytes and promotes T cell-mediated tumor regression. J. Exp. Med. 2019, 216, 2394–2411. [Google Scholar] [CrossRef] [PubMed]
- Mullins, S.R.; Vasilakos, J.P.; Deschler, K.; Grigsby, I.; Gillis, P.; John, J.; Elder, M.J.; Swales, J.; Timosenko, E.; Cooper, Z.; et al. Intratumoral immunotherapy with TLR7/8 agonist MEDI9197 modulates the tumor microenvironment leading to enhanced activity when combined with other immunotherapies. J. Immunother. Cancer 2019, 7, 244. [Google Scholar] [CrossRef]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiehagen, K.R.; Girgis, N.M.; Yamada, D.H.; Smith, A.A.; Chan, S.R.; Grewal, I.S.; Quigley, M.; Verona, R.I. Combination of CD40 agonism and CSF-1R blockade reconditions tumor-associated macrophages and drives potent antitumor immunity. Cancer Immunol. Res. 2017, 5, 1109–1121. [Google Scholar] [CrossRef] [Green Version]
- Schmid, M.C.; Avraamides, C.J.; Dippold, H.C.; Franco, I.; Foubert, P.; Ellies, L.G.; Acevedo, L.M.; Manglicmot, J.R.E.; Song, X.; Wrasidlo, W.; et al. Receptor tyrosine kinases and TLR/IL1Rs Unexpectedly activate myeloid cell PI3Kγ, A single convergent point promoting tumor inflammation and progression. Cancer Cell 2011, 19, 715–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kγ 3 is a molecular switch that controls immune suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Henau, O.; Rausch, M.; Winkler, D.; Campesato, L.F.; Liu, C.; Cymerman, D.H.; Budhu, S.; Ghosh, A.; Pink, M.; Tchaicha, J.; et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature 2016, 539, 443–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medler, T.R.; Murugan, D.; Horton, W.; Kumar, S.; Cotechini, T.; Forsyth, A.M.; Leyshock, P.; Leitenberger, J.J.; Kulesz-Martin, M.; Margolin, A.A.; et al. Complement C5a Fosters Squamous Carcinogenesis and Limits T Cell Response to Chemotherapy. Cancer Cell 2018, 34, 561–578.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zha, H.; Han, X.; Zhu, Y.; Yang, F.; Li, Y.; Li, Q.; Guo, B.; Zhu, B. Blocking C5aR signaling promotes the anti-tumor efficacy of PD-1/PD-L1 blockade. Oncoimmunology 2017, 6, e1349587. [Google Scholar] [CrossRef] [PubMed]
- Magrini, E.; Di Marco, S.; Mapelli, S.N.; Perucchini, C.; Pasqualini, F.; Donato, A.; Guevara Lopez, M.d.l.L.; Carriero, R.; Ponzetta, A.; Colombo, P.; et al. Complement activation promoted by the lectin pathway mediates C3aR-dependent sarcoma progression and immunosuppression. Nat. Cancer 2021, 2, 218–232. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [Green Version]
- Bleve, A.; Durante, B.; Sica, A.; Consonni, F.M. Lipid metabolism and cancer immunotherapy: Immunosuppressive myeloid cells at the crossroad. Int. J. Mol. Sci. 2020, 21, 5845. [Google Scholar] [CrossRef]
- Semba, H.; Takeda, N.; Isagawa, T.; Sugiura, Y.; Honda, K.; Wake, M.; Miyazawa, H.; Yamaguchi, Y.; Miura, M.; Jenkins, D.M.R.; et al. HIF-1α-PDK1 axis-induced active glycolysis plays an essential role in macrophage migratory capacity. Nat. Commun. 2016, 7, 11635. [Google Scholar] [CrossRef] [Green Version]
- Arts, R.J.W.; Plantinga, T.S.; Tuit, S.; Ulas, T.; Heinhuis, B.; Tesselaar, M.; Sloot, Y.; Adema, G.J.; Joosten, L.A.B.; Smit, J.W.A.; et al. Transcriptional and metabolic reprogramming induce an inflammatory phenotype in non-medullary thyroid carcinoma-induced macrophages. Oncoimmunology 2016, 5, e1229725. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.S.; Wang, H.; Li, X.; Chao, T.; Teav, T.; Christen, S.; DI Conza, G.; Cheng, W.C.; Chou, C.H.; Vavakova, M.; et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 2017, 18, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, E.M.; Menga, A.; Martín-Pérez, R.; Quinto, A.; Riera-Domingo, C.; De Tullio, G.; Hooper, D.C.; Lamers, W.H.; Ghesquière, B.; McVicar, D.W.; et al. Pharmacologic or Genetic Targeting of Glutamine Synthetase Skews Macrophages toward an M1-like Phenotype and Inhibits Tumor Metastasis. Cell Rep. 2017, 20, 1654–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogg, S.J.; Beavis, P.A.; Dawson, M.A.; Johnstone, R.W. Targeting the epigenetic regulation of antitumour immunity. Nat. Rev. Drug Discov. 2020, 19, 776–800. [Google Scholar] [CrossRef]
- Guerriero, J.L.; Sotayo, A.; Ponichtera, H.E.; Castrillon, J.A.; Pourzia, A.L.; Schad, S.; Johnson, S.F.; Carrasco, R.D.; Lazo, S.; Bronson, R.T.; et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature 2017, 543, 428–432. [Google Scholar] [CrossRef]
- Mola, S.; Pinton, G.; Erreni, M.; Corazzari, M.; Andrea, M.D.; Grolla, A.A.; Martini, V.; Moro, L.; Porta, C. Inhibition of the histone methyltransferase ezh2 enhances protumor monocyte recruitment in human mesothelioma spheroids. Int. J. Mol. Sci. 2021, 22, 4391. [Google Scholar] [CrossRef]
- Hamaidia, M.; Gazon, H.; Hoyos, C.; Hoffmann, G.B.; Louis, R.; Duysinx, B.; Willems, L. Inhibition of EZH2 methyltransferase decreases immunoediting of mesothelioma cells by autologous macrophages through a PD-1-dependent mechanism. JCI Insight 2019, 4, e128474. [Google Scholar] [CrossRef]
- Morel, K.L.; Sheahan, A.V.; Burkhart, D.L.; Baca, S.C.; Boufaied, N.; Liu, Y.; Qiu, X.; Cañadas, I.; Roehle, K.; Heckler, M.; et al. EZH2 inhibition activates a dsRNA–STING–interferon stress axis that potentiates response to PD-1 checkpoint blockade in prostate cancer. Nat. Cancer 2021, 2, 444–456. [Google Scholar] [CrossRef]
- Feng, M.; Jiang, W.; Kim, B.Y.S.; Zhang, C.C.; Fu, Y.X.; Weissman, I.L. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat. Rev. Cancer 2019, 19, 568–586. [Google Scholar] [CrossRef]
- Willingham, S.B.; Volkmer, J.P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Advani, R.; Flinn, I.; Popplewell, L.; Forero, A.; Bartlett, N.L.; Ghosh, N.; Kline, J.; Roschewski, M.; LaCasce, A.; Collins, G.P.; et al. CD47 Blockade by Hu5F9-G4 and Rituximab in Non-Hodgkin’s Lymphoma. New Engl. J. Med. 2018, 379, 1711–1721. [Google Scholar] [CrossRef]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Strauss, L.; Mahmoud, M.A.A.; Weaver, J.D.; Tijaro-Ovalle, N.M.; Christofides, A.; Wang, Q.; Pal, R.; Yuan, M.; Asara, J.; Patsoukis, N.; et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef]
- Liu, X.; Liu, L.; Ren, Z.; Yang, K.; Xu, H.; Luan, Y.; Fu, K.; Guo, J.; Peng, H.; Zhu, M.; et al. Dual Targeting of Innate and Adaptive Checkpoints on Tumor Cells Limits Immune Evasion. Cell Rep. 2018, 24, 2101–2111. [Google Scholar] [CrossRef] [Green Version]
- Weiskopf, K.; Jahchan, N.S.; Schnorr, P.J.; Cristea, S.; Ring, A.M.; Maute, R.L.; Volkmer, A.K.; Volkmer, J.P.; Liu, J.; Lim, J.S.; et al. CD47-blocking immunotherapies stimulate macrophage-mediated destruction of small-cell lung cancer. J. Clin. Investig. 2016, 126, 2610–2620. [Google Scholar] [CrossRef] [PubMed]
- Leijonhufvud, C.; Reger, R.; Segerberg, F.; Theorell, J.; Schlums, H.; Bryceson, Y.T.; Childs, R.W.; Carlsten, M. LIR-1 educates expanded human NK cells and defines a unique antitumor NK cell subset with potent antibody-dependent cellular cytotoxicity. Clin. Transl. Immunol. 2021, 10, e1346. [Google Scholar] [CrossRef] [PubMed]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Deng, L.; Hou, Y.; Meng, X.; Huang, X.; Rao, E.; Zheng, W.; Mauceri, H.; Mack, M.; Xu, M.; et al. Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat. Commun. 2017, 8, 1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karin, N.; Razon, H. The role of CCR5 in directing the mobilization and biological function of CD11b+Gr1+Ly6Clow polymorphonuclear myeloid cells in cancer. Cancer Immunol. Immunother. 2018, 67, 1949–1953. [Google Scholar] [CrossRef] [PubMed]
- Blattner, C.; Fleming, V.; Weber, R.; Himmelhan, B.; Altevogt, P.; Gebhardt, C.; Schulze, T.J.; Razon, H.; Hawila, E.; Wildbaum, G.; et al. CCR5+ myeloid-derived suppressor cells are enriched and activated in melanoma lesions. Cancer Res. 2018, 78, 157–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greene, S.; Robbins, Y.; Mydlarz, W.K.; Huynh, A.P.; Schmitt, N.C.; Friedman, J.; Horn, L.A.; Palena, C.; Schlom, J.; Maeda, D.Y.; et al. Inhibition of MDSC trafficking with SX-682, a CXCR1/2 inhibitor, enhances NK-cell immunotherapy in head and neck cancer models. Clin. Cancer Res. 2020, 26, 1420–1431. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Sun, H.; Wei, J.; Cen, B.; DuBois, R.N. CXCL1 is critical for premetastatic niche formation and metastasis in colorectal cancer. Cancer Res. 2017, 77, 3655–3665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.; Wang, J.; Wang, L.; Wen, J.; Guo, Y.; Qiao, W.; Zhou, J.; Xu, G.; Zhi, F. Phosphodiesterase-5 inhibition suppresses colonic inflammation-induced tumorigenesis via blocking the recruitment of MDSC. Am. J. Cancer Res. 2017, 7, 41–52. [Google Scholar]
- Fleming, V.; Hu, X.; Weber, R.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Targeting myeloid-derived suppressor cells to bypass tumor-induced immunosuppression. Front. Immunol. 2018, 9, 398. [Google Scholar] [CrossRef] [PubMed]
- Condamine, T.; Kumar, V.; Ramachandran, I.R.; Youn, J.I.; Celis, E.; Finnberg, N.; El-Deiry, W.S.; Winograd, R.; Vonderheide, R.H.; English, N.R.; et al. ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis. J. Clin. Investig. 2014, 124, 2626–2639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavazoie, M.F.; Pollack, I.; Tanqueco, R.; Ostendorf, B.N.; Reis, B.S.; Gonsalves, F.C.; Kurth, I.; Andreu-Agullo, C.; Derbyshire, M.L.; Posada, J.; et al. LXR/ApoE Activation Restricts Innate Immune Suppression in Cancer. Cell 2018, 172, 825–840.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelenay, S.; Van Der Veen, A.G.; Böttcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A.; et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, P.C.; Hernandez, C.P.; Quiceno, D.; Dubinett, S.M.; Zabaleta, J.; Ochoa, J.B.; Gilbert, J.; Ochoa, A.C. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J. Exp. Med. 2005, 202, 931–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Condamine, T.; Gabrilovich, D.I. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011, 32, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Veglia, F.; Tyurin, V.A.; Blasi, M.; De Leo, A.; Kossenkov, A.V.; Donthireddy, L.; To, T.K.J.; Schug, Z.; Basu, S.; Wang, F.; et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature 2019, 569, 73–78. [Google Scholar] [CrossRef]
- Kusmartsev, S.; Cheng, F.; Yu, B.; Nefedova, Y.; Sotomayor, E.; Lush, R.; Gabrilovich, D. All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res. 2003, 63, 4441–4449. [Google Scholar] [PubMed]
- Iclozan, C.; Antonia, S.; Chiappori, A.; Chen, D.T.; Gabrilovich, D. Therapeutic regulation of myeloid-derived suppressor cells and immune response to cancer vaccine in patients with extensive stage small cell lung cancer. Cancer Immunol. Immunother. 2013, 62, 909–918. [Google Scholar] [CrossRef] [Green Version]
- Bauer, R.; Udonta, F.; Wroblewski, M.; Ben-Batalla, I.; Santos, I.M.; Taverna, F.; Kuhlencord, M.; Gensch, V.; Päsler, S.; Vinckier, S.; et al. Blockade of myeloid-derived suppressor cell expansion with all-trans retinoic acid increases the efficacy of antiangiogenic therapy. Cancer Res. 2018, 78, 3220–3232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Xie, Y.; Xiong, Y.; Liu, S.; Qiu, C.; Zhu, Z.; Mao, H.; Yu, M.; Wang, X. TLR 7/8 agonist reverses oxaliplatin resistance in colorectal cancer via directing the myeloid-derived suppressor cells to tumoricidal M1-macrophages. Cancer Lett. 2020, 469, 173–185. [Google Scholar] [CrossRef]
- Panni, R.Z.; Sanford, D.E.; Belt, B.A.; Mitchem, J.B.; Worley, L.A.; Goetz, B.D.; Mukherjee, P.; Wang-Gillam, A.; Link, D.C.; Denardo, D.G.; et al. Tumor-induced STAT3 activation in monocytic myeloid-derived suppressor cells enhances stemness and mesenchymal properties in human pancreatic cancer. Cancer Immunol. Immunother. 2014, 63, 513–528. [Google Scholar] [CrossRef] [Green Version]
- Kortylewski, M.; Moreira, D. Myeloid cells as a target for oligonucleotide therapeutics: Turning obstacles into opportunities. Cancer Immunol. Immunother. 2017, 66, 979–988. [Google Scholar] [CrossRef]
- Al-Khami, A.A.; Zheng, L.; Del Valle, L.; Hossain, F.; Wyczechowska, D.; Zabaleta, J.; Sanchez, M.D.; Dean, M.J.; Rodriguez, P.C.; Ochoa, A.C. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology 2017, 6, e1344804. [Google Scholar] [CrossRef]
- Huang, S.; Wang, Z.; Zhou, J.; Huang, J.; Zhou, L.; Luo, J.; Wan, Y.Y.; Long, H.; Zhu, B. EZH2 inhibitor GSK126 suppresses antitumor immunity by driving production of myeloid-derived suppressor cells. Cancer Res. 2019, 79, 2009–2020. [Google Scholar] [CrossRef]
- Orillion, A.; Hashimoto, A.; Damayanti, N.; Shen, L.; Adelaiye-Ogala, R.; Arisa, S.; Chintala, S.; Ordentlich, P.; Kao, C.; Elzey, B.; et al. Entinostat neutralizes myeloid-derived suppressor cells and enhances the antitumor effect of PD-1 inhibition in murine models of lung and renal cell carcinoma. Clin. Cancer Res. 2017, 23, 5187–5201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Jaillon, S.; Garlanda, C.; Allavena, P. Tumor-associated myeloid cells: Diversity and therapeutic targeting. Cell. Mol. Immunol. 2021, 18, 566–578. [Google Scholar] [CrossRef]
- Edin, S.; Wikberg, M.L.; Oldenborg, P.A.; Palmqvist, R. Macrophages: Good guys in colorectal cancer. Oncoimmunology 2013, 2, e23038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Arabey, A.A.; Denizli, M.; Kanlikilicer, P.; Bayraktar, R.; Ivan, C.; Rashed, M.; Kabil, N.; Ozpolat, B.; Calin, G.A.; Salama, S.A.; et al. GATA3 as a master regulator for interactions of tumor-associated macrophages with high-grade serous ovarian carcinoma. Cell. Signal. 2020, 68, 109539. [Google Scholar] [CrossRef] [PubMed]
- Cannarile, M.A.; Weisser, M.; Jacob, W.; Jegg, A.M.; Ries, C.H.; Rüttinger, D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 2017, 5, 53. [Google Scholar] [CrossRef]
- Lewis, J.H.; Gelderblom, H.; van de Sande, M.; Stacchiotti, S.; Healey, J.H.; Tap, W.D.; Wagner, A.J.; Pousa, A.L.; Druta, M.; Lin, C.C.; et al. Pexidartinib Long-Term Hepatic Safety Profile in Patients with Tenosynovial Giant Cell Tumors. Oncologist 2021, 26, e863–e873. [Google Scholar] [CrossRef] [PubMed]
- Benner, B.; Good, L.; Quiroga, D.; Schultz, T.E.; Kassem, M.; Carson, W.E.; Cherian, M.A.; Sardesai, S.; Wesolowski, R. Pexidartinib, a novel small molecule csf-1r inhibitor in use for tenosynovial giant cell tumor: A systematic review of pre-clinical and clinical development. Drug Des. Devel. Ther. 2020, 14, 1693–1704. [Google Scholar] [CrossRef]
- Kuemmel, S.; Campone, M.; Loirat, D.; López López, R.; Beck, J.T.; De Laurentiis, M.; Im, S.-A.; Kim, S.-B.; Kwong, A.; Steger, G.G.; et al. A Randomized Phase II Study of Anti-CSF-1 Monoclonal Antibody Lacnotuzumab (MCS110) Combined with Gemcitabine and Carboplatin in Advanced Triple Negative Breast Cancer. Clin. Cancer Res. 2021, 28, 106–115. [Google Scholar] [CrossRef]
- Fei, L.; Ren, X.; Yu, H.; Zhan, Y. Targeting the CCL2/CCR2 Axis in Cancer Immunotherapy: One Stone, Three Birds? Front. Immunol. 2021, 12, 771210. [Google Scholar] [CrossRef] [PubMed]
- Pienta, K.J.; Machiels, J.P.; Schrijvers, D.; Alekseev, B.; Shkolnik, M.; Crabb, S.J.; Li, S.; Seetharam, S.; Puchalski, T.A.; Takimoto, C.; et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Investig. New Drugs 2013, 31, 760–768. [Google Scholar] [CrossRef]
- Nywening, T.M.; Wang-Gillam, A.; Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Cusworth, B.M.; Toriola, A.T.; Nieman, R.K.; Worley, L.A.; Yano, M.; et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: A single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016, 17, 651–662. [Google Scholar] [CrossRef] [Green Version]
- Aldinucci, D.; Borghese, C.; Casagrande, N. The ccl5/ccr5 axis in cancer progression. Cancers 2020, 12, 1765. [Google Scholar] [CrossRef]
- Scala, S. Molecular pathways: Targeting the CXCR4-CXCL12 Axis-Untapped potential in the tumor microenvironment. Clin. Cancer Res. 2015, 21, 4278–4285. [Google Scholar] [CrossRef] [Green Version]
- Argyle, D.; Kitamura, T. Targeting macrophage-recruiting chemokines as a novel therapeutic strategy to prevent the progression of solid tumors. Front. Immunol. 2018, 9, 2629. [Google Scholar] [CrossRef]
- Yusen, W.; Xia, W.; Shengjun, Y.; Shaohui, Z.; Hongzhen, Z. The expression and significance of tumor associated macrophages and CXCR4 in non-small cell lung cancer. J. BUON Off. J. Balk. Union Oncol. 2018, 23, 398–402. [Google Scholar]
- Ji, N.; Mukherjee, N.; Morales, E.E.; Tomasini, M.E.; Hurez, V.; Curiel, T.J.; Abate, G.; Hoft, D.F.; Zhao, X.R.; Gelfond, J.; et al. Percutaneous BCG enhances innate effector antitumor cytotoxicity during treatment of bladder cancer: A translational clinical trial. Oncoimmunology 2019, 8, e1614857. [Google Scholar] [CrossRef] [Green Version]
- Adams, S.; Kozhaya, L.; Martiniuk, F.; Meng, T.C.; Chiriboga, L.; Liebes, L.; Hochman, T.; Shuman, N.; Axelrod, D.; Speyer, J.; et al. Topical TLR7 agonist imiquimod can induce immune-mediated rejection of skin metastases in patients with breast cancer. Clin. Cancer Res. 2012, 18, 6748–6757. [Google Scholar] [CrossRef] [Green Version]
- Chi, H.; Li, C.; Zhao, F.S.; Zhang, L.; Ng, T.B.; Jin, G.; Sha, O. Anti-tumor activity of Toll-like receptor 7 agonists. Front. Pharmacol. 2017, 8, 304. [Google Scholar] [CrossRef] [PubMed]
- Ferris, R.L.; Saba, N.F.; Gitlitz, B.J.; Haddad, R.; Sukari, A.; Neupane, P.; Morris, J.C.; Misiukiewicz, K.; Bauman, J.E.; Fenton, M.; et al. Effect of adding motolimod to standard combination chemotherapy and cetuximab treatment of patients with squamous cell carcinoma of the head and neck the ACTIVE8 randomized clinical trial. JAMA Oncol. 2018, 4, 1583–1588. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.A.; Conkling, P.; Richards, D.A.; Nemunaitis, J.J.; Boyd, T.E.; Mita, A.C.; De La Bourdonnaye, G.; Wages, D.; Bexon, A.S. Antitumor activity and safety of combination therapy with the Toll-like receptor 9 agonist IMO-2055, erlotinib, and bevacizumab in advanced or metastatic non-small cell lung cancer patients who have progressed following chemotherapy. Cancer Immunol. Immunother. 2014, 63, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Vonderheide, R.H. CD40 Agonist Antibodies in Cancer Immunotherapy. Annu. Rev. Med. 2020, 71, 47–58. [Google Scholar] [CrossRef] [Green Version]
- O’Hara, M.H.; O’Reilly, E.M.; Varadhachary, G.; Wolff, R.A.; Wainberg, Z.A.; Ko, A.H.; Fisher, G.; Rahma, O.; Lyman, J.P.; Cabanski, C.R.; et al. CD40 agonistic monoclonal antibody APX005M (sotigalimab) and chemotherapy, with or without nivolumab, for the treatment of metastatic pancreatic adenocarcinoma: An open-label, multicentre, phase 1b study. Lancet Oncol. 2021, 22, 118–131. [Google Scholar] [CrossRef]
- Zhu, J.; Li, K.; Yu, L.; Chen, Y.; Cai, Y.; Jin, J.; Hou, T. Targeting phosphatidylinositol 3-kinase gamma (PI3Kγ): Discovery and development of its selective inhibitors. Med. Res. Rev. 2021, 41, 1599–1621. [Google Scholar] [CrossRef]
- Logtenberg, M.E.W.; Scheeren, F.A.; Schumacher, T.N. The CD47-SIRPα Immune Checkpoint. Immunity 2020, 52, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.P.; Takimoto, C.H.; Feng, D.D.; McKenna, K.; Gip, P.; Liu, J.; Volkmer, J.P.; Weissman, I.L.; Majeti, R. Therapeutic Targeting of the Macrophage Immune Checkpoint CD47 in Myeloid Malignancies. Front. Oncol. 2020, 9, 1380. [Google Scholar] [CrossRef]
- Feins, S.; Kong, W.; Williams, E.F.; Milone, M.C.; Fraietta, J.A. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am. J. Hematol. 2020, 94, S3–S9. [Google Scholar] [CrossRef] [Green Version]
- Carisma Therapeutics. Carisma Drives CAR-M Engineered Macrophage Cancer Therapy Forward. Available online: https://www.nature.com/articles/d43747-020-01096-y (accessed on 28 November 2021).
- Ostrand-Rosenberg, S.; Fenselau, C. Myeloid-Derived Suppressor Cells: Immune-Suppressive Cells That Impair Antitumor Immunity and Are Sculpted by Their Environment. J. Immunol. 2018, 200, 422–431. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Clavijo, P.E.; Robbins, Y.; Patel, P.; Friedman, J.; Greene, S.; Das, R.; Silvin, C.; Van Waes, C.; Horn, L.A.; et al. Inhibiting myeloid-derived suppressor cell trafficking enhances T cell immunotherapy. JCI Insight 2019, 4, e126853. [Google Scholar] [CrossRef] [Green Version]
- Elliott, L.A.; Doherty, G.A.; Sheahan, K.; Ryan, E.J. Human tumor-infiltrating myeloid cells: Phenotypic and functional diversity. Front. Immunol. 2017, 8, 86. [Google Scholar] [CrossRef]
- Alfaro, C.; Sanmamed, M.F.; Rodríguez-Ruiz, M.E.; Teijeira, Á.; Oñate, C.; González, Á.; Ponz, M.; Schalper, K.A.; Pérez-Gracia, J.L.; Melero, I. Interleukin-8 in cancer pathogenesis, treatment and follow-up. Cancer Treat. Rev. 2017, 60, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Obermajer, N.; Muthuswamy, R.; Odunsi, K.; Edwards, R.P.; Kalinski, P. PGE 2-induced CXCL 12 production and CXCR4 expression controls the accumulation of human MDSCs in ovarian cancer environment. Cancer Res. 2011, 71, 7463–7470. [Google Scholar] [CrossRef] [Green Version]
- Rivera, L.B.; Bergers, G. Intertwined regulation of angiogenesis and immunity by myeloid cells. Trends Immunol. 2015, 36, 240–249. [Google Scholar] [CrossRef] [Green Version]
- Koinis, F.; Vetsika, E.K.; Aggouraki, D.; Skalidaki, E.; Koutoulaki, A.; Gkioulmpasani, M.; Georgoulias, V.; Kotsakis, A. Effect of first-line treatment on myeloid-derived suppressor cells’ subpopulations in the peripheral blood of patients with non-small cell lung cancer. J. Thorac. Oncol. 2016, 11, 1263–1272. [Google Scholar] [CrossRef] [Green Version]
- Limagne, E.; Euvrard, R.; Thibaudin, M.; Rébé, C.; Derangère, V.; Chevriaux, A.; Boidot, R.; Végran, F.; Bonnefoy, N.; Vincent, J.; et al. Accumulation of MDSC and Th17 cells in patients with metastatic colorectal cancer predicts the efficacy of a FOLFOX-bevacizumab drug treatment regimen. Cancer Res. 2016, 76, 5241–5252. [Google Scholar] [CrossRef] [Green Version]
- Zitvogel, L.; Apetoh, L.; Ghiringhelli, F.; Kroemer, G. Immunological aspects of cancer chemotherapy. Nat. Rev. Immunol. 2008, 8, 59–73. [Google Scholar] [CrossRef]
- Eriksson, E.; Wenthe, J.; Irenaeus, S.; Loskog, A.; Ullenhag, G. Gemcitabine reduces MDSCs, tregs and TGFβ-1 while restoring the teff/treg ratio in patients with pancreatic cancer. J. Transl. Med. 2016, 14, 282. [Google Scholar] [CrossRef] [PubMed]
- Peereboom, D.M.; Alban, T.J.; Grabowski, M.M.; Alvarado, A.G.; Otvos, B.; Bayik, D.; Roversi, G.; McGraw, M.; Huang, P.; Mohammadi, A.M.; et al. Metronomic capecitabine as an immune modulator in glioblastoma patients reduces myeloid-derived suppressor cells. JCI Insight 2019, 4, e130748. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, S.; Baghdadi, M.; Tsuchikawa, T.; Wada, H.; Nakamura, T.; Abe, H.; Nakanishi, S.; Usui, Y.; Higuchi, K.; Takahashi, M.; et al. Chemotherapy-derived inflammatory responses accelerate the formation of immunosuppressive myeloid cells in the tissue microenvironment of human pancreatic cancer. Cancer Res. 2015, 75, 2629–2640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forghani, P.; Waller, E.K. Poly (I: C) modulates the immunosuppressive activity of myeloid-derived suppressor cells in a murine model of breast cancer. Breast Cancer Res. Treat. 2015, 153, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Migliorini, D.; Dutoit, V.; Allard, M.; Grandjean Hallez, N.; Marinari, E.; Widmer, V.; Philippin, G.; Corlazzoli, F.; Gustave, R.; Kreutzfeldt, M.; et al. Phase I/II trial testing safety and immunogenicity of the multipeptide IMA950/poly-ICLC vaccine in newly diagnosed adult malignant astrocytoma patients. Neuro. Oncol. 2019, 21, 923–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.; Park, C.S.; Lee, Y.R.; Im, S.A.; Song, S.; Lee, C.K. Resiquimod, a TLR7/8 agonist, promotes differentiation of myeloid-derived suppressor cells into macrophages and dendritic cells. Arch. Pharm. Res. 2014, 37, 1234–1240. [Google Scholar] [CrossRef]
- Chow, L.Q.M.; Morishima, C.; Eaton, K.D.; Baik, C.S.; Goulart, B.H.; Anderson, L.N.; Manjarrez, K.L.; Dietsch, G.N.; Bryan, J.K.; Hershberg, R.M.; et al. Phase Ib trial of the Toll-like receptor 8 agonist, motolimod (VTX-2337), combined with cetuximab in patients with recurrent or metastatic SCCHN. Clin. Cancer Res. 2017, 23, 2442–2450. [Google Scholar] [CrossRef] [Green Version]
- Shayan, G.; Kansy, B.A.; Gibson, S.P.; Srivastava, R.M.; Bryan, J.K.; Bauman, J.E.; Ohr, J.; Kim, S.; Duvvuri, U.; Clump, D.A.; et al. Phase Ib study of immune biomarker modulation with neoadjuvant cetuximab and TLR8 stimulation in head and neck cancer to overcome suppressive myeloid signals. Clin. Cancer Res. 2018, 24, 62–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vollmer, J.; Krieg, A.M. Immunotherapeutic applications of CpG oligodeoxynucleotide TLR9 agonists. Adv. Drug Deliv. Rev. 2009, 61, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; Hu, G.; Cai, X. The success and the challenge of all-trans retinoic acid in the treatment of cancer. Crit. Rev. Food Sci. Nutr. 2019, 59, S71–S80. [Google Scholar] [CrossRef] [PubMed]
- Lo-Coco, F.; Avvisati, G.; Vignetti, M.; Thiede, C.; Orlando, S.M.; Iacobelli, S.; Ferrara, F.; Fazi, P.; Cicconi, L.; Di Bona, E.; et al. Retinoic Acid and Arsenic Trioxide for Acute Promyelocytic Leukemia. New Engl. J. Med. 2013, 369, 111–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nefedova, Y.; Fishman, M.; Sherman, S.; Wang, X.; Beg, A.A.; Gabrilovich, D.I. Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res. 2007, 67, 11021–11028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirza, N.; Fishman, M.; Fricke, I.; Dunn, M.; Neuger, A.M.; Frost, T.J.; Lush, R.M.; Antonia, S.; Gabrilovich, D.I. All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res. 2006, 66, 9299–9307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tobin, R.P.; Jordan, K.R.; Robinson, W.A.; Davis, D.; Borges, V.F.; Gonzalez, R.; Lewis, K.D.; McCarter, M.D. Targeting myeloid-derived suppressor cells using all-trans retinoic acid in melanoma patients treated with Ipilimumab. Int. Immunopharmacol. 2018, 63, 282–291. [Google Scholar] [CrossRef]
- Reilley, M.J.; McCoon, P.; Cook, C.; Lyne, P.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; et al. STAT3 antisense oligonucleotide AZD9150 in a subset of patients with heavily pretreated lymphoma: Results of a phase 1b trial. J. Immunother. Cancer 2018, 6, 119. [Google Scholar] [CrossRef] [Green Version]
- Eruslanov, E.; Daurkin, I.; Ortiz, J.; Vieweg, J.; Kusmartsev, S. Pivotal Advance: Tumor-mediated induction of myeloid-derived suppressor cells and M2-polarized macrophages by altering intracellular PGE 2 catabolism in myeloid cells. J. Leukoc. Biol. 2010, 88, 839–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prima, V.; Kaliberova, L.N.; Kaliberov, S.; Curiel, D.T.; Kusmartsev, S. COX2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc. Natl. Acad. Sci. USA 2017, 114, 1117–1122. [Google Scholar] [CrossRef] [Green Version]
- Veltman, J.D.; Lambers, M.E.H.; van Nimwegen, M.; Hendriks, R.W.; Hoogsteden, H.C.; Aerts, J.G.J.V.; Hegmans, J.P.J.J. COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. Celecoxib influences MDSC function. BMC Cancer 2010, 10, 464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Take, Y.; Koizumi, S.; Nagahisa, A. Prostaglandin E Receptor 4 Antagonist in Cancer Immunotherapy: Mechanisms of Action. Front. Immunol. 2020, 11, 324. [Google Scholar] [CrossRef]
- Rotella, D.P. Phosphodiesterase 5 inhibitors: Current status and potential applications. Nat. Rev. Drug Discov. 2002, 1, 674–682. [Google Scholar] [CrossRef]
- Hassel, J.C.; Jiang, H.; Bender, C.; Winkler, J.; Sevko, A.; Shevchenko, I.; Halama, N.; Dimitrakopoulou-Strauss, A.; Haefeli, W.E.; Jäger, D.; et al. Tadalafil has biologic activity in human melanoma. Results of a pilot trial with Tadalafil in patients with metastatic Melanoma (TaMe). Oncoimmunology 2017, 6, e1326440. [Google Scholar] [CrossRef] [Green Version]
- Weed, D.T.; Vella, J.L.; Reis, I.M.; De La Fuente, A.C.; Gomez, C.; Sargi, Z.; Nazarian, R.; Califano, J.; Borrello, I.; Serafini, P. Tadalafil reduces myeloid-derived suppressor cells and regulatory t cells and promotes tumor immunity in patients with head and neck squamous cell carcinoma. Clin. Cancer Res. 2015, 21, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, A.; Fukumoto, T.; Zhang, R.; Gabrilovich, D. Selective targeting of different populations of myeloid-derived suppressor cells by histone deacetylase inhibitors. Cancer Immunol. Immunother. 2020, 69, 1929–1936. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Zou, J.; Li, S.; Topper, M.J.; Tao, Y.; Zhang, H.; Jiao, X.; Xie, W.; Kong, X.; Vaz, M.; et al. Epigenetic therapy inhibits metastases by disrupting premetastatic niches. Nature 2020, 579, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Janne, P.A.; Opyrchal, M.; Hafez, N.; Raez, L.E.; Gabrilovich, D.I.; Wang, F.; Trepel, J.B.; Lee, M.J.; Yuno, A.; et al. Entinostat plus Pembrolizumab in Patients with Metastatic NSCLC Previously Treated with Anti–PD-(L)1 Therapy. Clin. Cancer Res. 2021, 27, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, L.; Chen, X.; Li, L.; Li, Y.; Ping, Y.; Huang, L.; Yue, D.; Zhang, Z.; Wang, F.; et al. CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-β-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology 2017, 6, e1320011. [Google Scholar] [CrossRef] [Green Version]
- Theate, I.; Van Baren, N.; Pilotte, L.; Moulin, P.; Larrieu, P.; Renauld, J.C.; Herve, C.; Gutierrez-Roelens, I.; Marbaix, E.; Sempoux, C.; et al. Extensive profiling of the expression of the indoleamine 2,3-dioxygenase 1 protein in normal and tumoral human tissues. Cancer Immunol. Res. 2015, 3, 161–172. [Google Scholar] [CrossRef] [Green Version]
- Meireson, A.; Devos, M.; Brochez, L. IDO Expression in Cancer: Different Compartment, Different Functionality? Front. Immunol. 2020, 11, 531491. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Pico de Coaña, Y.; Poschke, I.; Gentilcore, G.; Mao, Y.; Nyström, M.; Hansson, J.; Masucci, G.V.; Kiessling, R. Ipilimumab treatment results in an early decrease in the frequency of circulating granulocytic myeloid-derived suppressor cells as well as their Arginase1 production. Cancer Immunol. Res. 2013, 1, 158–162. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wang, L.; Li, J.; Fan, Z.; Yang, L.; Zhang, Z.; Zhang, C.; Yue, D.; Qin, G.; Zhang, T.; et al. Metformin-induced reduction of CD39 and CD73 blocks myeloid-derived suppressor cell activity in patients with ovarian cancer. Cancer Res. 2018, 78, 1779–1791. [Google Scholar] [CrossRef] [Green Version]
- Lathers, D.M.R.; Clark, J.I.; Achille, N.J.; Young, M.R.I. Phase 1B study to improve immune responses in head and neck cancer patients using escalating doses of 25-hydroxyvitamm D3. Cancer Immunol. Immunother. 2004, 53, 422–430. [Google Scholar] [CrossRef] [PubMed]

| Strategy | Target | Drug Name | Combined Therapy | Disease | Clinical Trial | Clinical Benefit Rate |
|---|---|---|---|---|---|---|
| TAM depletion | CSF1/CSF1R | Pexidartinib | Single agent | Tenosynovial Giant Cell Tumor (TGCT) | NCT02371369 | Overall response: 53% |
| Paclitaxel | Advanced solid tumors | NCT01525602 | Clinical benefit: 40% | |||
| Single agent | Acute Myeloid Leukemia | NCT01349049 | Overall response: 21% | |||
| Durvalumab | Advanced Pancreatic and Colorectal Cancer | NCT02777710 | No results posted | |||
| Eribulin | Metastatic Breast Cancer | NCT01596751 | Not yet reported | |||
| PLX7486 | Single agent | Advanced solid tumors, TGCT | NCT01804530 | No results posted | ||
| BLZ945 | Spartalizumab | Advanced solid tumors | NCT02829723 | Not yet reported | ||
| Edicotinib | Daratumumab | Advanced Prostate Cancer | NCT03177460 | Not yet reported | ||
| ARRY-382 | Pembrolizumab | Advanced Solid Tumors | NCT02880371 | No results posted | ||
| IMC-CS4 | GVAX, Pembrolizumab | Pancreatic Cancer | NCT03153410 | Not yet reported | ||
| Durvalumab, Tremelimumab | Advanced Solid Tumors | NCT02718911 | Disease Control: 33.3% | |||
| Vemurafenib, Cobimetinib | Melanoma | NCT03101254 | Not yet reported | |||
| Emactuzumab | Atezolizumab | Advanced Solid Tumors | NCT02323191 | No results posted | ||
| Paclitaxel | Advanced Solid Tumors | NCT01494688 | Overall Response: 71% | |||
| Bevacizumab, Paclitaxel | Ovarian, Fallopian Tube or Peritoneal Cancer | NCT02923739 | Not yet reported | |||
| Cabiralizumab | Single agent | Tenosynovial Giant Cell Tumor | NCT02471716 | Not yet reported | ||
| Nivolumab | Advanced Solid Tumors | NCT02526017 | Not yet reported | |||
| Nivolumab, chemotherapies | Advanced Pancreatic Cancer | NCT03336216 | Not yet reported | |||
| Sotigalimab, Nivolumab | Melanoma, NSC Lung, Renal Cell Carcinoma | NCT03502330 | Not yet reported | |||
| Vimseltinib | Avelumab | Advanced or Metastatic Sarcomas | NCT04242238 | Not yet reported | ||
| Single agent | Tenosynovial Giant Cell Tumor | NCT05059262 | Not yet reported | |||
| AMG 820 | Pembrolizumab | Advanced Solid Tumor Cancer | NCT02713529 | Overall Response: 34% | ||
| Axatilimab | Durvalumab | Solid Tumors | NCT03238027 | Not yet reported | ||
| Durvalumab | Unresectable Cholangiocarcinoma | NCT04301778 | Not yet reported | |||
| MCS110 | Spartalizumab | Breast and Pancreatic Cancer, Melanoma | NCT02807844 | Overall Response: 27% | ||
| Carboplatin, Gemcitabine | Advanced Triple-Negative Breast Cancer | NCT02435680 | Clinical benefit: 29.4% | |||
| Dabrafenib, Trametinib | Melanoma | NCT03455764 | Not yet reported | |||
| TPX-0022 | Single agent | Advanced Solid Tumor | NCT03993873 | Not yet reported | ||
| Whole cell | Biphosphonates | Single agents | Primary Breast Cancer | NCT00127205 | Overall survival: 92.4% | |
| Denosumab | Metastatic Breast Cancer | NCT00091832 | Not yet reported | |||
| Caspase 8 | Trabectedin | Low-dose radiotherapy | Advanced/Metastatic Sarcomas | NCT05131386 | Not yet reported | |
| Olaratumab | Advanced Soft-tissue Sarcoma | NCT03985722 | Not yet reported | |||
| Single agent | Malignant Pleural Mesothelioma | NCT02194231 | Not yet reported | |||
| Inhibition of TAM recruitment | CCR2/CCL2 | Carlumab | Single agent | Metastatic Castrate-Resistant Prostate Cancer | NCT00992186 | Stable disease: 2.4% |
| Single agent | Solid Tumors | NCT00537368 | No results posted | |||
| Chemotherapies | Solid Tumors | NCT01204996 | Overall response: 38% | |||
| Plozalizumab | Single agent | Bone Metastatic Solid Tumors | NCT01015560 | Overall response: 14% | ||
| ICIs | Advanced Melanoma | NCT02723006 | Interrupted | |||
| PF-04136309 | Nab-paclitaxel, Gemcitabine | Metastatic Pancreatic Ductal Adenocarcinoma | NCT02732938 | Objective response: 23% | ||
| FOLFIRINOX | Pancreatic Neoplasms | NCT01413022 | Objective response: 49% | |||
| CCX872-B | Single agent | Pancreatic Adenocarcinoma | NCT02345408 | Overall survival: 29% | ||
| CCR2-CCR5 | BMS-813160 | Nivolumab, Chemotherapies | Pancreatic Ductal Adenocarcinoma | NCT03496662 | Not yet reported | |
| Nivolumab, GVAX | Pancreatic Ductal Adenocarcinoma | NCT03767582 | Not yet reported | |||
| Chemotherapy, Nivolumab | Advanced Solid Tumors | NCT03184870 | Not yet reported | |||
| Nivolumab, BMS-986253 | NSC Lung and Hepatocellular Carcinoma | NCT04123379 | Not yet reported | |||
| CCR5/CCL5 | Maraviroc | Pembrolizumab | Metastatic Colorectal Cancer | NCT03274804 | Disease Control: 5.3% | |
| Ipilimumab, Nivolumab | Metastatic Colorectal and Pancreatic Cancer | NCT04721301 | Not yet reported | |||
| Vicriviroc | Pembrolizumab | Advanced Colorectal Cancers | NCT03631407 | No results posted | ||
| Leronlimab | Single agent | Advanced Solid Tumors | NCT04504942 | Not yet reported | ||
| Single agent | Metastatic Triple-Negative Breast Carcinoma | NCT04313075 | Not yet reported | |||
| Carboplatin | Metastatic Triple-Negative Breast Carcinoma | NCT03838367 | Not yet reported | |||
| CXCR4/CXCL12 | LY2510924 | Sunitinib | Metastatic Renal Cell Carcinoma | NCT01391130 | Insufficient Efficacy | |
| Carboplatin, Etoposide | Extensive Stage Small Cell Lung Carcinoma | NCT01439568 | Insufficient Efficacy | |||
| Durvalumab | Solid Tumors | NCT02737072 | Interrupted | |||
| Motixafortide | Cemiplimab, Chemotherapy | Pancreatic Adenocarcinoma | NCT04543071 | Not yet reported | ||
| Pembrolizumab | Metastatic Pancreatic Cancer | NCT02907099 | Not yet reported | |||
| Pembrolizumab, Onivyde® | Metastatic Pancreatic Cancer | NCT02826486 | Disease Control: 77% | |||
| Plerixafor | Cemiplimab | Metastatic Pancreatic Cancer | NCT04177810 | Not yet reported | ||
| Single agent | Pancreatic, Ovarian and CRC Cancers | NCT02179970 | Stable disease: 57% | |||
| Pembrolizumab | Head and Neck Cancer | NCT04058145 | Interrupted |
| Strategy | Target | Drug Name | Combined Therapy | Disease | Clinical Trial |
|---|---|---|---|---|---|
| Reprogramming TAM polarization | TLRs | GSK1795091 | GSK3174998, Pembrolizumab | Advanced Solid Tumors | NCT03447314 |
| Imiquimod | 5-fluorouracil | Squamous Cell Carcinoma | NCT03370406 | ||
| Abraxane | Advanced Breast Cancer | NCT00821964 | |||
| Cyclophosphamide, Radiotherapy | Breast Cancer with Skin Metastases | NCT01421017 | |||
| Single agent | Breast Cancer with Skin Metastases | NCT00899574 | |||
| 852A | Single agent | Unresectable Metastatic Cutaneous Melanoma | NCT00189332 | ||
| Single agent | Breast, Ovarian, Endometrial, Cervical Cancers | NCT00319748 | |||
| Resiquimod | gp100 and MAGE3 peptide vaccine | Melanoma | NCT00960752 | ||
| Motolimod | Durvalumab, Doxorubicin | Recurrent, Platinum-resistant Ovarian Cancer | NCT02431559 | ||
| Nivolumab | Head and Neck Cancer | NCT03906526 | |||
| Cetuximab | Metastatic Head and Neck Squamous Carcinoma | NCT01836029 | |||
| IMO-2055 | FOLFIRI, Cetuximab | Colorectal Cancer | NCT00719199 | ||
| Erlotinib, Bevacizumab | Non-Small Cell Lung Cancer | NCT00633529 | |||
| Single agent | Clear Cell Renal Carcinoma | NCT00729053 | |||
| Tilsotolimod | Ipilimumab | Metastatic Melanoma | NCT03445533 | ||
| Single agent | Malignant Melanoma | NCT04126876 | |||
| Ipilimumab, Pembrolizumab | Metastatic Melanoma | NCT02644967 | |||
| CMP-001 | Pembrolizumab | Head and Neck Squamous Cell Carcinoma | NCT04633278 | ||
| Nivolumab | Advanced Melanoma | NCT04698187 | |||
| Stereotactic body radiotherapy | Early-Stage Triple Negative Breast Cancer | NCT04807192 | |||
| Atezolizumab, Radiotherapy | Non-Small Cell Lung Cancer | NCT03438318 | |||
| CD40 | Selicrelumab | Atezolizumab | Advanced Solid Tumors | NCT02304393 | |
| Nab-paclitaxel, Gemcitabine | Pancreatic Cancer | NCT02588443 | |||
| Emactuzumab | Advanced Solid Tumors | NCT02760797 | |||
| SEA-CD40 | Pembrolizumab, Carboplatin | Melanoma, Non-Small Cell Lung Cancer | NCT04993677 | ||
| Pembrolizumab, Gemcitabine | Advanced Solid Tumors | NCT02376699 | |||
| Sotigalimab | Doxorubicin | Soft Tissue Sarcoma | NCT03719430 | ||
| Pembrolizumab | Metastatic Melanoma | NCT02706353 | |||
| Single agent | Pediatric CNS Tumors | NCT03389802 | |||
| CP-870,893 | Tremelimumab | Metastatic Melanoma | NCT01103635 | ||
| Paclitaxel + Carboplatin | Advanced Solid Tumors | NCT00607048 | |||
| CDX-1140 | Pembrolizumab, Chemotherapy | Advanced Solid Tumors | NCT03329950 | ||
| ABBV-428 | Nivolumab | Advanced Solid Tumors | NCT02955251 | ||
| PI3Kγ | Eganelisib | Nivolumab | Advanced Solid Tumors | NCT02637531 | |
| Etrumadenant, doxorubicin, paclitaxel | Triple-Negative Breast and Ovarian Cancer | NCT03719326 | |||
| Nivolumab | Advanced Urothelial Carcinoma | NCT03980041 | |||
| HDACs | Tucidinostat | Tislelizumab | Metastatic Urothelial Carcinoma | NCT04562311 | |
| Cisplatin | Metastatic Triple-negative Breast Cancer | NCT04192903 | |||
| Toripalimab | Advanced Cervical Cancer | NCT04651127 | |||
| STAT3 | TTI-101 | Single agent | Advanced Solid Tumors | NCT03195699 | |
| Re-activation of phagocytosis | CD47/SIRP1α | Magrolimab | Cetuximab | Advanced Solid Tumors | NCT02953782 |
| Avelumab | Ovarian Cancer | NCT03558139 | |||
| Dinutuximab | Neuroblastoma, Osteosarcoma | NCT04751383 | |||
| Docetaxel | Advanced Solid Tumors | NCT04827576 | |||
| Pactiltaxel, Nab-paclitaxel | Metastatic Triple-Negative Breast Cancer | NCT04958785 | |||
| Pembrolizumab, Chemotherapies | Head and Neck Squamous Cell Carcinoma | NCT04854499 | |||
| TTI-621 | Rituximab, Nivolumab | Solid Tumors | NCT02663518 | ||
| ICIs, Radiation, T-Vec | Advanced Solid Tumors | NCT02890368 | |||
| Doxorubicin | Metastatic High-Grade Leiomyosarcoma | NCT04996004 | |||
| CC-95251 | Rituximab, Cetuximab | Advanced Solid and Hematologic Cancer | NCT03783403 | ||
| CC-90002 | Rituximab | Advanced Solid and Hematologic Cancer | NCT02367196 | ||
| STI-6643 | Single agent | Advanced Solid Tumors | NCT04900519 | ||
| Genetically engineering TAM | HER2-directed CAR-M | CT-0508 | Single agent | HER2-overexpressing Solid Tumors | NCT04660929 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bleve, A.; Consonni, F.M.; Porta, C.; Garlatti, V.; Sica, A. Evolution and Targeting of Myeloid Suppressor Cells in Cancer: A Translational Perspective. Cancers 2022, 14, 510. https://doi.org/10.3390/cancers14030510
Bleve A, Consonni FM, Porta C, Garlatti V, Sica A. Evolution and Targeting of Myeloid Suppressor Cells in Cancer: A Translational Perspective. Cancers. 2022; 14(3):510. https://doi.org/10.3390/cancers14030510
Chicago/Turabian StyleBleve, Augusto, Francesca Maria Consonni, Chiara Porta, Valentina Garlatti, and Antonio Sica. 2022. "Evolution and Targeting of Myeloid Suppressor Cells in Cancer: A Translational Perspective" Cancers 14, no. 3: 510. https://doi.org/10.3390/cancers14030510
APA StyleBleve, A., Consonni, F. M., Porta, C., Garlatti, V., & Sica, A. (2022). Evolution and Targeting of Myeloid Suppressor Cells in Cancer: A Translational Perspective. Cancers, 14(3), 510. https://doi.org/10.3390/cancers14030510