
Inflammation, Fibrosis and Cancer: Mechanisms, Therapeutic Options and Challenges



Abstract
:Simple Summary
Abstract
1. Introduction
2. Tissue Inflammation
2.1. Inflammation and Tissue Fibrosis
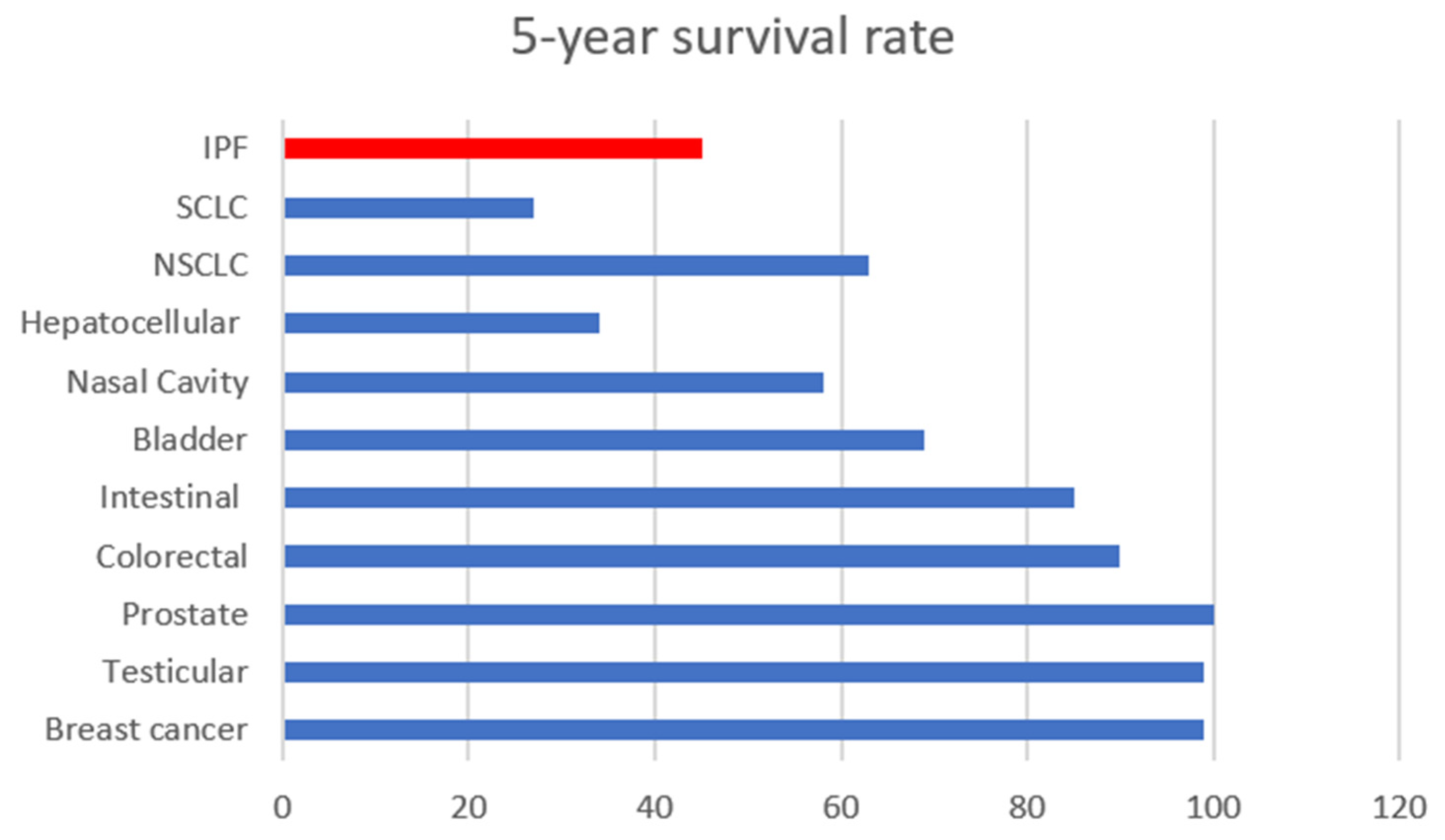
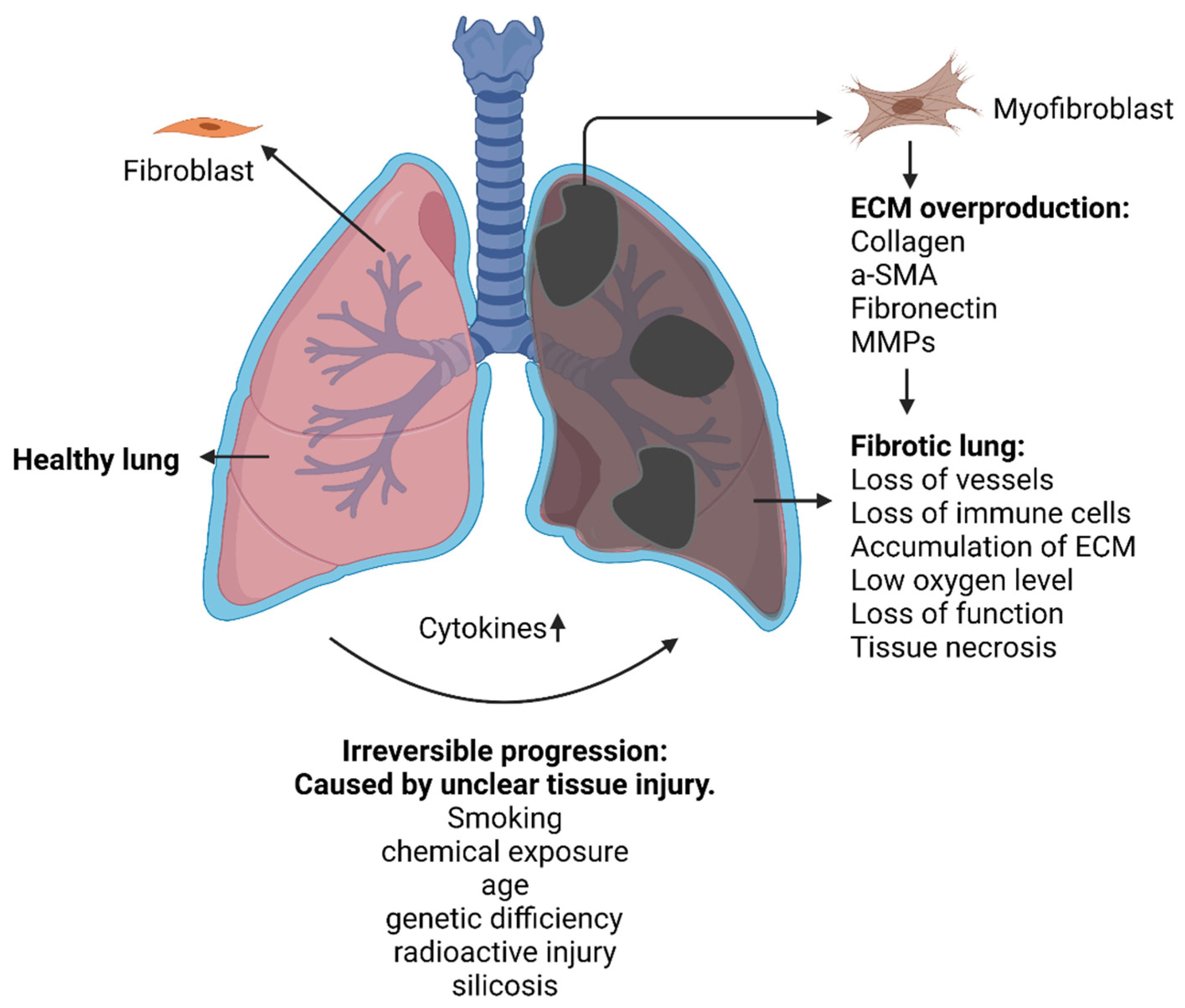
2.1.1. Idiopathic Pulmonary Fibrosis (IPF)
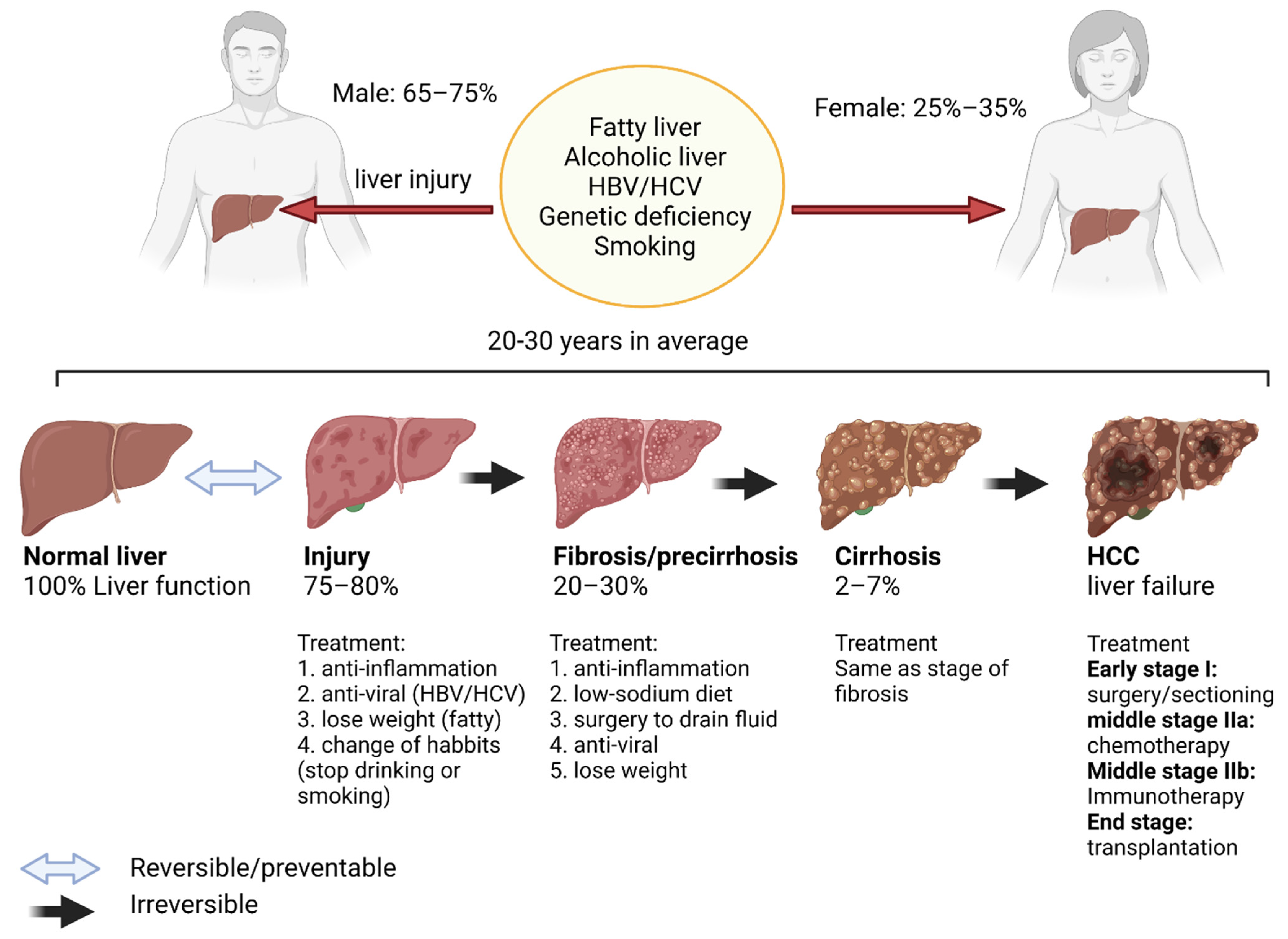
2.1.2. Liver Cirrhosis
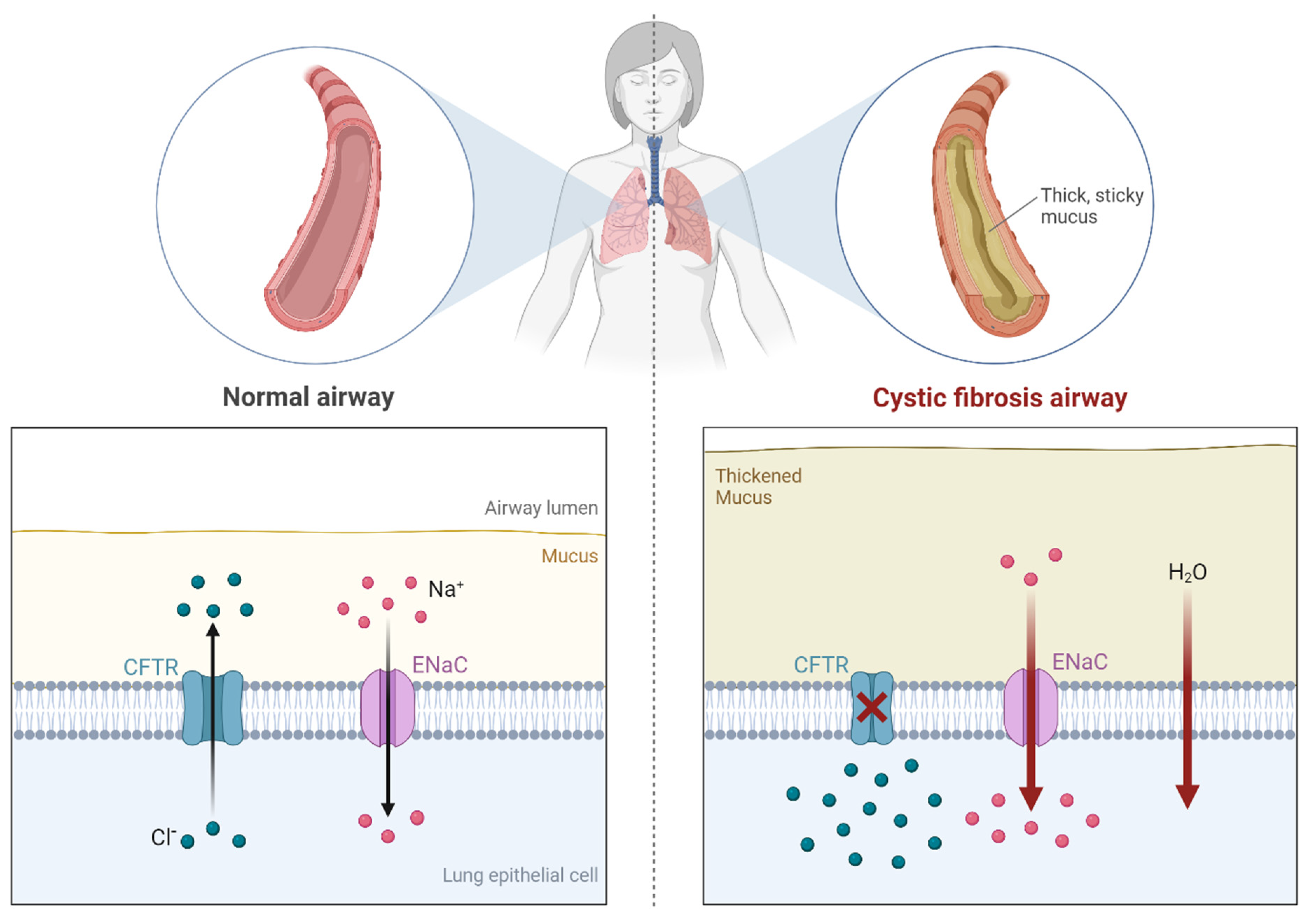
2.1.3. Cystic Fibrosis
2.1.4. Drug-Induced Pulmonary Fibrosis
2.1.5. Radiation-Induced Fibrosis
2.2. Inflammation and Cancer
2.2.1. Cancer Types Linked to Chronic Tissue Inflammation
2.2.2. Sources of Inflammation That May Lead to Cancers
Infections
Chemical Exposure
Autoimmune Conditions
3. Major Inflammatory Pathways in Tumor Progression and Tissue Fibrosis
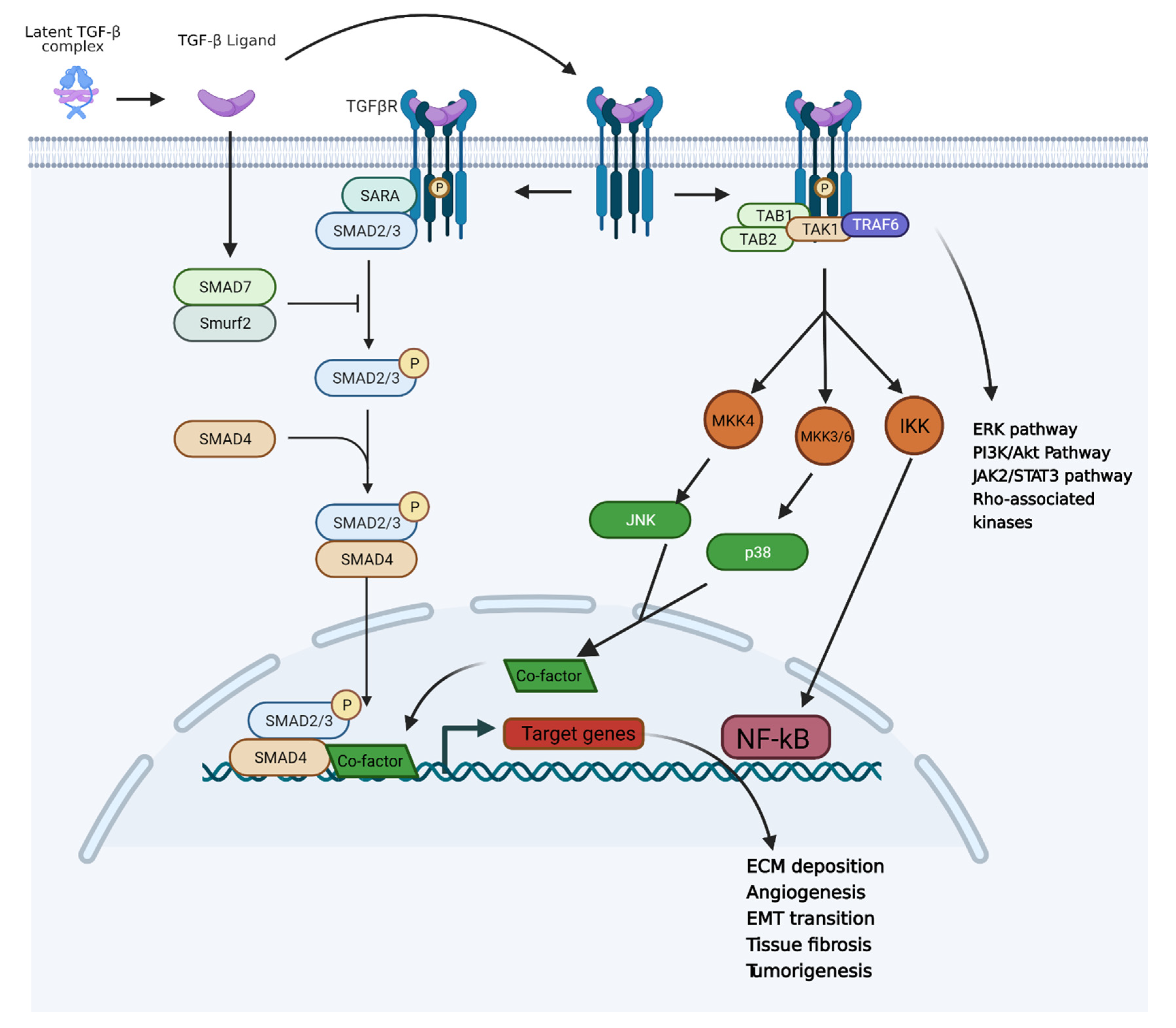
3.1. TGF-β Pathway and Inflammation-Induced Carcinogenesis
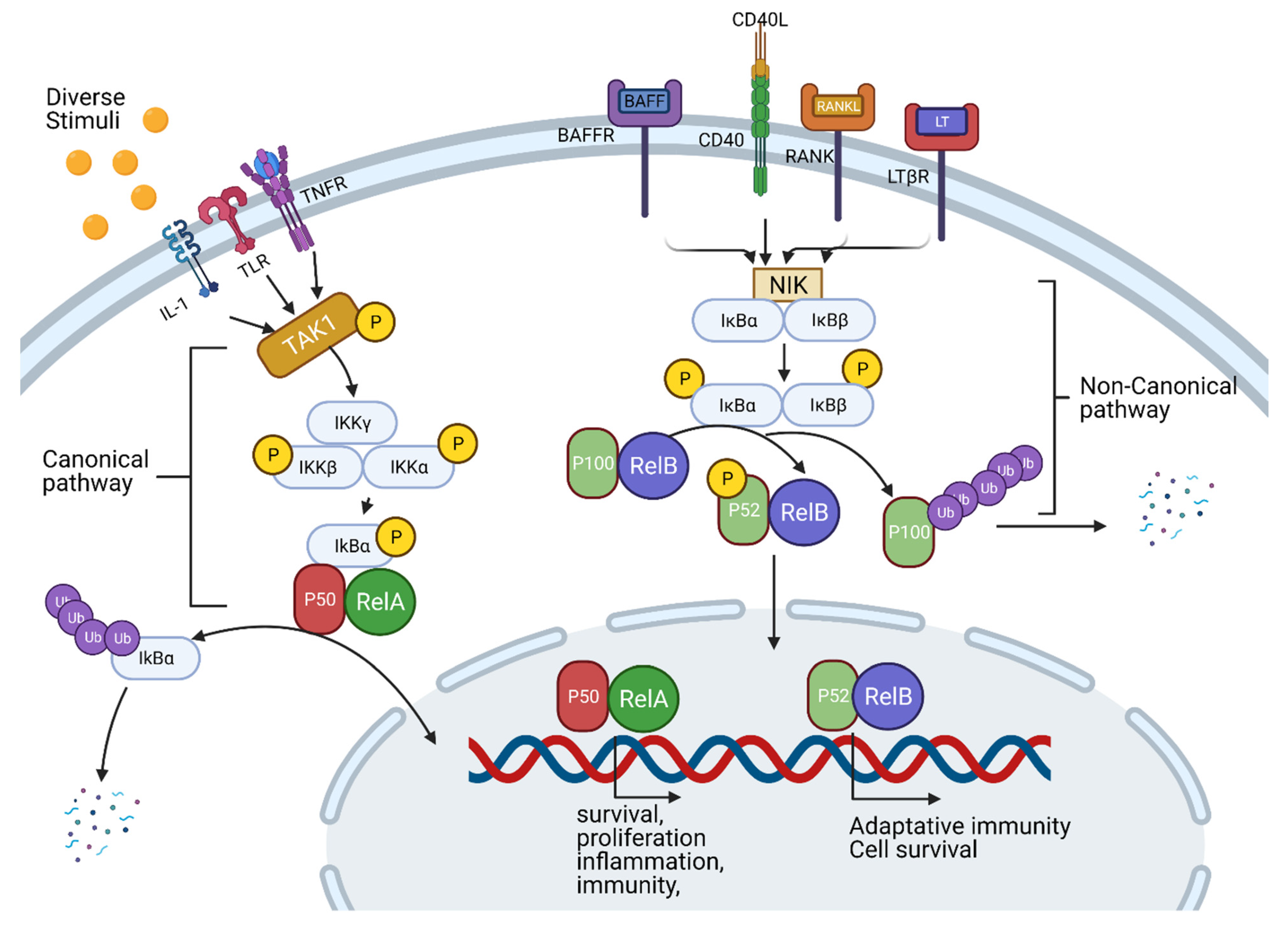
3.2. TNF-Alpha, NF-κB Pathway and Inflammation
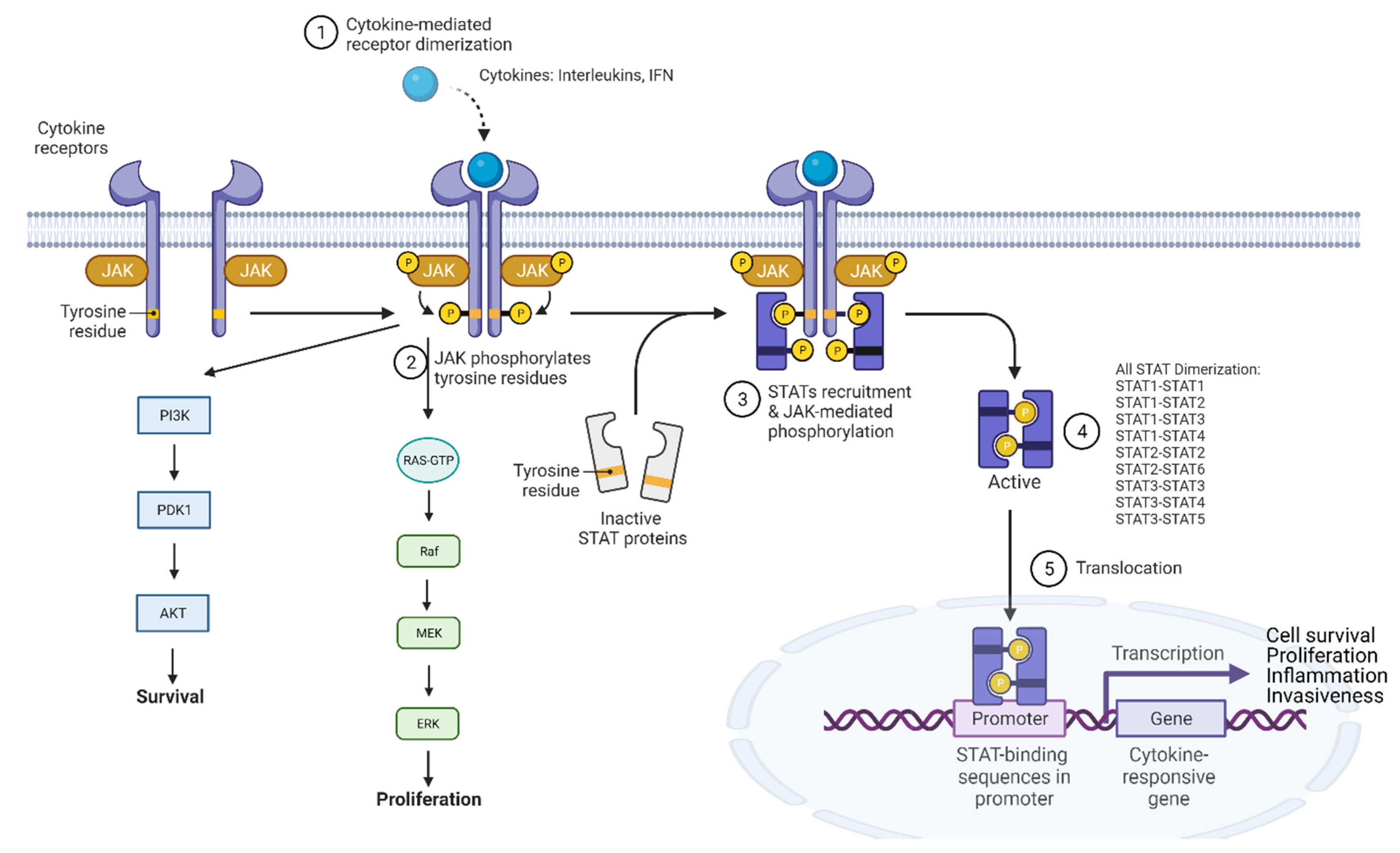
3.3. JAK-STAT Pathway and Inflammation
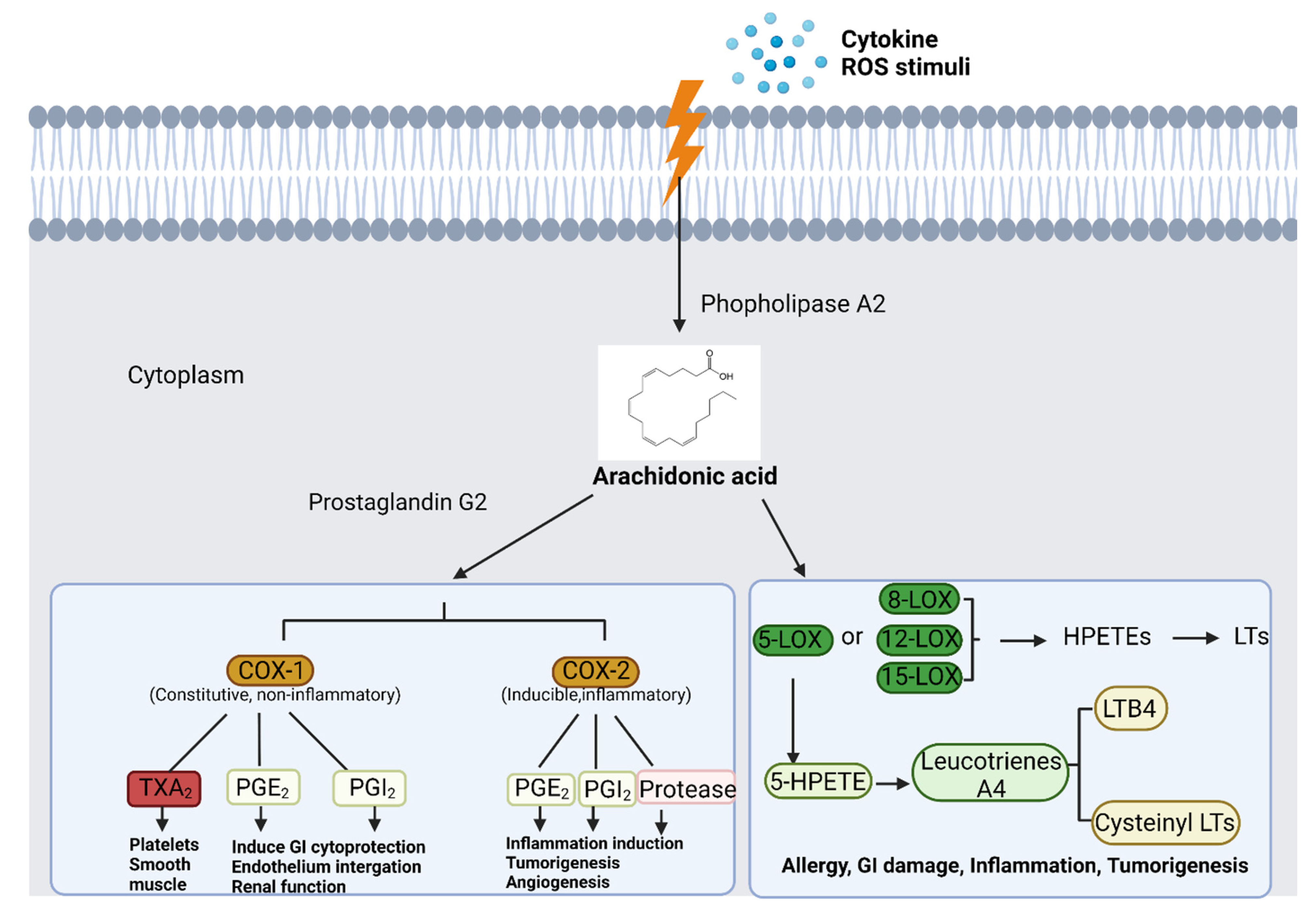
3.4. Arachidonic Acid Metabolism Pathway and Inflammation
3.5. Epigenetic Dysfunction
4. Current Medical Challenges and Novel Solutions for Targeting Inflammation Pathways
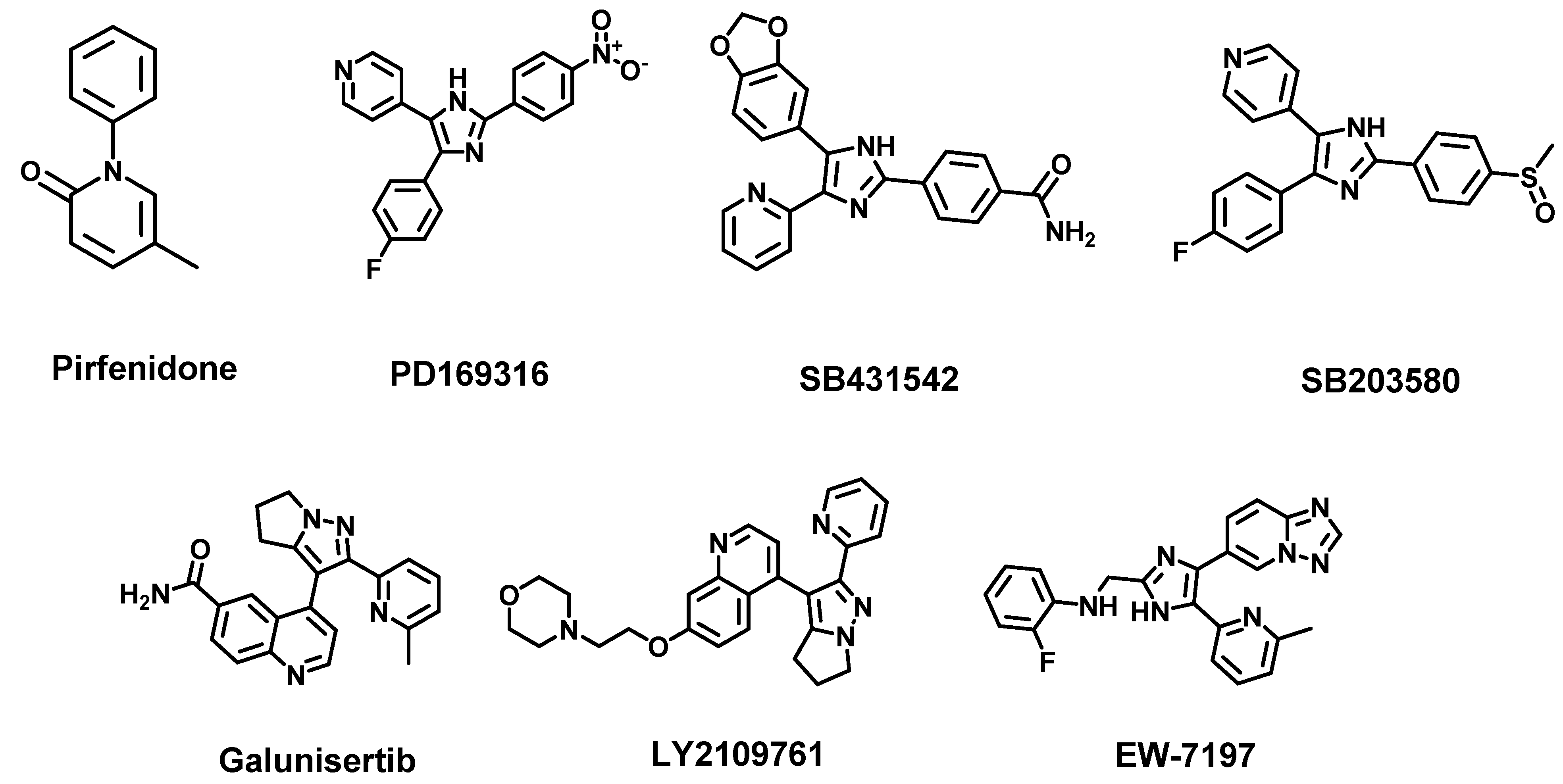
4.1. TGF-β Pathway
4.2. Challenges with NF-κB Pathway Inhibition
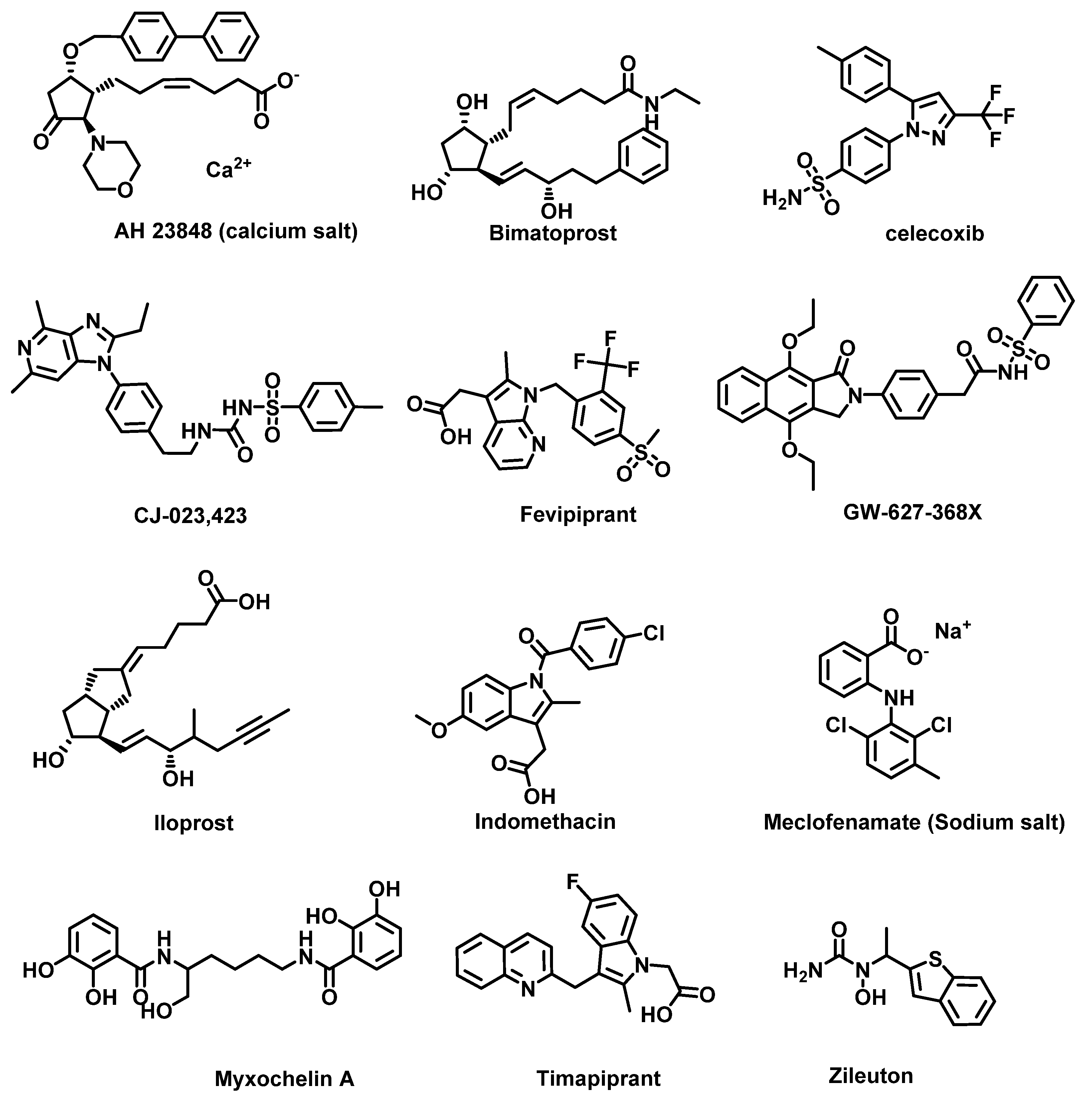
4.3. Challenges with COX Inhibition
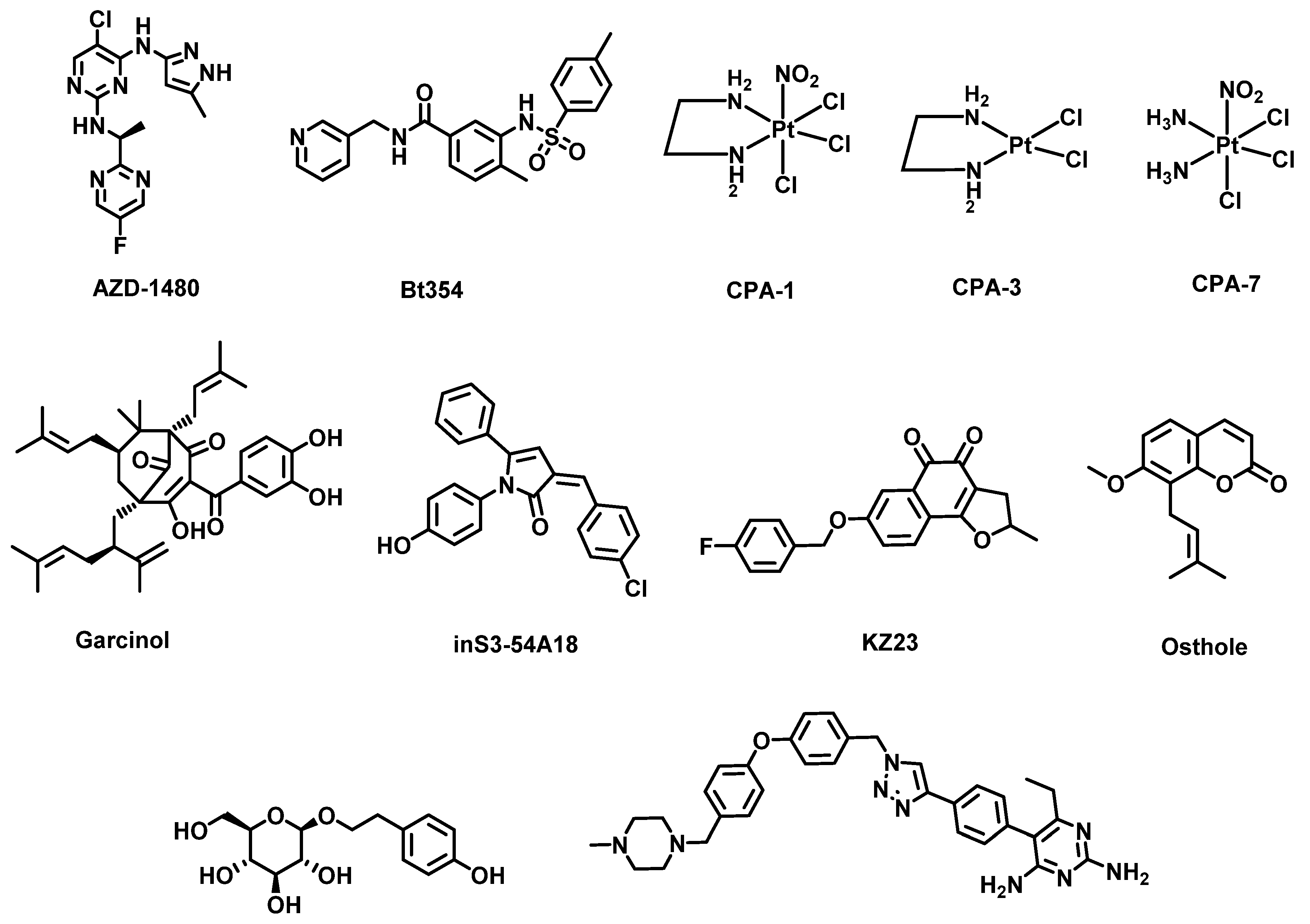
4.4. Challenges with STAT3 Inhibition and Solution
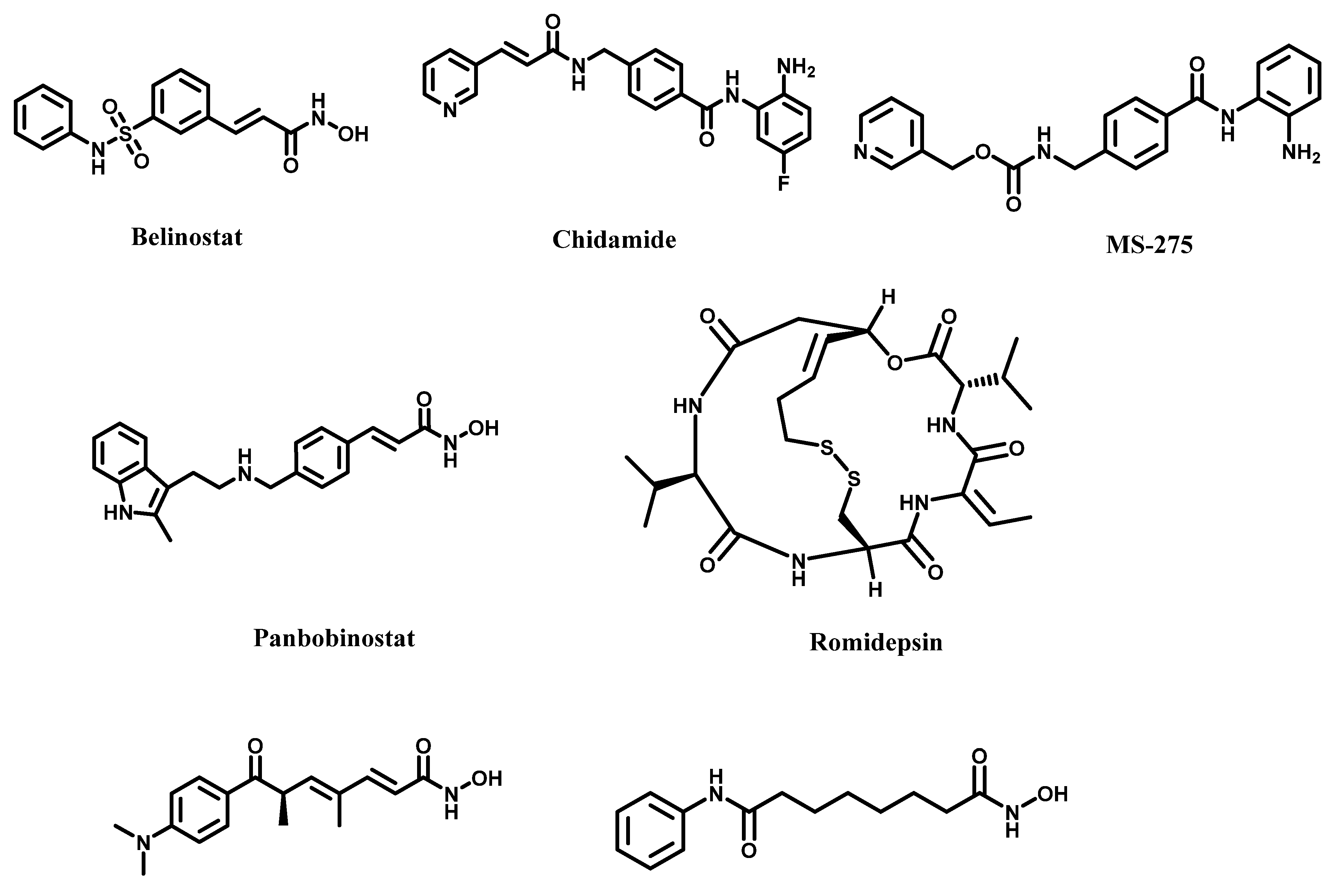
4.5. Challenges with HDAC Inhibition
5. Conclusions and Future Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Greten, F.R.; Grivennikov, S.I. Inflammation and cancer: Triggers, mechanisms, and consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Baby, D.; Rajguru, J.P.; Patil, P.B.; Thakkannavar, S.S.; Pujari, V.B. Inflammation and cancer. Ann. Afr. Med. 2019, 18, 121. [Google Scholar] [CrossRef] [PubMed]
- Vancheri, C.; du Bois, R.M. A progression-free end-point for idiopathic pulmonary fibrosis trials: Lessons from cancer. Eur. Respir. J. 2013, 41, 262–269. [Google Scholar] [CrossRef] [Green Version]
- Kuraishy, A.; Karin, M.; Grivennikov, S.I. Tumor promotion via injury-and death-induced inflammation. Immunity 2011, 35, 467–477. [Google Scholar] [CrossRef] [Green Version]
- Elliot, A.; Myllymäki, H.; Feng, Y. Inflammatory Responses during Tumour Initiation: From Zebrafish Transgenic Models of Cancer to Evidence from Mouse and Man. Cells 2020, 9, 1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borsig, L.; Wolf, M.J.; Roblek, M.; Lorentzen, A.; Heikenwalder, M. Inflammatory chemokines and metastasis—Tracing the accessory. Oncogene 2014, 33, 3217–3224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Kinoshita, T.; Goto, T. Molecular mechanisms of pulmonary fibrogenesis and its progression to lung cancer: A review. Int. J. Mol. Sci. 2019, 20, 1461. [Google Scholar] [CrossRef] [Green Version]
- Ballester, B.; Milara, J.; Cortijo, J. Idiopathic pulmonary fibrosis and lung cancer: Mechanisms and molecular targets. Int. J. Mol. Sci. 2019, 20, 593. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, S.; Saito, A.; Nagase, T. YAP/TAZ signaling as a molecular link between fibrosis and cancer. Int. J. Mol. Sci. 2018, 19, 3674. [Google Scholar] [CrossRef] [Green Version]
- Vancheri, C.; Failla, M.; Crimi, N.; Raghu, G. Idiopathic pulmonary fibrosis: A disease with similarities and links to cancer biology. Eur. Respir. J. 2010, 35, 496–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandler, C.; Liu, T.; Buckanovich, R.; Coffman, L.G. The double edge sword of fibrosis in cancer. Transl. Res. 2019, 209, 55–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinko, D.; Diakos, C.I.; Clarke, S.J.; Charles, K.A. Cancer-related systemic inflammation: The challenges and therapeutic opportunities for personalized medicine. Clin. Pharmacol. Ther. 2017, 102, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Mack, M. Inflammation and fibrosis. Matrix Biol. 2018, 68, 106–121. [Google Scholar] [CrossRef]
- Thomas, T.P.; Grisanti, L.A. The dynamic interplay between cardiac inflammation and fibrosis. Front. Physiol. 2020, 11, 529075. [Google Scholar] [CrossRef] [PubMed]
- Ritter, B.; Greten, F.R. Modulating inflammation for cancer therapy. J. Exp. Med. 2019, 216, 1234–1243. [Google Scholar] [CrossRef]
- Mancini, M.L.; Sonis, S.T. Mechanisms of cellular fibrosis associated with cancer regimen-related toxicities. Front. Pharmacol. 2014, 5, 51. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.K.; Khare, P.; Obaid, A.; Conlon, K.P.; Basrur, V.; DePinho, R.A.; Venuprasad, K. SUMOylation of ROR-γt inhibits IL-17 expression and inflammation via HDAC2. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ranneh, Y.; Ali, F.; Akim, A.M.; Hamid, H.A.; Khazaai, H.; Fadel, A. Crosstalk between reactive oxygen species and pro-inflammatory markers in developing various chronic diseases: A review. Appl. Biol. Chem. 2017, 60, 327–338. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.-M.; An, J. Cytokines, inflammation and pain. Int. Anesthesiol. Clin. 2007, 45, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Midwood, K.S.; Williams, L.V.; Schwarzbauer, J.E. Tissue repair and the dynamics of the extracellular matrix. Int. J. Biochem. Cell Biol. 2004, 36, 1031–1037. [Google Scholar] [CrossRef] [PubMed]
- Herrera, J.; Henke, C.A.; Bitterman, P.B. Extracellular matrix as a driver of progressive fibrosis. J. Clin. Investig. 2018, 128, 45–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liaskou, E.; Wilson, D.V.; Oo, Y.H. Innate immune cells in liver inflammation. Mediat. Inflamm. 2012, 2012, 949157. [Google Scholar] [CrossRef] [Green Version]
- Culley, F.J. Natural killer cells in infection and inflammation of the lung. Immunology 2009, 128, 151–163. [Google Scholar] [CrossRef]
- Davies, A.J.; Rinaldi, S.; Costigan, M.; Oh, S.B. Cytotoxic immunity in peripheral nerve injury and pain. Front. Neurosci. 2020, 14, 142. [Google Scholar] [CrossRef]
- Akl, H.; Vervloessem, T.; Kiviluoto, S.; Bittremieux, M.; Parys, J.B.; De Smedt, H.; Bultynck, G. A dual role for the anti-apoptotic Bcl-2 protein in cancer: Mitochondria versus endoplasmic reticulum. Biochim. Biophys. Acta 2014, 1843, 2240–2252. [Google Scholar] [CrossRef] [Green Version]
- Schneider, G.; Henrich, A.; Greiner, G.; Wolf, V.; Lovas, A.; Wieczorek, M.; Wagner, T.; Reichardt, S.; von Werder, A.; Schmid, R.M.; et al. Cross talk between stimulated NF-kappaB and the tumor suppressor p53. Oncogene 2010, 29, 2795–2806. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.L.; Morgan, T.R. The natural history of hepatitis C virus (HCV) infection. Int. J. Med. Sci. 2006, 3, 47–52. [Google Scholar] [CrossRef] [Green Version]
- Sauleda, J.; Nunez, B.; Sala, E.; Soriano, J.B. Idiopathic Pulmonary Fibrosis: Epidemiology, Natural History, Phenotypes. Med. Sci. 2018, 6, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navaratnam, V.; Fleming, K.M.; West, J.; Smith, C.J.; Jenkins, R.G.; Fogarty, A.; Hubbard, R.B. The rising incidence of idiopathic pulmonary fibrosis in the U.K. Thorax 2011, 66, 462–467. [Google Scholar] [CrossRef] [Green Version]
- Jeganathan, N.; Smith, R.A.; Sathananthan, M. Mortality trends of idiopathic pulmonary fibrosis in the United States from 2004 through 2017. Chest 2021, 159, 228–238. [Google Scholar] [CrossRef]
- The National Institute of Health. Idiopathic Pulmonary Fibrosis. 2020. Available online: https://www.nhlbi.nih.gov/health-topics/idiopathic-pulmonary-fibrosis (accessed on 20 July 2021).
- Fernandez Perez, E.R.; Daniels, C.E.; Schroeder, D.R.; St Sauver, J.; Hartman, T.E.; Bartholmai, B.J.; Yi, E.S.; Ryu, J.H. Incidence, prevalence, and clinical course of idiopathic pulmonary fibrosis: A population-based study. Chest 2010, 137, 129–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaunisto, J.; Salomaa, E.-R.; Hodgson, U.; Kaarteenaho, R.; Kankaanranta, H.; Koli, K.; Vahlberg, T.; Myllärniemi, M. Demographics and survival of patients with idiopathic pulmonary fibrosis in the FinnishIPF registry. ERJ Open Res. 2019, 5, 00170–02018. [Google Scholar] [CrossRef]
- American Cancer Society. Cancer Statistics Center. Available online: http://cancerstatisticscenter.cancer.org (accessed on 9 November 2021).
- Huard, J.; Li, Y.; Fu, F.H. Muscle injuries and repair: Current trends in research. J. Bone Jt. Surg. Am. 2002, 84, 822–832. [Google Scholar] [CrossRef]
- Heinemeier, K.M.; Olesen, J.L.; Haddad, F.; Langberg, H.; Kjaer, M.; Baldwin, K.M.; Schjerling, P. Expression of collagen and related growth factors in rat tendon and skeletal muscle in response to specific contraction types. J. Physiol. 2007, 582, 1303–1316. [Google Scholar] [CrossRef]
- Wolters, P.J.; Collard, H.R.; Jones, K.D. Pathogenesis of idiopathic pulmonary fibrosis. Annu. Rev. Pathol. 2014, 9, 157–179. [Google Scholar] [CrossRef] [Green Version]
- Kramann, R.; DiRocco, D.P.; Humphreys, B.D. Understanding the origin, activation and regulation of matrix-producing myofibroblasts for treatment of fibrotic disease. J. Pathol. 2013, 231, 273–289. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Jiang, Y.G.; Luo, Y.; He, D.l.; Li, X.; Zhang, L.l.; Peng, T.; Li, M.C.; Lin, Y.H. Role of Wnt/β-catenin signaling pathway in epithelial-mesenchymal transition of human prostate cancer induced by hypoxia-inducible factor-1α. Int. J. Urol. 2007, 14, 1034–1039. [Google Scholar] [CrossRef] [PubMed]
- Grande, M.; Franzen, A.; Karlsson, J.-O.; Ericson, L.E.; Heldin, N.-E.; Nilsson, M. Transforming growth factor-β and epidermal growth factor synergistically stimulate epithelial to mesenchymal transition (EMT) through a MEK-dependent mechanism in primary cultured pig thyrocytes. J. Cell Sci. 2002, 115, 4227–4236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonnylal, S.; Shi-Wen, X.; Leoni, P.; Naff, K.; Van Pelt, C.S.; Nakamura, H.; Leask, A.; Abraham, D.; Bou-Gharios, G.; De Crombrugghe, B. Selective expression of connective tissue growth factor in fibroblasts in vivo promotes systemic tissue fibrosis. Arthritis Rheum. 2010, 62, 1523–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strutz, F.; Zeisberg, M.; Ziyadeh, F.N.; Yang, C.-Q.; Kalluri, R.; Müller, G.A.; Neilson, E.G.; Renziehausen, A.; Sisic, Z. Role of basic fibroblast growth factor-2 in epithelial-mesenchymal transformation. Kidney Int. 2002, 61, 1714–1728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Gregorio, J.; Robuffo, I.; Spalletta, S.; Giambuzzi, G.; De Iuliis, V.; Toniato, E.; Martinotti, S.; Conti, P.; Flati, V. The epithelial-to-mesenchymal transition as a possible therapeutic target in fibrotic disorders. Front. Cell Dev. Biol. 2020, 8, 607483. [Google Scholar] [CrossRef] [PubMed]
- Piera-Velazquez, S.; Li, Z.; Jimenez, S.A. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am. J. Pathol. 2011, 179, 1074–1080. [Google Scholar] [CrossRef]
- Ginhoux, F.; Guilliams, M. Tissue-resident macrophage ontogeny and homeostasis. Immunity 2016, 44, 439–449. [Google Scholar] [CrossRef]
- van Furth, R.; Cohn, Z.A. The origin and kinetics of mononuclear phagocytes. J. Exp. Med. 1968, 128, 415–435. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Scott, C.L.; Henri, S.; Guilliams, M. Mononuclear phagocytes of the intestine, the skin, and the lung. Immunol. Rev. 2014, 262, 9–24. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y.; Wu, G.; Xiong, W.; Gu, W.; Wang, C.-Y. Macrophages: Friend or foe in idiopathic pulmonary fibrosis? Respir. Res. 2018, 19, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sepanlou, S.G.; Safiri, S.; Bisignano, C.; Ikuta, K.S.; Merat, S.; Saberifiroozi, M.; Poustchi, H.; Tsoi, D.; Colombara, D.V.; Abdoli, A. The global, regional, and national burden of cirrhosis by cause in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 245–266. [Google Scholar] [CrossRef] [Green Version]
- Poh, Z.; Goh, B.-B.G.; Chang, P.-E.J.; Tan, C.-K. Rates of cirrhosis and hepatocellular carcinoma in chronic hepatitis B and the role of surveillance: A 10-year follow-up of 673 patients. Eur. J. Gastroenterol. Hepatol. 2015, 27, 638. [Google Scholar] [CrossRef] [Green Version]
- Sangro, B.; Sarobe, P.; Hervás-Stubbs, S.; Melero, I. Advances in immunotherapy for hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 525–543. [Google Scholar] [CrossRef] [PubMed]
- Brian, P.; O’Sullivan, S.D.F. Cystic fibrosis. Lancet 2009, 373, 1891–1904. [Google Scholar] [CrossRef]
- Elborn, J.S.; Shale, D.; Britton, J. Cystic fibrosis: Current survival and population estimates to the year 2000. Thorax 1991, 46, 881–885. [Google Scholar] [CrossRef] [Green Version]
- Thabut, G.; Mal, H. Outcomes after lung transplantation. J. Thorac. Dis. 2017, 9, 2684. [Google Scholar] [CrossRef] [Green Version]
- Stoltz, D.A.; Meyerholz, D.K.; Welsh, M.J. Origins of cystic fibrosis lung disease. N. Engl. J. Med. 2015, 372, 351–362. [Google Scholar] [CrossRef] [Green Version]
- Reprinted from “Cystic Fibrosis Airway”, by BioRender.com. Available online: https://app.biorender.com/biorender-templates (accessed on 9 November 2021).
- Hoy, S.M. Elexacaftor/ivacaftor/tezacaftor: First approval. Drugs 2019, 79, 2001–2007. [Google Scholar] [CrossRef]
- Veit, G.; Roldan, A.; Hancock, M.A.; Da Fonte, D.F.; Xu, H.; Hussein, M.; Frenkiel, S.; Matouk, E.; Velkov, T.; Lukacs, G.L. Allosteric folding correction of F508del and rare CFTR mutants by elexacaftor-tezacaftor-ivacaftor (Trikafta) combination. JCI Insight 2020, 5, e139983. [Google Scholar] [CrossRef]
- Burcham, P.; Sarzynski, L.; Khalfoun, S.; Novak, K.J.; Miller, J.C.; Tumin, D.; Hayes, D. Immunosuppression drug therapy in lung transplantation for cystic fibrosis. Pediatric Drugs 2017, 19, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Maisonneuve, P.; FitzSimmons, S.C.; Neglia, J.P.; Campbell, P.W., III; Lowenfels, A.B. Cancer risk in nontransplanted and transplanted cystic fibrosis patients: A 10-year study. J. Natl. Cancer Inst. 2003, 95, 381–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobelska-Dubiel, N.; Klincewicz, B.; Cichy, W. Liver disease in cystic fibrosis. Prz. Gastroenterol. 2014, 9, 136. [Google Scholar] [CrossRef] [PubMed]
- Flass, T.; Narkewicz, M.R. Cirrhosis and other liver disease in cystic fibrosis. J. Cyst. Fibros. 2013, 12, 116–124. [Google Scholar] [CrossRef] [Green Version]
- Schwaiblmair, M.; Behr, W.; Haeckel, T.; Markl, B.; Foerg, W.; Berghaus, T. Drug induced interstitial lung disease. Open Respir. Med. J. 2012, 6, 63–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ripley, B.A.; Kelil, T.; Gill, R.R. Deciphering drug-induced interstitial lung disease: A mechanistic approach. Appl. Radiol. 2016, 45, 9. [Google Scholar]
- Cooper, J.A., Jr.; Zitnik, R.J.; Matthay, R.A. Mechanisms of drug-induced pulmonary disease. Annu. Rev. Med. 1988, 39, 395–404. [Google Scholar] [CrossRef]
- Delaney, G.; Jacob, S.; Featherstone, C.; Barton, M. The role of radiotherapy in cancer treatment: Estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer 2005, 104, 1129–1137. [Google Scholar] [CrossRef]
- Barnett, G.C.; West, C.M.; Dunning, A.M.; Elliott, R.M.; Coles, C.E.; Pharoah, P.D.; Burnet, N.G. Normal tissue reactions to radiotherapy: Towards tailoring treatment dose by genotype. Nat. Rev. Cancer 2009, 9, 134–142. [Google Scholar] [CrossRef] [Green Version]
- Burger, A.; Loffler, H.; Bamberg, M.; Rodemann, H.P. Molecular and cellular basis of radiation fibrosis. Int. J. Radiat. Biol. 1998, 73, 401–408. [Google Scholar] [CrossRef]
- Lawrence, T.S.; Robertson, J.M.; Anscher, M.S.; Jirtle, R.L.; Ensminger, W.D.; Fajardo, L.F. Hepatic toxicity resulting from cancer treatment. Int. J. Radiat. Oncol. Biol. Phys. 1995, 31, 1237–1248. [Google Scholar] [CrossRef]
- Sciubba, J.J.; Goldenberg, D. Oral complications of radiotherapy. Lancet Oncol. 2006, 7, 175–183. [Google Scholar] [CrossRef]
- Whelan, T.J.; Pignol, J.P.; Levine, M.N.; Julian, J.A.; MacKenzie, R.; Parpia, S.; Shelley, W.; Grimard, L.; Bowen, J.; Lukka, H.; et al. Long-term results of hypofractionated radiation therapy for breast cancer. N. Engl. J. Med. 2010, 362, 513–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, V. Radiation pneumonitis and pulmonary fibrosis in non-small-cell lung cancer: Pulmonary function, prediction, and prevention. Int. J. Radiat. Oncol. Biol. Phys. 2005, 63, 5–24. [Google Scholar] [CrossRef] [PubMed]
- Turkkan, G.; Willems, Y.; Hendriks, L.E.L.; Mostard, R.; Conemans, L.; Gietema, H.A.; Mitea, C.; Peeters, S.; De Ruysscher, D. Idiopathic pulmonary fibrosis: Current knowledge, future perspectives and its importance in radiation oncology. Radiother. Oncol. 2021, 155, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Giaj-Levra, N.; Sciascia, S.; Fiorentino, A.; Fersino, S.; Mazzola, R.; Ricchetti, F.; Roccatello, D.; Alongi, F. Radiotherapy in patients with connective tissue diseases. Lancet Oncol. 2016, 17, e109–e117. [Google Scholar] [CrossRef]
- De Ruysscher, D.; Niedermann, G.; Burnet, N.G.; Siva, S.; Lee, A.W.M.; Hegi-Johnson, F. Radiotherapy toxicity. Nat. Rev. Dis. Primers 2019, 5, 13. [Google Scholar] [CrossRef]
- Hiniker, S.M.; Sodji, Q.; Quon, A.; Gutkin, P.M.; Arksey, N.; Graves, E.E.; Chin, F.T.; Maxim, P.G.; Diehn, M.; Loo, B.W., Jr. FLT-PET-CT for the Detection of Disease Recurrence After Stereotactic Ablative Radiotherapy or Hyperfractionation for Thoracic Malignancy: A Prospective Pilot Study. Front. Oncol. 2019, 9, 467. [Google Scholar] [CrossRef]
- Kartikasari, A.E.; Huertas, C.S.; Mitchell, A.; Plebanski, M. Tumor-Induced Inflammatory Cytokines and the Emerging Diagnostic Devices for Cancer Detection and Prognosis. Front. Oncol. 2021, 11, 692142. [Google Scholar] [CrossRef]
- Oft, M. IL-10: Master switch from tumor-promoting inflammation to antitumor immunity. Cancer Immunol. Res. 2014, 2, 194–199. [Google Scholar] [CrossRef] [Green Version]
- Francescone, R.; Hou, V.; Grivennikov, S.I. Cytokines, IBD, and colitis-associated cancer. Inflamm. Bowel Dis. 2015, 21, 409–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Kim, D.S.; Shim, T.S.; Lim, C.M.; Koh, Y.; Lee, S.D.; Kim, W.S.; Kim, W.D.; Lee, J.S.; Song, K.S. Lung cancer in patients with idiopathic pulmonary fibrosis. Eur. Respir. J. 2001, 17, 1216–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karampitsakos, T.; Tzilas, V.; Tringidou, R.; Steiropoulos, P.; Aidinis, V.; Papiris, S.A.; Bouros, D.; Tzouvelekis, A. Lung cancer in patients with idiopathic pulmonary fibrosis. Pulm. Pharm. 2017, 45, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Multhoff, G.; Molls, M.; Radons, J. Chronic inflammation in cancer development. Front. Immunol. 2011, 2, 98. [Google Scholar] [CrossRef] [Green Version]
- Pietras, E.M. Inflammation: A key regulator of hematopoietic stem cell fate in health and disease. Blood 2017, 130, 1693–1698. [Google Scholar] [CrossRef] [Green Version]
- Fabregat, I.; Caballero-Díaz, D. Transforming growth factor-β-induced cell plasticity in liver fibrosis and hepatocarcinogenesis. Front. Oncol. 2018, 8, 357. [Google Scholar] [CrossRef] [Green Version]
- Arrese, M.; Hernandez, A.; Astete, L.; Estrada, L.; Cabello-Verrugio, C.; Cabrera, D. TGF-beta and hepatocellular carcinoma: When a friend becomes an enemy. Curr. Protein Pept. Sci. 2018, 19, 1172–1179. [Google Scholar] [CrossRef]
- Vescovo, T.; Refolo, G.; Vitagliano, G.; Fimia, G.M.; Piacentini, M. Molecular mechanisms of hepatitis C virus-induced hepatocellular carcinoma. Clin. Microbiol. Infect. 2016, 22, 853–861. [Google Scholar] [CrossRef] [Green Version]
- Salih, B.A. Helicobacter pylori infection in developing countries: The burden for how long? Saudi J. Gastroenterol. 2009, 15, 201–207. [Google Scholar] [CrossRef]
- Wroblewski, L.E.; Peek, R.M., Jr.; Wilson, K.T. Helicobacter pylori and gastric cancer: Factors that modulate disease risk. Clin. Microbiol. Rev. 2010, 23, 713–739. [Google Scholar] [CrossRef] [Green Version]
- Teoh, C.M.; Tan, S.S.; Tran, T. Integrins as Therapeutic Targets for Respiratory Diseases. Curr. Mol. Med. 2015, 15, 714–734. [Google Scholar] [CrossRef] [PubMed]
- Phan, S.H. Biology of fibroblasts and myofibroblasts. Proc. Am. Thorac. Soc. 2008, 5, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Salton, F.; Volpe, M.C.; Confalonieri, M. Epithelial(-)Mesenchymal Transition in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Medicina 2019, 55, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valencia, J.C.; Egbukichi, N.; Erwin-Cohen, R.A. Autoimmunity and cancer, the paradox comorbidities challenging therapy in the context of preexisting autoimmunity. J. Interf. Cytokine Res. 2019, 39, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.; Kroger, C.J.; Tisch, R.M. Type 1 diabetes: A chronic anti-self-inflammatory response. Front. Immunol. 2017, 8, 1898. [Google Scholar] [CrossRef] [Green Version]
- Magruder, J.T.; Elahi, D.; Andersen, D.K. Diabetes and pancreatic cancer: Chicken or egg? Pancreas 2011, 40, 339–351. [Google Scholar] [CrossRef]
- Lakatos, P.L.; Lakatos, L. Risk for colorectal cancer in ulcerative colitis: Changes, causes and management strategies. World J. Gastroenterol. WJG 2008, 14, 3937. [Google Scholar] [CrossRef] [Green Version]
- Durnian, J.M.; Stewart, R.M.; Tatham, R.; Batterbury, M.; Kaye, S.B. Cyclosporin-A associated malignancy. Clin. Ophthalmol. 2007, 1, 421–430. [Google Scholar]
- Poniatowski, Ł.A.; Wojdasiewicz, P.; Gasik, R.; Szukiewicz, D. Transforming growth factor Beta family: Insight into the role of growth factors in regulation of fracture healing biology and potential clinical applications. Mediat. Inflamm. 2015, 2015, 137823. [Google Scholar] [CrossRef] [Green Version]
- Munger, J.S.; Sheppard, D. Cross talk among TGF-β signaling pathways, integrins, and the extracellular matrix. Cold Spring Harb. Perspect. Biol. 2011, 3, a005017. [Google Scholar] [CrossRef] [Green Version]
- Annes, J.P.; Chen, Y.; Munger, J.S.; Rifkin, D.B. Integrin αVβ6-mediated activation of latent TGF-β requires the latent TGF-β binding protein-1. J. Cell Biol. 2004, 165, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Lyons, R.M.; Keski-Oja, J.; Moses, H.L. Proteolytic activation of latent transforming growth factor-beta from fibroblast-conditioned medium. J. Cell Biol. 1988, 106, 1659–1665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Q.; Stamenkovic, I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000, 14, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Barcellos-Hoff, M.H.; Dix, T.A. Redox-mediated activation of latent transforming growth factor-beta 1. Mol. Endocrinol. 1996, 10, 1077–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raugi, G.J.; Olerud, J.E.; Gown, A.M. Thrombospondin in early human wound tissue. J. Investig. Dermatol. 1987, 89, 551–554. [Google Scholar] [CrossRef] [Green Version]
- Wipff, P.J.; Hinz, B. Integrins and the activation of latent transforming growth factor beta1—An intimate relationship. Eur. J. Cell Biol. 2008, 87, 601–615. [Google Scholar] [CrossRef]
- Verrecchia, F.; Mauviel, A. Transforming growth factor-beta signaling through the Smad pathway: Role in extracellular matrix gene expression and regulation. J. Investig. Dermatol. 2002, 118, 211–215. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.E. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, F.; ten Dijke, P. Signaling interplay between transforming growth factor-beta receptor and PI3K/AKT pathways in cancer. Trends Biochem. Sci. 2013, 38, 612–620. [Google Scholar] [CrossRef]
- Ota, T.; Fujii, M.; Sugizaki, T.; Ishii, M.; Miyazawa, K.; Aburatani, H.; Miyazono, K. Targets of transcriptional regulation by two distinct type I receptors for transforming growth factor-β in human umbilical vein endothelial cells. J. Cell. 2002, 193, 299–318. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, P.A.; McCarty, J.H. TGF-β Activation and Signaling in Angiogenesis. In Physiologic and Pathologic Angiogenesis-Signaling Mechanisms and Targeted Therapy; IntechOpen: Rijeka, Croatia, 2017; Available online: https://www.intechopen.com/chapters/53316 (accessed on 15 August 2021). [CrossRef] [Green Version]
- Lyden, D.; Young, A.Z.; Zagzag, D.; Yan, W.; Gerald, W.; O’Reilly, R.; Bader, B.L.; Hynes, R.O.; Zhuang, Y.; Manova, K. Id1 and Id3 are required for neurogenesis, angiogenesis and vascularization of tumour xenografts. Nature 1999, 401, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Akhurst, R.J.; Hata, A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef] [Green Version]
- Colak, S.; ten Dijke, P. Targeting TGF-β signaling in cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef]
- Stylianopoulos, T.; Jain, R.K. Combining two strategies to improve perfusion and drug delivery in solid tumors. Proc. Natl. Acad. Sci. USA 2013, 110, 18632–18637. [Google Scholar] [CrossRef] [Green Version]
- Lohr, M.; Schmidt, C.; Ringel, J.; Kluth, M.; Muller, P.; Nizze, H.; Jesnowski, R. Transforming growth factor-beta1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Res. 2001, 61, 550–555. [Google Scholar]
- Walker, R.A. The complexities of breast cancer desmoplasia. Breast Cancer Res 2001, 3, 143–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, C.; Zeldin, Y.; Baratz, M.E.; Kathju, S.; Satish, L. Investigating the effects of Pirfenidone on TGF-β1 stimulated non-SMAD signaling pathways in Dupuytren’s disease-derived fibroblasts. BMC Musculoskelet. Disord. 2019, 20, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hostettler, K.; Zhong, J.; Tamm, M.; Lardinois, D.; Roth, M. Effect of pirfenidone on TGF-β-induced pro-fibrotic effects in primary human lung cells derived from patients with idiopathic pulmonary fibrosis. Eur. Respir. J. 2014, 44. [Google Scholar]
- Miura, Y.; Saito, T.; Tanaka, T.; Takoi, H.; Yatagai, Y.; Inomata, M.; Nei, T.; Saito, Y.; Gemma, A.; Azuma, A. Reduced incidence of lung cancer in patients with idiopathic pulmonary fibrosis treated with pirfenidone. Respir. Investig. 2018, 56, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Orner, B.P.; Huang, T.; Hinck, A.P.; Kiessling, L.L. Peptide ligands that use a novel binding site to target both TGF-β receptors. Mol. BioSystems 2010, 6, 2392–2402. [Google Scholar] [CrossRef] [Green Version]
- Boswell, S.; Sharif, S.; Alisa, A.; Pereira, S.P.; Williams, R.; Behboudi, S. Induction of latency-associated peptide (transforming growth factor-β1) expression on CD4+ T cells reduces Toll-like receptor 4 ligand-induced tumour necrosis factor-α production in a transforming growth factor-β-dependent manner. Immunology 2011, 133, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Boye, A.; Kan, H.; Wu, C.; Jiang, Y.; Yang, X.; He, S.; Yang, Y. MAPK inhibitors differently modulate TGF-β/Smad signaling in HepG2 cells. Tumor Biol. 2015, 36, 3643–3651. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Shen, S.; Verma, I.M. NF-kappaB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [Green Version]
- Catz, S.D.; Johnson, J.L. Transcriptional regulation of bcl-2 by nuclear factor κB and its significance in prostate cancer. Oncogene 2001, 20, 7342–7351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micheau, O.; Lens, S.; Gaide, O.; Alevizopoulos, K.; Tschopp, J. NF-kappaB signals induce the expression of c-FLIP. Mol. Cell. Biol. 2001, 21, 5299–5305. [Google Scholar] [CrossRef] [Green Version]
- Xie, T.X.; Xia, Z.; Zhang, N.; Gong, W.; Huang, S. Constitutive NF-kappaB activity regulates the expression of VEGF and IL-8 and tumor angiogenesis of human glioblastoma. Oncol. Rep. 2010, 23, 725–732. [Google Scholar]
- Huber, M.A.; Azoitei, N.; Baumann, B.; Grunert, S.; Sommer, A.; Pehamberger, H.; Kraut, N.; Beug, H.; Wirth, T. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J. Clin. Investig. 2004, 114, 569–581. [Google Scholar] [CrossRef] [Green Version]
- Nan, J.; Du, Y.; Chen, X.; Bai, Q.; Wang, Y.; Zhang, X.; Zhu, N.; Zhang, J.; Hou, J.; Wang, Q.; et al. TPCA-1 is a direct dual inhibitor of STAT3 and NF-kappaB and regresses mutant EGFR-associated human non-small cell lung cancers. Mol. Cancer. Ther. 2014, 13, 617–629. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.H.; Roh, E.; Lee, H.Y.; Lee, I.J.; Ahn, B.; Jung, S.H.; Lee, H.; Han, S.B.; Kim, Y. Benzoxathiole derivative blocks lipopolysaccharide-induced nuclear factor-kappaB activation and nuclear factor-kappaB-regulated gene transcription through inactivating inhibitory kappaB kinase beta. Mol. Pharmacol. 2008, 73, 1309–1318. [Google Scholar] [CrossRef]
- Burke, J.R.; Pattoli, M.A.; Gregor, K.R.; Brassil, P.J.; MacMaster, J.F.; McIntyre, K.W.; Yang, X.; Iotzova, V.S.; Clarke, W.; Strnad, J.; et al. BMS-345541 is a highly selective inhibitor of I kappa B kinase that binds at an allosteric site of the enzyme and blocks NF-kappa B-dependent transcription in mice. J. Biol. Chem. 2003, 278, 1450–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, A.; Konno, M.; Muto, S.; Kambe, N.; Morii, E.; Nakahata, T.; Itai, A.; Matsuda, H. A novel NF-kappaB inhibitor, IMD-0354, suppresses neoplastic proliferation of human mast cells with constitutively activated c-kit receptors. Blood 2005, 105, 2324–2331. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Rhee, M.H.; Kim, E.; Cho, J.Y. BAY 11-7082 is a broad-spectrum inhibitor with anti-inflammatory activity against multiple targets. Mediat. Inflamm. 2012, 2012, 416036. [Google Scholar] [CrossRef] [PubMed]
- Saadane, A.; Masters, S.; DiDonato, J.; Li, J.; Berger, M. Parthenolide inhibits IkappaB kinase, NF-kappaB activation, and inflammatory response in cystic fibrosis cells and mice. Am. J. Respir. Cell Mol. Biol. 2007, 36, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, Z.H.; Wong, H.R.; Odoms, K.; Deitch, E.A.; Szabo, C.; Vizi, E.S.; Hasko, G. Proteasome inhibitors induce inhibitory kappa B (I kappa B) kinase activation, I kappa B alpha degradation, and nuclear factor kappa B activation in HT-29 cells. Mol. Pharmacol. 2004, 65, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Lecker, S.; Post, M.J.; Hietaranta, A.J.; Li, J.; Volk, R.; Li, M.; Sato, K.; Saluja, A.K.; Steer, M.L.; et al. Inhibition of ubiquitin-proteasome pathway-mediated I kappa B alpha degradation by a naturally occurring antibacterial peptide. J. Clin. Investig. 2000, 106, 439–448. [Google Scholar] [CrossRef]
- Kisselev, A.F.; Goldberg, A.L. Proteasome inhibitors: From research tools to drug candidates. Chem. Biol. 2001, 8, 739–758. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Peng, H.; Du, Q.; Lin, W.; Liu, Y. GYY4137, a hydrogen sulfidereleasing molecule, inhibits the inflammatory response by suppressing the activation of nuclear factorkappa B and mitogenactivated protein kinases in Coxsackie virus B3infected rat cardiomyocytes. Mol. Med. Rep. 2015, 11, 1837–1844. [Google Scholar] [CrossRef]
- Chen, K.M.; Spratt, T.E.; Stanley, B.A.; De Cotiis, D.A.; Bewley, M.C.; Flanagan, J.M.; Desai, D.; Das, A.; Fiala, E.S.; Amin, S.; et al. Inhibition of nuclear factor-kappaB DNA binding by organoselenocyanates through covalent modification of the p50 subunit. Cancer Res. 2007, 67, 10475–10483. [Google Scholar] [CrossRef] [Green Version]
- Miller, S.C.; Huang, R.; Sakamuru, S.; Shukla, S.J.; Attene-Ramos, M.S.; Shinn, P.; Van Leer, D.; Leister, W.; Austin, C.P.; Xia, M. Identification of known drugs that act as inhibitors of NF-κB signaling and their mechanism of action. Biochem. Pharmacol. 2010, 79, 1272–1280. [Google Scholar] [CrossRef] [Green Version]
- Aghai, Z.H.; Kumar, S.; Farhath, S.; Kumar, M.A.; Saslow, J.; Nakhla, T.; Eydelman, R.; Strande, L.; Stahl, G.; Hewitt, C. Dexamethasone suppresses expression of Nuclear Factor-kappaB in the cells of tracheobronchial lavage fluid in premature neonates with respiratory distress. Pediatric Res. 2006, 59, 811–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, Z.; Kampfrath, T.; Sun, Q.; Parthasarathy, S.; Rajagopalan, S. Evidence that α-lipoic acid inhibits NF-κB activation independent of its antioxidant function. Inflamm. Res. 2011, 60, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Kastrati, I.; Siklos, M.I.; Calderon-Gierszal, E.L.; El-Shennawy, L.; Georgieva, G.; Thayer, E.N.; Thatcher, G.R.; Frasor, J. Dimethyl fumarate inhibits the nuclear factor κB pathway in breast cancer cells by covalent modification of p65 protein. J. Biol. Chem. 2016, 291, 3639–3647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakshmanan, K.; Byran, G.; Bandlamudi, S.; Krishnamurthy, P.T. The Role of STAT3 Signaling in Different Types of Cancers: A Comprehensive Review. Curr. Enzym. Inhib. 2020, 16, 189–198. [Google Scholar] [CrossRef]
- Michalska, A.; Blaszczyk, K.; Wesoly, J.; Bluyssen, H.A. A positive feedback amplifier circuit that regulates interferon (IFN)-stimulated gene expression and controls type I and type II IFN responses. Front. Immunol. 2018, 9, 1135. [Google Scholar] [CrossRef] [Green Version]
- Steen, H.C.; Gamero, A.M. STAT2 phosphorylation and signaling. Jak-Stat. 2013, 2, e25790. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Cheung, S.T. STAT3: An emerging therapeutic target for hepatocellular carcinoma. Cancers 2019, 11, 1646. [Google Scholar] [CrossRef] [Green Version]
- Dutta, P.; Sabri, N.; Li, J.; Li, W.X. Role of STAT3 in lung cancer. Jak-Stat. 2014, 3, e999503. [Google Scholar] [CrossRef]
- Robinson, R.L.; Sharma, A.; Bai, S.; Heneidi, S.; Lee, T.J.; Kodeboyina, S.K.; Patel, N.; Sharma, S. Comparative STAT3-regulated gene expression profile in renal cell carcinoma subtypes. Front. Oncol. 2019, 9, 72. [Google Scholar] [CrossRef] [Green Version]
- Liang, R.; Chen, X.; Chen, L.; Wan, F.; Chen, K.; Sun, Y.; Zhu, X. STAT3 signaling in ovarian cancer: A potential therapeutic target. J. Cancer 2020, 11, 837. [Google Scholar] [CrossRef] [Green Version]
- Seffens, A.; Herrera, A.; Tegla, C.; Buus, T.B.; Hymes, K.B.; Ødum, N.; Geskin, L.J.; Koralov, S.B. STAT3 dysregulation in mature T and NK cell lymphomas. Cancers 2019, 11, 1711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.H.; Qin, L.; Li, X. Role of STAT3 signaling pathway in breast cancer. Cell Commun. Signal. 2020, 18, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Mai, H.; Peng, J.; Zhou, B.; Hou, J.; Jiang, D. STAT4: An immunoregulator contributing to diverse human diseases. Int. J. Biol. Sci. 2020, 16, 1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rädler, P.D.; Wehde, B.L.; Wagner, K.-U. Crosstalk between STAT5 activation and PI3K/AKT functions in normal and transformed mammary epithelial cells. Mol. Cell. Endocrinol. 2017, 451, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Delgoffe, G.M.; Vignali, D.A. STAT heterodimers in immunity: A mixed message or a unique signal? Jak-Stat 2013, 2, e23060. [Google Scholar] [CrossRef] [Green Version]
- Blease, K.; Schuh, J.M.; Jakubzick, C.; Lukacs, N.W.; Kunkel, S.L.; Joshi, B.H.; Puri, R.K.; Kaplan, M.H.; Hogaboam, C.M. Stat6-deficient mice develop airway hyperresponsiveness and peribronchial fibrosis during chronic fungal asthma. Am. J. Pathol. 2002, 160, 481–490. [Google Scholar] [CrossRef] [Green Version]
- Valladao, A.C.; Frevert, C.W.; Koch, L.K.; Campbell, D.J.; Ziegler, S.F. STAT6 regulates the development of eosinophilic versus neutrophilic asthma in response to Alternaria alternata. J. Immunol. 2016, 197, 4541–4551. [Google Scholar] [CrossRef] [Green Version]
- Walford, H.H.; Doherty, T.A. STAT6 and lung inflammation. Jak-stat 2013, 2, e25301. [Google Scholar] [CrossRef] [Green Version]
- Sherman, M.A. The role of STAT6 in mast cell IL-4 production. Immunol. Rev. 2001, 179, 48–56. [Google Scholar] [CrossRef]
- Song, T.L.; Nairismägi, M.-L.; Laurensia, Y.; Lim, J.-Q.; Tan, J.; Li, Z.-M.; Pang, W.-L.; Kizhakeyil, A.; Wijaya, G.-C.; Huang, D.-C. Oncogenic activation of the STAT3 pathway drives PD-L1 expression in natural killer/T-cell lymphoma. Blood J. Am. Soc. Hematol. 2018, 132, 1146–1158. [Google Scholar] [CrossRef] [Green Version]
- Teng, T.S.; Lin, B.; Manser, E.; Ng, D.C.H.; Cao, X. Stat3 promotes directional cell migration by regulating Rac1 activity via its activator βPIX. J. Cell Sci. 2009, 122, 4150–4159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Han, Z.C. STAT3: A critical transcription activator in angiogenesis. Med. Res. Rev. 2008, 28, 185–200. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ji, M.; Zhang, S.; Xue, N.; Xu, H.; Lin, S.; Chen, X. Bt354 as a new STAT3 signaling pathway inhibitor against triple negative breast cancer. J. Drug Target. 2018, 26, 920–930. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Yin, C.; Zhang, Y.; Guo, G.; Zhao, C.; Wang, O.; Xiang, Y.; Zhang, X.; Liang, G. Osthole inhibits triple negative breast cancer cells by suppressing STAT3. J. Exp. Clin. Cancer Res. 2018, 37, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yu, W.; Cai, G.; Zhu, J.; Zhang, C.; Li, S.; Guo, J.; Yin, G.; Chen, C.; Kong, L. A new synthetic derivative of cryptotanshinone KYZ3 as STAT3 inhibitor for triple-negative breast cancer therapy. Cell Death Dis. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sethi, G.; Chatterjee, S.; Rajendran, P.; Li, F.; Shanmugam, M.K.; Wong, K.F.; Kumar, A.P.; Senapati, P.; Behera, A.K.; Hui, K.M. Inhibition of STAT3 dimerization and acetylation by garcinol suppresses the growth of human hepatocellular carcinoma in vitro and in vivo. Mol. Cancer 2014, 13, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Lim, E.J.; Hong, D.Y.; Park, J.H.; Joung, Y.H.; Darvin, P.; Kim, S.Y.; Na, Y.M.; Hwang, T.S.; Ye, S.-K.; Moon, E.-S. Methylsulfonylmethane suppresses breast cancer growth by down-regulating STAT3 and STAT5b pathways. PLoS ONE 2012, 7, e33361. [Google Scholar] [CrossRef]
- Turkson, J.; Zhang, S.; Palmer, J.; Kay, H.; Stanko, J.; Mora, L.B.; Sebti, S.; Yu, H.; Jove, R. Inhibition of constitutive signal transducer and activator of transcription 3 activation by novel platinum complexes with potent antitumor activity. Mol. Cancer Ther. 2004, 3, 1533–1542. [Google Scholar]
- Huang, W.; Dong, Z.; Chen, Y.; Wang, F.; Wang, C.; Peng, H.; He, Y.; Hangoc, G.; Pollok, K.; Sandusky, G. Small-molecule inhibitors targeting the DNA-binding domain of STAT3 suppress tumor growth, metastasis and STAT3 target gene expression in vivo. Oncogene 2016, 35, 783–792. [Google Scholar] [CrossRef]
- Kang, D.Y.; Sp, N.; Kim, D.H.; Joung, Y.H.; Lee, H.G.; Park, Y.M.; Yang, Y.M. Salidroside inhibits migration, invasion and angiogenesis of MDA-MB 231 TNBC cells by regulating EGFR/Jak2/STAT3 signaling via MMP2. Int. J. Oncol. 2018, 53, 877–885. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Payero, B.; Taylor, S.; Oyelere, A.K. Discovery of novel STAT3 DNA binding domain inhibitors. Future Med. Chem. 2021, 13, 1253–1269. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, M.; Rosenberg, D.W. Multifaceted roles of PGE2 in inflammation and cancer. Semin. Immunopathol. 2013, 35, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Austin, S.C.; Rocca, B.; Koller, B.H.; Coffman, T.M.; Grosser, T.; Lawson, J.A.; FitzGerald, G.A. Role of prostacyclin in the cardiovascular response to thromboxane A2. Science 2002, 296, 539–541. [Google Scholar] [CrossRef] [PubMed]
- Dorris, S.L.; Peebles, R.S. PGI2 as a regulator of inflammatory diseases. Mediat. Inflamm. 2012, 2012, 926968. [Google Scholar] [CrossRef] [Green Version]
- Smyth, E.M. Thromboxane and the thromboxane receptor in cardiovascular disease. Clin. Lipidol. 2010, 5, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Schrör, K. Thromboxane A2 and platelets as mediators of coronary arterial vasoconstriction in myocardial ischaemia. Eur. Heart J. 1990, 11, 27–34. [Google Scholar] [CrossRef]
- Ohmori, M.; Kuzuya, T.; Kodama, K.; Nanto, S.; Kamada, T.; Tada, M. Thromboxane A2 as an enhancing factor of coronary vasospasticity in variant angina. Jpn. Circ. J. 1987, 51, 495–502. [Google Scholar] [CrossRef]
- Ekambaram, P.; Lambiv, W.; Cazzolli, R.; Ashton, A.W.; Honn, K.V. The thromboxane synthase and receptor signaling pathway in cancer: An emerging paradigm in cancer progression and metastasis. Cancer Metastasis Rev. 2011, 30, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Parida, S.; Pal, I.; Parekh, A.; Thakur, B.; Bharti, R.; Das, S.; Mandal, M. GW627368X inhibits proliferation and induces apoptosis in cervical cancer by interfering with EP4/EGFR interactive signaling. Cell Death Dis. 2016, 7, e2154. [Google Scholar] [CrossRef] [Green Version]
- Nakao, K.; Murase, A.; Ohshiro, H.; Okumura, T.; Taniguchi, K.; Murata, Y.; Masuda, M.; Kato, T.; Okumura, Y.; Takada, J. CJ-023,423, a novel, potent and selective prostaglandin EP4 receptor antagonist with antihyperalgesic properties. J. Pharmacol. Exp. Ther. 2007, 322, 686–694. [Google Scholar] [CrossRef]
- Brittain, R.; Boutal, L.; Carter, M.; Coleman, R.; Collington, E.; Geisow, H.; Hallett, P.; Hornby, E.; Humphrey, P.; Jack, D. AH23848: A thromboxane receptor-blocking drug that can clarify the pathophysiologic role of thromboxane A2. Circulation 1985, 72, 1208–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peskar, B.M. Role of cyclooxygenase isoforms in gastric mucosal defence. J. Physiol. 2001, 95, 3–9. [Google Scholar] [CrossRef]
- Gupta, R.A.; Tejada, L.V.; Tong, B.J.; Das, S.K.; Morrow, J.D.; Dey, S.K.; DuBois, R.N. Cyclooxygenase-1 is overexpressed and promotes angiogenic growth factor production in ovarian cancer. Cancer Res. 2003, 63, 906–911. [Google Scholar] [PubMed]
- Haakensen, V.D.; Bjøro, T.; Lüders, T.; Riis, M.; Bukholm, I.K.; Kristensen, V.N.; Troester, M.A.; Homen, M.M.; Ursin, G.; Børresen-Dale, A.-L. Serum estradiol levels associated with specific gene expression patterns in normal breast tissue and in breast carcinomas. BMC Cancer 2011, 11, 332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sales, K.J.; Katz, A.A.; Howard, B.; Soeters, R.P.; Millar, R.P.; Jabbour, H.N. Cyclooxygenase-1 is up-regulated in cervical carcinomas: Autocrine/paracrine regulation of cyclooxygenase-2, prostaglandin e receptors, and angiogenic factors by cyclooxygenase-1. Cancer Res. 2002, 62, 424–432. [Google Scholar] [PubMed]
- Harris, R.E.; Casto, B.C.; Harris, Z.M. Cyclooxygenase-2 and the inflammogenesis of breast cancer. World J. Clin. Oncol. 2014, 5, 677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirschenbaum, A.; Liu, X.-H.; Yao, S.; Levine, A.C. The role of cyclooxygenase-2 in prostate cancer. Urology 2001, 58, 127–131. [Google Scholar] [CrossRef]
- Chen, H.; Cai, W.; Chu, E.; Tang, J.; Wong, C.; Wong, S.; Sun, W.; Liang, Q.; Fang, J.; Sun, Z. Hepatic cyclooxygenase-2 overexpression induced spontaneous hepatocellular carcinoma formation in mice. Oncogene 2017, 36, 4415–4426. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Qu, L.; Yan, S. Cyclooxygenase-2 promotes tumor growth and suppresses tumor immunity. Cancer Cell Int. 2015, 15, 106. [Google Scholar] [CrossRef] [Green Version]
- Gately, S.; Li, W.W. Multiple roles of COX-2 in tumor angiogenesis: A target for antiangiogenic therapy. Semin. Oncol. 2004, 31, 2–11. [Google Scholar] [CrossRef]
- Guo, Z.; Jiang, J.-H.; Zhang, J.; Yang, H.-J.; Yang, F.-Q.; Qi, Y.-P.; Zhong, Y.-P.; Su, J.; Yang, R.-R.; Li, L.-Q. COX-2 promotes migration and invasion by the side population of cancer stem cell-like hepatocellular carcinoma cells. Medicine 2015, 94, e1806. [Google Scholar] [CrossRef] [PubMed]
- Puratchikody, A.; Umamaheswari, A.; Irfan, N.; Sinha, S.; Manju, S.; Ramanan, M.; Ramamoorthy, G.; Doble, M. A novel class of tyrosine derivatives as dual 5-LOX and COX-2/mPGES1 inhibitors with PGE 2 mediated anticancer properties. New J. Chem. 2019, 43, 834–846. [Google Scholar] [CrossRef]
- Henderson, W.R. The role of leukotrienes in inflammation. Ann. Intern. Med. 1994, 121, 684–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetzl, E.; Goldman, D.; Naccache, P.; Sha’Afi, R.; Pickett, W. Mediation of leukocyte components of inflammatory reactions by lipoxygenase products of arachidonic acid. Adv. Prostaglandin Thromboxane Leukot. Res. 1982, 9, 273–282. [Google Scholar]
- Wisastra, R.; Dekker, F.J. Inflammation, cancer and oxidative lipoxygenase activity are intimately linked. Cancers 2014, 6, 1500–1521. [Google Scholar] [CrossRef] [Green Version]
- Wasilewicz, M.P.; Kołodziej, B.; Bojułko, T.; Kaczmarczyk, M.; Sulżyc-Bielicka, V.; Bielicki, D.; Ciepiela, K. Overexpression of 5-lipoxygenase in sporadic colonic adenomas and a possible new aspect of colon carcinogenesis. Int. J. Colorectal Dis. 2010, 25, 1079–1085. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Grignon, D.J.; Chbihi, T.; Zacharek, A.; Chen, Y.Q.; Sakr, W.; Porter, A.T.; Crissman, J.D.; Pontes, J.E.; Powell, I.J. Elevated 12-lipoxygenase mRNA expression correlates with advanced stage and poor differentiation of human prostate cancer. Urology 1995, 46, 227–237. [Google Scholar] [CrossRef]
- Nithipatikom, K.; Isbell, M.A.; See, W.A.; Campbell, W.B. Elevated 12-and 20-hydroxyeicosatetraenoic acid in urine of patients with prostatic diseases. Cancer Lett. 2006, 233, 219–225. [Google Scholar] [CrossRef]
- Shrimanker, R.; Borg, K.; Connolly, C.; Thulborn, S.; Cane, J.; Xue, L.; Hynes, G.; Hinks, T.; Kots, M.; Girardello, L.; et al. Late Breaking Abstract-Effect of timapiprant, a DP2 antagonist, on airway inflammation in severe eosinophilic asthma. Eur. Respir. J. 2019, 54, RCT3784. [Google Scholar]
- Lai, Y.-J.; Pullamsetti, S.S.; Dony, E.; Weissmann, N.; Butrous, G.; Banat, G.-A.; Ghofrani, H.A.; Seeger, W.; Grimminger, F.; Schermuly, R.T. Role of the prostanoid EP4 receptor in iloprost-mediated vasodilatation in pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2008, 178, 188–196. [Google Scholar] [CrossRef] [Green Version]
- Bateman, E.D.; Guerreros, A.G.; Brockhaus, F.; Holzhauer, B.; Pethe, A.; Kay, R.A.; Townley, R.G. Fevipiprant, an oral prostaglandin DP2 receptor (CRTh2) antagonist, in allergic asthma uncontrolled on low-dose inhaled corticosteroids. Eur. Respir. J. 2017, 50, 1700670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, N.; Dehabadi, M.H.; Nair, S.; Quartilho, A.; Bunce, C.; Reekie, I.; Obikpo, R. Efficacy and safety of bimatoprost in glaucoma and ocular hypertension in non-responder patients. Int. J. Ophthalmol. 2017, 10, 1251. [Google Scholar] [PubMed]
- Ghlichloo, I.; Gerriets, V. Nonsteroidal Anti-Inflammatory Drugs (NSAIDs); StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Eliasson, O.; Densmore, M.J.; Scherzer, H.H.; DeGraff Jr, A.C. The effect of sodium meclofenamate in premenstrual asthma: A controlled clinical trial. J. Allergy Clin. Immunol. 1987, 79, 909–918. [Google Scholar] [CrossRef]
- Dahlen, B.; Nizankowska, E.; Szczeklik, A.; Zetterstrom, O.; Bochenek, G.; Kumlin, M.; Mastalerz, L.; Pinis, G.; Swanson, L.J.; Boodhoo, T.I.; et al. Benefits from adding the 5-lipoxygenase inhibitor zileuton to conventional therapy in aspirin-intolerant asthmatics. Am. J. Respir. Crit. Care Med. 1998, 157, 1187–1194. [Google Scholar] [CrossRef]
- Sester, A.; Winand, L.; Pace, S.; Hiller, W.; Werz, O.; Nett, M. Myxochelin-and Pseudochelin-Derived Lipoxygenase Inhibitors from a Genetically Engineered Myxococcus xanthus Strain. J. Nat. Prod. 2019, 82, 2544–2549. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Seto, E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witt, O.; Deubzer, H.E.; Milde, T.; Oehme, I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2009, 277, 8–21. [Google Scholar] [CrossRef]
- Liu, N.; He, S.; Ma, L.; Ponnusamy, M.; Tang, J.; Tolbert, E.; Bayliss, G.; Zhao, T.C.; Yan, H.; Zhuang, S. Blocking the class I histone deacetylase ameliorates renal fibrosis and inhibits renal fibroblast activation via modulating TGF-beta and EGFR signaling. PLoS ONE 2013, 8, e54001. [Google Scholar] [CrossRef]
- Zhu, H.; Shan, L.; Schiller, P.W.; Mai, A.; Peng, T. Histone deacetylase-3 activation promotes tumor necrosis factor-α (TNF-α) expression in cardiomyocytes during lipopolysaccharide stimulation. J. Biol. Chem. 2010, 285, 9429–9436. [Google Scholar] [CrossRef] [Green Version]
- Ziesche, E.; Kettner-Buhrow, D.; Weber, A.; Wittwer, T.; Jurida, L.; Soelch, J.; Müller, H.; Newel, D.; Kronich, P.; Schneider, H. The coactivator role of histone deacetylase 3 in IL-1-signaling involves deacetylation of p65 NF-κB. Nucleic Acids Res. 2013, 41, 90–109. [Google Scholar] [CrossRef] [Green Version]
- Durham, B.S.; Grigg, R.; Wood, I.C. Inhibition of histone deacetylase 1 or 2 reduces microglia activation through a gene expression independent mechanism. bioRxiv 2017, 107649. [Google Scholar] [CrossRef]
- Yoon, S.; Kang, G.; Eom, G.H. HDAC inhibitors: Therapeutic potential in fibrosis-associated human diseases. Int. J. Mol. Sci. 2019, 20, 1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barter, M.J.; Pybus, L.; Litherland, G.J.; Rowan, A.D.; Clark, I.M.; Edwards, D.R.; Cawston, T.E.; Young, D.A. HDAC-mediated control of ERK-and PI3K-dependent TGF-β-induced extracellular matrix-regulating genes. Matrix Biol. 2010, 29, 602–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.H.; Oh, S.W.; Kang, M.S.; Kwon, H.J.; Oh, G.T.; Kim, D.Y. Trichostatin A attenuates airway inflammation in mouse asthma model. Clin. Exp. Allergy 2005, 35, 89–96. [Google Scholar] [CrossRef]
- Cui, S.-N.; Chen, Z.-Y.; Yang, X.-B.; Chen, L.; Yang, Y.-Y.; Pan, S.-W.; Wang, Y.-X.; Xu, J.-Q.; Zhou, T.; Xiao, H.-R. Trichostatin A modulates the macrophage phenotype by enhancing autophagy to reduce inflammation during polymicrobial sepsis. Int. Immunopharmacol. 2019, 77, 105973. [Google Scholar] [CrossRef]
- Zhuang, S. Regulation of STAT signaling by acetylation. Cell. Signal. 2013, 25, 1924–1931. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Álvarez, A.; Llorente-Izquierdo, C.; Mayoral, R.; Agra, N.; Boscá, L.; Casado, M.; Martín-Sanz, P. Evaluation of epigenetic modulation of cyclooxygenase-2 as a prognostic marker for hepatocellular carcinoma. Oncogenesis 2012, 1, e23. [Google Scholar] [CrossRef] [Green Version]
- Gryder, B.E.; Sodji, Q.H.; Oyelere, A.K. Targeted cancer therapy: Giving histone deacetylase inhibitors all they need to succeed. Future Med. Chem. 2012, 4, 505–524. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Richon, V.; Ni, X.; Talpur, R.; Duvic, M. Selective induction of apoptosis by histone deacetylase inhibitor SAHA in cutaneous T-cell lymphoma cells: Relevance to mechanism of therapeutic action. J. Investig. Dermatol. 2005, 125, 1045–1052. [Google Scholar] [CrossRef] [Green Version]
- Rashidi, A.; Cashen, A.F. Belinostat for the treatment of relapsed or refractory peripheral T-cell lymphoma. Future Oncol. 2015, 11, 1659–1664. [Google Scholar] [CrossRef]
- Chan, T.S.; Tse, E.; Kwong, Y.-L. Chidamide in the treatment of peripheral T-cell lymphoma. OncoTargets Ther. 2017, 10, 347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, C.; Rahman, F.; Piekarz, R.; Peer, C.; Frye, R.; Robey, R.W.; Gardner, E.R.; Figg, W.D.; Bates, S.E. Romidepsin: A new therapy for cutaneous T-cell lymphoma and a potential therapy for solid tumors. Expert Rev. Anticancer. Ther. 2010, 10, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Raedler, L.A. Farydak (Panobinostat): First HDAC inhibitor approved for patients with relapsed multiple myeloma. Am. Health Drug Benefits 2016, 9, 84. [Google Scholar] [PubMed]
- Zhang, L.; Zhang, J.; Jiang, Q.; Zhang, L.; Song, W. Zinc binding groups for histone deacetylase inhibitors. J. Enzym. Inhib. Med. Chem. 2018, 33, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Ononye, S.N.; VanHeyst, M.D.; Oblak, E.Z.; Zhou, W.; Ammar, M.; Anderson, A.C.; Wright, D.L. Tropolones as lead-like natural products: The development of potent and selective histone deacetylase inhibitors. ACS Med. Chem. Lett. 2013, 4, 757–761. [Google Scholar] [CrossRef] [Green Version]
- Godoy, L.D.; Lucas, J.E.; Bender, A.J.; Romanick, S.S.; Ferguson, B.S. Targeting the epigenome: Screening bioactive compounds that regulate histone deacetylase activity. Mol. Nutr. Food Res. 2017, 61, 1600744. [Google Scholar] [CrossRef] [Green Version]
- Patil, V.; Sodji, Q.H.; Kornacki, J.R.; Mrksich, M.; Oyelere, A.K. 3-Hydroxypyridin-2-thione as novel zinc binding group for selective histone deacetylase inhibition. J. Med. Chem. 2013, 56, 3492–3506. [Google Scholar] [CrossRef] [Green Version]
- Sodji, Q.H.; Patil, V.; Kornacki, J.R.; Mrksich, M.; Oyelere, A.K. Synthesis and structure–activity relationship of 3-hydroxypyridine-2-thione-based histone deacetylase inhibitors. J. Med. Chem. 2013, 56, 9969–9981. [Google Scholar] [CrossRef]
- Methot, J.L.; Chakravarty, P.K.; Chenard, M.; Close, J.; Cruz, J.C.; Dahlberg, W.K.; Fleming, J.; Hamblett, C.L.; Hamill, J.E.; Harrington, P. Exploration of the internal cavity of histone deacetylase (HDAC) with selective HDAC1/HDAC2 inhibitors (SHI-1: 2). Bioorganic Med. Chem. Lett. 2008, 18, 973–978. [Google Scholar] [CrossRef]
- Li, Y.; Wang, F.; Chen, X.; Wang, J.; Zhao, Y.; Li, Y.; He, B. Zinc-dependent deacetylase (HDAC) inhibitors with different zinc binding groups. Curr. Top. Med. Chem. 2019, 19, 223–241. [Google Scholar] [CrossRef]
- Greenwood, S.O.; Chan, A.E.; Hansen, D.F.; Marson, C.M. Potent non-hydroxamate inhibitors of histone deacetylase-8: Role and scope of an isoindolin-2-yl linker with an α-amino amide as the zinc-binding unit. Bioorganic Med. Chem. Lett. 2020, 30, 126926. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, R.; Redente, E.F.; Thakur, A.; Riches, D.W.; Kompella, U.B. Local delivery of biodegradable pirfenidone nanoparticles ameliorates bleomycin-induced pulmonary fibrosis in mice. Nanotechnology 2012, 23, 505101. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zheng, L.; Yuan, Q.; Zhen, G.; Crane, J.L.; Zhou, X.; Cao, X. Transforming growth factor-β in stem cells and tissue homeostasis. Bone Res. 2018, 6, 1–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takai, E.; Tsukimoto, M.; Kojima, S. TGF-β1 downregulates COX-2 expression leading to decrease of PGE2 production in human lung cancer A549 cells, which is involved in fibrotic response to TGF-β1. PLoS ONE 2013, 8, e76346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderton, M.J.; Mellor, H.R.; Bell, A.; Sadler, C.; Pass, M.; Powell, S.; Steele, S.J.; Roberts, R.R.; Heier, A. Induction of heart valve lesions by small-molecule ALK5 inhibitors. Toxicol. Pathol. 2011, 39, 916–924. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, A.F.; Ten Dijke, P.; Zhu, H.-J. On-Target Anti-TGF-β Therapies Are Not Succeeding in Clinical Cancer Treatments: What Are Remaining Challenges? Front. Cell Dev. Biol. 2020, 8, 605. [Google Scholar] [CrossRef]
- Fisher, M.; Nathan, S.D.; Hill, C.; Marshall, J.; Dejonckheere, F.; Thuresson, P.-O.; Maher, T.M. Predicting life expectancy for pirfenidone in idiopathic pulmonary fibrosis. J. Manag. Care Spec. Pharm. 2017, 23, S17–S24. [Google Scholar] [CrossRef]
- Jiang, J.-H.; Deng, P. Discovery of new inhibitors of transforming growth factor-beta type 1 receptor by utilizing docking and structure-activity relationship analysis. Int. J. Mol. Sci. 2019, 20, 4090. [Google Scholar] [CrossRef] [Green Version]
- Inman, G.J.; Nicolás, F.J.; Callahan, J.F.; Harling, J.D.; Gaster, L.M.; Reith, A.D.; Laping, N.J.; Hill, C.S. SB-431542 is a potent and specific inhibitor of transforming growth factor-β superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 2002, 62, 65–74. [Google Scholar] [CrossRef]
- Melisi, D.; Ishiyama, S.; Sclabas, G.M.; Fleming, J.B.; Xia, Q.; Tortora, G.; Abbruzzese, J.L.; Chiao, P.J. LY2109761, a novel transforming growth factor β receptor type I and type II dual inhibitor, as a therapeutic approach to suppressing pancreatic cancer metastasis. Mol. Cancer Ther. 2008, 7, 829–840. [Google Scholar] [CrossRef] [Green Version]
- Herbertz, S.; Sawyer, J.S.; Stauber, A.J.; Gueorguieva, I.; Driscoll, K.E.; Estrem, S.T.; Cleverly, A.L.; Desaiah, D.; Guba, S.C.; Benhadji, K.A. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des. Dev. Ther. 2015, 9, 4479. [Google Scholar]
- Tauriello, D.B.; Daniel, M.M.; Joan, A.; Batlle, G.; Riera Escalé, E.A. TGFβ Inhibitor and Prodrugs. International Patent Application No. PCT/EP2019/082215; WO/2020/104648, 28 May 2020. [Google Scholar]
- Wu, B. Discovery of Novel Drugs with Tissue and Cell-Type Selectivity for Cancer and Inflammation. Ph.D. Thesis, Georgia Institute of Technology, Atlanta, GA, USA, 26 April 2021. [Google Scholar]
- Zeligs, K.P.; Neuman, M.K.; Annunziata, C.M. Molecular pathways: The balance between cancer and the immune system challenges the therapeutic specificity of targeting nuclear factor-κB signaling for cancer treatment. Clin. Cancer Res. 2016, 22, 4302–4308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, M.A.; Power, D.G.; Kindler, H.L.; Holen, K.D.; Kemeny, M.M.; Ilson, D.H.; Tang, L.; Capanu, M.; Wright, J.J.; Kelsen, D.P. A multicenter, phase II study of bortezomib (PS-341) in patients with unresectable or metastatic gastric and gastroesophageal junction adenocarcinoma. Investig. New Drugs 2011, 29, 1475–1481. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, L.; Hsu, S.C.; Davidson, B.; Birrer, M.J.; Kohn, E.C.; Annunziata, C.M. Activation of NF-κB signaling by inhibitor of NF-κB kinase β increases aggressiveness of ovarian cancer. Cancer Res. 2010, 70, 4005–4014. [Google Scholar] [CrossRef] [Green Version]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef] [Green Version]
- Linker, R.A.; Lee, D.-H.; Ryan, S.; van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011, 134, 678–692. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, A.; Kang, M.-I.; Watai, Y.; Tong, K.I.; Shibata, T.; Uchida, K.; Yamamoto, M. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol. Cell. Biol. 2006, 26, 221–229. [Google Scholar] [CrossRef] [Green Version]
- Saidu, N.E.B.; Kavian, N.; Leroy, K.; Jacob, C.; Nicco, C.; Batteux, F.; Alexandre, J. Dimethyl fumarate, a two-edged drug: Current status and future directions. Med. Res. Rev. 2019, 39, 1923–1952. [Google Scholar] [CrossRef]
- Choi, B.-M.; Kim, S.-M.; Park, T.-K.; Li, G.; Hong, S.-J.; Park, R.; Chung, H.-T.; Kim, B.-R. Piperine protects cisplatin-induced apoptosis via heme oxygenase-1 induction in auditory cells. J. Nutr. Biochem. 2007, 18, 615–622. [Google Scholar] [CrossRef]
- Linker, R.A.; Gold, R. Dimethyl fumarate for treatment of multiple sclerosis: Mechanism of action, effectiveness, and side effects. Curr. Neurol. Neurosci. Rep. 2013, 13, 1–7. [Google Scholar] [CrossRef]
- Yan, H.; Duan, X.; Pan, H.; Holguin, N.; Rai, M.F.; Akk, A.; Springer, L.E.; Wickline, S.A.; Sandell, L.J.; Pham, C.T. Suppression of NF-κB activity via nanoparticle-based siRNA delivery alters early cartilage responses to injury. Procl. Natl. Acad. Sci. USA 2016, 113, E6199–E6208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, M.; Salisbury, R.L.; Maurer, E.I.; Hussain, S.M.; Sulentic, C.E. Gold nanoparticles induce transcriptional activity of NF-κB in a B-lymphocyte cell line. Nanoscale 2013, 5, 3747–3756. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Khan, M.J. Nano-gold displayed anti-inflammatory property via NF-kB pathways by suppressing COX-2 activity. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1149–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-P.; Chen, C.-T.; Liu, T.-P.; Chien, F.-C.; Wu, S.-H.; Chen, P.; Mou, C.-Y. Catcher in the rel: Nanoparticles-antibody conjugate as NF-κB nuclear translocation blocker. Biomaterials 2020, 246, 119997. [Google Scholar] [CrossRef]
- Li, W.; Cao, Y.; Xu, J.; Wang, Y.; Li, W.; Wang, Q.; Hu, Z.; Hao, Y.; Hu, L.; Sun, Y. YAP transcriptionally regulates COX-2 expression and GCCSysm-4 (G-4), a dual YAP/COX-2 inhibitor, overcomes drug resistance in colorectal cancer. J. Exp. Clin. Cancer Res. 2017, 36, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.L.; Fan, Z.Q.; Jiang, H.D.; Qu, J.M. Selective Cox-2 inhibitor celecoxib induces epithelial-mesenchymal transition in human lung cancer cells via activating MEK-ERK signaling. Carcinogenesis 2013, 34, 638–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, C.T.; Dannenberg, A.J.; Subbaramaiah, K.; Dickler, M.N.; Moasser, M.M.; Seidman, A.D.; D’Andrea, G.M.; Theodoulou, M.; Panageas, K.S.; Norton, L. Phase II study of celecoxib and trastuzumab in metastatic breast cancer patients who have progressed after prior trastuzumab-based treatments. Clin. Cancer Res. 2004, 10, 4062–4067. [Google Scholar] [CrossRef] [Green Version]
- Solomon, S.D.; McMurray, J.J.; Pfeffer, M.A.; Wittes, J.; Fowler, R.; Finn, P.; Anderson, W.F.; Zauber, A.; Hawk, E.; Bertagnolli, M. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N. Engl. J. Med. 2005, 352, 1071–1080. [Google Scholar] [CrossRef] [Green Version]
- Caldwell, B.; Aldington, S.; Weatherall, M.; Shirtcliffe, P.; Beasley, R. Risk of cardiovascular events and celecoxib: A systematic review and meta-analysis. J. R. Soc. Med. 2006, 99, 132–140. [Google Scholar] [CrossRef]
- Chavez, M.L.; DeKorte, C.J. Valdecoxib: A review. Clin. Ther. 2003, 25, 817–851. [Google Scholar] [CrossRef]
- Perrone, M.G.; Scilimati, A.; Simone, L.; Vitale, P. Selective COX-1 inhibition: A therapeutic target to be reconsidered. Curr. Med. Chem. 2010, 17, 3769–3805. [Google Scholar] [CrossRef] [PubMed]
- Brune, K. Safety of anti-inflammatory treatment—New ways of thinking. Rheumatology 2004, 43, i16–i20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, A.; Yang, Z.; Shen, Y.; Zhou, J.; Shen, Q. Transcription factor STAT3 as a novel molecular target for cancer prevention. Cancers 2014, 6, 926–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.-H.; Vakili, M.R.; Molavi, O.; Morrissey, Y.; Wu, C.; Paiva, I.; Soleimani, A.H.; Sanaee, F.; Lavasanifar, A.; Lai, R. Decoration of anti-CD38 on nanoparticles carrying a STAT3 inhibitor can improve the therapeutic efficacy against myeloma. Cancers 2019, 11, 248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doheny, D.; Sirkisoon, S.; Carpenter, R.L.; Aguayo, N.R.; Regua, A.T.; Anguelov, M.; Manore, S.G.; Arrigo, A.; Jalboush, S.A.; Wong, G.L. Combined inhibition of JAK2-STAT3 and SMO-GLI1/tGLI1 pathways suppresses breast cancer stem cells, tumor growth, and metastasis. Oncogene 2020, 39, 6589–6605. [Google Scholar] [CrossRef]
- Zerdes, I.; Wallerius, M.; Sifakis, E.G.; Wallmann, T.; Betts, S.; Bartish, M.; Tsesmetzis, N.; Tobin, N.P.; Coucoravas, C.; Bergh, J. STAT3 activity promotes programmed-death ligand 1 expression and suppresses immune responses in breast cancer. Cancers 2019, 11, 1479. [Google Scholar] [CrossRef] [Green Version]
- Ashizawa, T.; Iizuka, A.; Maeda, C.; Tanaka, E.; Kondou, R.; Miyata, H.; Sugino, T.; Kawata, T.; Deguchi, S.; Mitsuya, K. Impact of combination therapy with anti-PD-1 blockade and a STAT3 inhibitor on the tumor-infiltrating lymphocyte status. Immunol. Lett. 2019, 216, 43–50. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Janus kinase (JAK) inhibitors in the treatment of inflammatory and neoplastic diseases. Pharmacol. Res. 2016, 111, 784–803. [Google Scholar] [CrossRef]
- Nezamololama, N.; Fieldhouse, K.; Metzger, K.; Gooderham, M. Emerging systemic JAK inhibitors in the treatment of atopic dermatitis: A review of abrocitinib, baricitinib, and upadacitinib. Drugs Context 2020, 9. [Google Scholar] [CrossRef]
- Perry, A.S.; Watson, R.W.G.; Lawler, M.; Hollywood, D. The epigenome as a therapeutic target in prostate cancer. Nat. Rev. Urol. 2010, 7, 668. [Google Scholar] [CrossRef]
- Freese, K.; Seitz, T.; Dietrich, P.; Lee, S.M.; Thasler, W.E.; Bosserhoff, A.; Hellerbrand, C. Histone deacetylase expressions in hepatocellular carcinoma and functional effects of histone deacetylase inhibitors on liver cancer cells in vitro. Cancers 2019, 11, 1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coradini, D.; Zorzet, S.; Rossin, R.; Scarlata, I.; Pellizzaro, C.; Turrin, C.; Bello, M.; Cantoni, S.; Speranza, A.; Sava, G. Inhibition of hepatocellular carcinomas in vitro and hepatic metastases in vivo in mice by the histone deacetylase inhibitor HA-But. Clin. Cancer Res. 2004, 10, 4822–4830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svechnikova, I.; Gray, S.G.; Kundrotiene, J.; Ponthan, F.; Kogner, P.; Ekström, T.J. Apoptosis and tumor remission in liver tumor xenografts by 4-phenylbutyrate. Int. J. Oncol. 2003, 22, 579–588. [Google Scholar] [PubMed]
- Tapadar, S.; Fathi, S.; Wu, B.; Sun, C.Q.; Raji, I.; Moore, S.G.; Arnold, R.S.; Gaul, D.A.; Petros, J.A.; Oyelere, A.K. Liver-targeting class I selective histone deacetylase inhibitors potently suppress hepatocellular tumor growth as standalone agents. Cancers 2020, 12, 3095. [Google Scholar] [CrossRef]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination therapy with histone deacetylase inhibitors (HDACi) for the treatment of cancer: Achieving the full therapeutic potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef] [Green Version]
- Conte, M.; De Palma, R.; Altucci, L. HDAC inhibitors as epigenetic regulators for cancer immunotherapy. Int. J. Biochem. Cell Biol. 2018, 98, 65–74. [Google Scholar] [CrossRef]
- Kroesen, M.; Gielen, P.; Brok, I.C.; Armandari, I.; Hoogerbrugge, P.M.; Adema, G.J. HDAC inhibitors and immunotherapy; a double edged sword? Oncotarget 2014, 5, 6558. [Google Scholar] [CrossRef] [Green Version]
- Bae, J.; Hideshima, T.; Tai, Y.-T.; Song, Y.; Richardson, P.; Raje, N.; Munshi, N.C.; Anderson, K.C. Histone deacetylase (HDAC) inhibitor ACY241 enhances anti-tumor activities of antigen-specific central memory cytotoxic T lymphocytes against multiple myeloma and solid tumors. Leukemia 2018, 32, 1932–1947. [Google Scholar] [CrossRef]
- Briere, D.; Sudhakar, N.; Woods, D.M.; Hallin, J.; Engstrom, L.D.; Aranda, R.; Chiang, H.; Sodré, A.L.; Olson, P.; Weber, J.S. The class I/IV HDAC inhibitor mocetinostat increases tumor antigen presentation, decreases immune suppressive cell types and augments checkpoint inhibitor therapy. Cancer Immunol. Immunother. 2018, 67, 381–392. [Google Scholar] [CrossRef]
- Woods, D.M.; Sodré, A.L.; Villagra, A.; Sarnaik, A.; Sotomayor, E.M.; Weber, J. HDAC inhibition upregulates PD-1 ligands in melanoma and augments immunotherapy with PD-1 blockade. Cancer Immunol. Res. 2015, 3, 1375–1385. [Google Scholar] [CrossRef] [Green Version]
- Chung, Y.-L.; Lee, M.-Y.; Wang, A.-J.; Yao, L.-F. A therapeutic strategy uses histone deacetylase inhibitors to modulate the expression of genes involved in the pathogenesis of rheumatoid arthritis. Mol. Ther. 2003, 8, 707–717. [Google Scholar] [CrossRef]
- Lyu, X.; Hu, M.; Peng, J.; Zhang, X.; Sanders, Y.Y. HDAC inhibitors as antifibrotic drugs in cardiac and pulmonary fibrosis. Ther. Adv. Chronic Dis. 2019, 10, 2040622319862697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adcock, I. HDAC inhibitors as anti-inflammatory agents. Br. J. Pharmacol. 2007, 150, 829–831. [Google Scholar] [CrossRef] [PubMed]
- Iwata, K.; Tomita, K.; Sano, H.; Fujii, Y.; Yamasaki, A.; Shimizu, E. Trichostatin A, a histone deacetylase inhibitor, down-regulates interleukin-12 transcription in SV-40-transformed lung epithelial cells. Cell. Immunol. 2002, 218, 26–33. [Google Scholar] [CrossRef]
- Weiss, U.; Möller, M.; Husseini, S.A.; Manderscheid, C.; Häusler, J.; Geisslinger, G.; Niederberger, E. Inhibition of HDAC Enzymes Contributes to Differential Expression of Pro-Inflammatory Proteins in the TLR-4 Signaling Cascade. Int. J. Mol. Sci. 2020, 21, 8943. [Google Scholar] [CrossRef] [PubMed]
- Lau, M.-T.; Wong, A.S.; Leung, P.C. Gonadotropins induce tumor cell migration and invasion by increasing cyclooxygenases expression and prostaglandin E2 production in human ovarian cancer cells. Endocrinology 2010, 151, 2985–2993. [Google Scholar] [CrossRef] [Green Version]
- Scilimati, A.; Ferorelli, S.; Iaselli, M.C.; Miciaccia, M.; Pati, M.L.; Fortuna, C.G.; Aleem, A.M.; Marnett, L.J.; Perrone, M.G. Targeting COX-1 by mofezolac-based fluorescent probes for ovarian cancer detection. Eur. J. Med. Chem. 2019, 179, 16–25. [Google Scholar] [CrossRef]
- Koromilas, A.E.; Sexl, V. The tumor suppressor function of STAT1 in breast cancer. Jak-Stat. 2013, 2, e23353. [Google Scholar] [CrossRef]
- Liu, S.; Imani, S.; Deng, Y.; Pathak, J.L.; Wen, Q.; Chen, Y.; Wu, J. Targeting IFN/STAT1 Pathway as a Promising Strategy to Overcome Radioresistance. OncoTargets Ther. 2020, 13, 6037. [Google Scholar] [CrossRef]
- Hou, Y.; Li, X.; Li, Q.; Xu, J.; Yang, H.; Xue, M.; Niu, G.; Zhuo, S.; Mu, K.; Wu, G. STAT 1 facilitates oestrogen receptor α transcription and stimulates breast cancer cell proliferation. J. Cell. Mol. Med. 2018, 22, 6077–6086. [Google Scholar] [CrossRef]
- Tian, X.; Guan, W.; Zhang, L.; Sun, W.; Zhou, D.; Lin, Q.; Ren, W.; Nadeem, L.; Xu, G. Physical interaction of STAT1 isoforms with TGF-β receptors leads to functional crosstalk between two signaling pathways in epithelial ovarian cancer. J. Exp. Clin. Cancer Res. 2018, 37, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inflammation Conditions | Cancern Type | Inducer |
|---|---|---|
| Asbestosis | Lung carcinoma | Silica |
| Chronic bronchitis | Lung carcinoma | Silica |
| IPF | Lung carcinoma | Unclear |
| Tuberculosis | Lung carcinoma | Mycobacterium tuberculosis |
| Liver cirrhosis | HCC | Hepatitis infection, alcoholic, genetic |
| IBD, Crohn’s disease, chronic ulcerative colitis | Colorectal cancer | Gut pathogens |
| Chronic gastric inflammation | Gastric cancer | Helicobacter pylori |
| Reflux oesophagitis, Barrett’s oesophagus | Oesophageal carcinoma | Gastric acids |
| Skin inflammation | Melanoma | UV light |
| Chronic pancreatitis, hereditary pancreatitis | Pancreatic carcinoma | Alcohol, gene mutation |
| Schistosomiasis | Bladder carcinoma | Gram-uropathogens |
| Cervicitis | Cervical cancer | Human papilloma virus |
| Chronic prostatitis | Prostate cancer | Bacterial infection |
| Sialadenitis | Salivary gland carcinoma | Bacterial infection |
| Sjögrensyndrome, Hashimoto’s thyroiditis | MALT lymphoma | unclear |
| Gingivitis, lichen planus | Oral squamous cell carcinoma | Bacterial infection |
| Chronic cholecystitis | Gall bladder cancer | Bacteria, gall bladder stones |
| STAT Protein Types | Stimulators (Inflammation) | Stimulators (Anti-Inflammation) | Heterodimerization |
|---|---|---|---|
| STAT1 | Type I IFN Type II IFN IL-6 | IL-10 IL-27 IL-35 | STAT2 STAT3 STAT4 |
| STAT2 | Type I IFN | STAT1 STAT6 | |
| STAT3 | IL-2 IL-5 IL-6 IL-23 MCSF GCSF Type-II IFN | IL-10 IL-27 | STAT1 STAT4 STAT5 |
| STAT4 | IL-12 IL-23 | IL-35 | STAT1 STAT3 |
| STAT5 | IL-2, IL-9, IL-15 IL-21 MCSF GCSF | STAT3 | |
| STAT6 | Type I IFN IL-3, IL-4, IL-13 | STAT2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, B.; Sodji, Q.H.; Oyelere, A.K. Inflammation, Fibrosis and Cancer: Mechanisms, Therapeutic Options and Challenges. Cancers 2022, 14, 552. https://doi.org/10.3390/cancers14030552
Wu B, Sodji QH, Oyelere AK. Inflammation, Fibrosis and Cancer: Mechanisms, Therapeutic Options and Challenges. Cancers. 2022; 14(3):552. https://doi.org/10.3390/cancers14030552
Chicago/Turabian StyleWu, Bocheng, Quaovi H. Sodji, and Adegboyega K. Oyelere. 2022. "Inflammation, Fibrosis and Cancer: Mechanisms, Therapeutic Options and Challenges" Cancers 14, no. 3: 552. https://doi.org/10.3390/cancers14030552
APA StyleWu, B., Sodji, Q. H., & Oyelere, A. K. (2022). Inflammation, Fibrosis and Cancer: Mechanisms, Therapeutic Options and Challenges. Cancers, 14(3), 552. https://doi.org/10.3390/cancers14030552