
Expression of CTLA-4 and CD86 Antigens and Epstein-Barr Virus Reactivation in Chronic Lymphocytic Leukemia—Any Link with Known Prognostic Factors?

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Preparation of Material
2.3. Immunophenotyping
2.4. Leukemia Cell Genotyping
2.5. DNA Isolation and Calculation of EBV Load
2.6. Measurement of EBV-Specific Antibodies in Serum
2.7. Measurement of Lactic Dehydrogenase and Beta-2 Microglobulin Concentrations in Serum
2.8. Statistical Analysis
3. Results
4. Discussion
5. Limitations of the Study
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rozman, C.; Montserrat, E. Chronic Lymphocytic Leukemia. N. Engl. J. Med. 1995, 333, 1052–1057. [Google Scholar] [CrossRef] [Green Version]
- Ghia, P.; Ferreri, A.M.; Galigaris-Cappio, F. Chronic Lymphocytic Leukemia. Crit. Rev. Oncol. Hematol. 2007, 64, 234–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulte, D.; Castro, F.A.; Jansen, L.; Luttmann, S.; Holleczek, B.; Nennecke, A.; Ressing, M.; Katalinic, A.; Brenner, H. Trends in Survival of Chronic Lymphocytic Leukemia Patients in Germany and the USA in the First Decade of the Twenty-First Century. J. Hematol. Oncol. 2016, 9, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghia, P.; Hallek, M. Management of Chronic Lymphocytic Leukemia. Haematologica 2014, 99, 965–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Küppers, R. B Cells under Influence: Transformation of B Cells by Epstein-Barr Virus. Nat. Rev. Immunol. 2003, 3, 801–812. [Google Scholar] [CrossRef]
- De Roos, A.J.; Martinez-Maza, O.; Jerome, K.R.; Mirick, D.K.; Kopecky, K.J.; Madeleine, M.M.; Magpantay, L.; Edlefsen, K.L.; LaCroix, A.Z. Investigation of Epstein-Barr Virus as a Potential Cause of b-Cell Non-Hodgkin Lymphoma in a Prospective Cohort. Cancer Epidemiol. Biomark. Prev. 2013, 22, 1747–1755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, N.; Falster, M.O.; Vajdic, C.M.; de Sanjose, S.; Martínez-Maza, O.; Bracci, P.M.; Melbye, M.; Smedby, K.E.; Engels, E.A.; Turner, J.; et al. Self-Reported History of Infections and the Risk of Non-Hodgkin Lymphoma: An InterLymph Pooled Analysis. Int. J. Cancer 2012, 131, 2342–2348. [Google Scholar] [CrossRef] [Green Version]
- García-Barchino, M.J.; Sarasquete, M.E.; Panizo, C.; Morscio, J.; Martinez, A.; Alcoceba, M.; Fresquet, V.; Gonzalez-Farre, B.; Paiva, B.; Young, K.H.; et al. Richter Transformation Driven by Epstein–Barr Virus Reactivation during Therapy-Related Immunosuppression in Chronic Lymphocytic Leukemia. J. Pathol. 2018, 245, 61–73. [Google Scholar] [CrossRef]
- Giussani, E.; Binatti, A.; Calabretto, G.; Gasparini, V.R.; Teramo, A.; Vicenzetto, C.; Barilà, G.; Facco, M.; Coppe, A.; Semenzato, G.; et al. Lack of Viral Load Within Chronic Lymphoproliferative Disorder of Natural Killer Cells: What Is Outside the Leukemic Clone? Front. Oncol. 2021, 10, 3064. [Google Scholar] [CrossRef]
- Zambello, R.; Loughran, T.P.; Trentin, L.; Rassu, M.; Facco, M.; Bortolin, M.; Nash, R.; Agostini, C.; Semenzato, G. Spontaneous Resolution of P58/EB6 Antigen Restricted NK-Type Lymphoproliferative Disease of Granular Lymphocytes: Role of Epstein Barr Virus Infection. Br. J. Haematol. 1997, 99, 215–221. [Google Scholar] [CrossRef]
- Grywalska, E.; Roliński, J.; Pasiarski, M.; Korona-Glowniak, I.; Maj, M.; Surdacka, A.; Grafka, A.; Stelmach-Gołdyś, A.; Zgurski, M.; Góźdź, S.; et al. High Viral Loads of Epstein-Barr Virus DNA in Peripheral Blood of Patients with Chronic Lymphocytic Leukemia Associated with Unfavorable Prognosis. PLoS ONE 2015, 10, e0140178. [Google Scholar] [CrossRef] [Green Version]
- Grywalska, E.; Pasiarski, M.; Sosnowska-Pasiarska, B.; Macek, P.; Rolińska, A.; Samardakiewicz, M.; Ludian, J.; Góźdź, S.; Roliński, J. Programmed Cell Death 1 Expression and Epstein-Barr Virus Infection in Chronic Lymphocytic Leukemia: A Prospective Cohort Study. Cancer Manag. Res. 2019, 11, 7605. [Google Scholar] [CrossRef] [Green Version]
- McKinney, E.F.; Lee, J.C.; Jayne, D.R.W.; Lyons, P.A.; Smith, K.G.C. T-Cell Exhaustion, Co-Stimulation and Clinical Outcome in Autoimmunity and Infection. Nature 2015, 523, 612–616. [Google Scholar] [CrossRef] [Green Version]
- Frydecka, I.; Kosmaczewska, A. Antygen CTLA−4-Negatywny Regulator Procesu Aktywacji Limfocytów T CTLA−4-a Negative Regulator of T Cell Activation. Clin. Exp. Med. 2003, 12, 791–799. [Google Scholar]
- Boulougouris, G.; McLeod, J.D.; Patel, Y.I.; Ellwood, C.N.; Walker, L.S.K.; Sansom, D.M. Positive and Negative Regulation of Human T Cell Activation Mediated by the CTLA-4/CD28 Ligand CD80. J. Immunol. 1998, 161, 3919–3924. [Google Scholar]
- Frydecka, I.; Kosmaczewska, A.; Bocko, D.; Ciszak, L.; Wolowiec, D.; Kuliczkowski, K.; Kochanowska, I. Alterations of the Expression of T-Cell-Related Costimulatory CD28 and Downregulatory CD152 (CTLA-4) Molecules in Patients with B-Cell Chronic Lymphocytic Leukemia. Br. J. Cancer 2004, 90, 2042. [Google Scholar] [CrossRef] [Green Version]
- Lindsten, T.; Lee, K.P.; Harris, E.S.; Petryniak, B.; Craighead, N.; Reynolds, P.J.; Lombard, D.B.; Freeman, G.J.; Nadler, L.M.; Gray, G.S. Characterization of CTLA-4 Structure and Expression on Human T Cells. J. Immunol. 1993, 151, 3489–3499. [Google Scholar]
- Chen, W.; Jin, W.; Wahl, S.M. Engagement of Cytotoxic T Lymphocyte–Associated Antigen 4 (CTLA-4) Induces Transforming Growth Factor β (TGF-β) Production by Murine CD4+ T Cells. J. Exp. Med. 1998, 188, 1849. [Google Scholar] [CrossRef] [Green Version]
- Carreno, B.M.; Bennett, F.; Chau, T.A.; Ling, V.; Luxenberg, D.; Jussif, J.; Baroja, M.L.; Madrenas, J. CTLA-4 (CD152) Can Inhibit T Cell Activation by Two Different Mechanisms Depending on Its Level of Cell Surface Expression. J. Immunol. 2000, 165, 1352–1356. [Google Scholar] [CrossRef] [Green Version]
- Blair, P.J.; Riley, J.L.; Levine, B.L.; Lee, K.P.; Craighead, N.; Francomano, T.; Perfetto, S.J.; Gray, G.S.; Carreno, B.M.; June, C.H. Cutting Edge: CTLA-4 Ligation Delivers a Unique Signal to Resting Human CD4 T Cells That Inhibits Interleukin-2 Secretion but Allows Bcl-XL Induction. J. Immunol. 1998, 160, 12–15. [Google Scholar]
- Brunner, M.C.; Chambers, C.A.; Chan, F.K.-M.; Hanke, J.; Winoto, A.; Allison, J.P. CTLA-4-Mediated Inhibition of Early Events of T Cell Proliferation. J. Immunol. 1999, 162, 5813–5820. [Google Scholar] [PubMed]
- Greenwald, R.; Oosterwegel, M.; Woude, D.; Kubal, A.; Mandelbrot, D.; Boussiotis, V.; Sharpe, A. CTLA-4 Regulates Cell Cycle Progression during a Primary Immune Response. Eur. J. Immunol. 2002, 32, 366–373. [Google Scholar] [CrossRef]
- Masteller, E.L.; Chuang, E.; Mullen, A.C.; Reiner, S.L.; Thompson, C.B. Structural Analysis of CTLA-4 Function In Vivo. J. Immunol. 2000, 164, 5319–5327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costello, T.; Mallet, F.; Sainty, D.; Maraninchi, D.; Gastaut, J.-A.; Olive, D. Regulation of CD80/B7-1 and CD86/B7-2 Molecule Expression in Human Primary Acute Myeloid Leukemia and Their Role in Allogenic Immune Recognition. Eur. J. Immunol. 1998, 28, 90–103. [Google Scholar] [CrossRef]
- Fernández-Ruiz, E.; Somoza, C.; Sánchez-Madrid, F.; Lanier, L.L. CD28/CTLA-4 Ligands: The Gene Encoding CD86 (B70/B7.2) Maps to the Same Region as CD80 (B7/B7.1) Gene in Human Chromosome 3q13-q23. Eur. J. Immunol. 1995, 25, 1453–1456. [Google Scholar] [CrossRef] [PubMed]
- Hock, B.D.; Starling, G.C.; Patton, W.N.; Salm, N.; Bond, K.; McArthur, L.T.; McKenzie, J.L. Identification of a Circulating Soluble Form of CD80: Levels in Patients with Hematological Malignancies. Leuk. Lymphoma 2009, 45, 2111–2118. [Google Scholar] [CrossRef] [PubMed]
- Scrivener, S.; Kaminski, E.R.; Demaine, A.; Prentice, A.G. Analysis of the Expression of Critical Activation/Interaction Markers on Peripheral Blood T Cells in B-Cell Chronic Lymphocytic Leukemia: Evidence of Immune Dysregulation. Br. J. Haematol. 2001, 112, 959–964. [Google Scholar] [CrossRef]
- Pavkovic, M.; Georgievski, B.; Cevreska, L.; Spiroski, M.; Efremov, D.G. CTLA-4 Exon 1 Polymorphism in Patients with Autoimmune Blood Disorders. Am. J. Hematol. 2003, 72, 147–149. [Google Scholar] [CrossRef]
- Kosmaczewska, A.; Ciszak, L.; Suwalska, K.; Wolowiec, D.; Frydecka, I. CTLA-4 Overexpression in CD19+/CD5+ Cells Correlates with the Level of Cell Cycle Regulators and Disease Progression in B-CLL Patients. Leukemia 2005, 19, 301–304. [Google Scholar] [CrossRef] [Green Version]
- Raziorrouh, B.; Ulsenheimer, A.; Schraut, W.; Heeg, M.; Kurktschiev, P.; Zachoval, R.; Jung, M.; Thimme, R.; Neumannhaefelin, C.; Horster, S.; et al. Inhibitory Molecules That Regulate Expansion and Restoration of HCV-Specific CD4+ T Cells in Patients with Chronic Infection. Gastroenterology 2011, 141, 1422–1431. [Google Scholar] [CrossRef]
- Comin-Anduix, B.; Lee, Y.; Jalil, J.; Algazi, A.; de la Rocha, P.; Camacho, L.H.; Bozon, V.A.; Bulanhagui, C.A.; Seja, E.; Villanueva, A.; et al. Detailed Analysis of Immunologic Effects of the Cytotoxic T Lymphocyte-Associated Antigen 4-Blocking Monoclonal Antibody Tremelimumab in Peripheral Blood of Patients with Melanoma. J. Transl. Med. 2008, 6, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Suarez, N.; Alfaro, C.; Dubrot, J.; Palazon, A.; Bolaños, E.; Erro, L.; Hervas-Stubbs, S.; Martinez-Forero, I.; Morales-Kastresana, A.; Martin-Algarra, S.; et al. Synergistic Effects of CTLA-4 Blockade with Tremelimumab and Elimination of Regulatory T Lymphocytes in Vitro and in Vivo. Int. J. Cancer 2010, 129, 374–386. [Google Scholar] [CrossRef]
- Parks, C.G.; Cooper, G.S.; Hudson, L.L.; Dooley, M.A.; Treadwell, E.L.; St. Clair, E.W.; Gilkeson, G.S.; Pandey, J.P. Association of Epstein-Barr Virus with Systemic Lupus Erythematosus: Effect Modification by Race, Age, and Cytotoxic T Lymphocyte-Associated Antigen 4 Genotype. Arthritis Rheum. 2005, 52, 1148–1159. [Google Scholar] [CrossRef]
- Hallek, M. Chronic Lymphocytic Leukemia: 2013 Update on Diagnosis, Risk Stratification and Treatment. Am. J. Hematol. 2013, 88, 803–816. [Google Scholar] [CrossRef]
- Hus, I.; Podhorecka, M.; Bojarska-Junak, A.; Roliński, J.; Schmitt, M.; Sieklucka, M.; Wąsik-Szczepanek, E.; Dmoszyńska, A. The Clinical Significance of ZAP-70 and CD38 Expression in B-Cell Chronic Lymphocytic Leukemia. Ann. Oncol. 2006, 17, 683–690. [Google Scholar] [CrossRef]
- Bartik, M.M.; Welker, D.; Kay, N.E. Impairments in Immune Cell Function in B Cell Chronic Lymphocytic Leukemia. Semin. Oncol. 1998, 25, 27–33. [Google Scholar]
- Pistillo, M.P.; Tazzari, P.L.; Palmisano, G.L.; Pierri, I.; Bolognesi, A.; Ferlito, F.; Capanni, P.; Polito, L.; Ratta, M.; Pileri, S.; et al. CTLA-4 Is Not Restricted to the Lymphoid Cell Lineage and Can Function as a Target Molecule for Apoptosis Induction of Leukemic Cells. Blood 2003, 101, 202–209. [Google Scholar] [CrossRef] [Green Version]
- Wolowiec, D.; Ciszak, L.; Kosmaczewska, A.; Bocko, D.; Teodorowska, R.; Frydecka, I.; Kuliczkowski, K. Cell Cycle Regulatory Proteins and Apoptosis in B-Cell Chronic Lymphocytic Leukemia. Haematologica 2002, 86, 1296–1304. [Google Scholar]
- Suwalska, K.; Pawlak, E.; Karabon, L.; Tomkiewicz, A.; Dobosz, T.; Urbaniak-Kujda, D.; Kuliczkowski, K.; Wolowiec, D.; Jedynak, A.; Frydecka, I. Association Studies of CTLA-4, CD28, and ICOS Gene Polymorphisms with B-Cell Chronic Lymphocytic Leukemia in the Polish Population. Hum. Immunol. 2008, 69, 193–201. [Google Scholar] [CrossRef]
- Mittal, A.K.; Chaturvedi, N.K.; Rohlfsen, R.A.; Gupta, P.; Joshi, A.D.; Hegde, G.V.; Bociek, R.G.; Joshi, S.S. Role of CTLA4 in the Proliferation and Survival of Chronic Lymphocytic Leukemia. PLoS ONE 2013, 8, e70352. [Google Scholar] [CrossRef] [Green Version]
- Joshi, A.D.; Hegde, G.V.; Dickinson, J.D.; Mittal, A.K.; Lynch, J.C.; Eudy, J.D.; Armitage, J.O.; Bierman, P.J.; Bociek, R.G.; Devetten, M.P.; et al. ATM, CTLA4, MNDA, and HEM1 in High versus Low CD38-Expressing B-Cell Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2007, 13, 5295–5304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grandjenette, C.; Kennel, A.; Faure, G.C.; Béné, M.C.; Feugier, P. Expression of Functional Toll-like Receptors by B-Chronic Lymphocytic Leukemia Cells. Haematologica 2007, 92, 1279–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crozat, K.; Beutler, B. TLR7: A New Sensor of Viral Infection. Proc. Natl. Acad. Sci. USA 2004, 101, 6835–6836. [Google Scholar] [CrossRef] [Green Version]
- Spaner, D.E.; Masellis, A. Toll-like Receptor Agonists in the Treatment of Chronic Lymphocytic Leukemia. Leukemia 2007, 21, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Tomic, J.; White, D.; Shi, Y.; Mena, J.; Hammond, C.; He, L.; Miller, R.L.; Spaner, D.E. Sensitization of IL-2 Signaling through TLR-7 Enhances B Lymphoma Cell Immunogenicity. J. Immunol. 2006, 176, 3830–3839. [Google Scholar] [CrossRef] [PubMed]
- Valente, R.M.; Ehlers, E.; Xu, D.; Ahmad, H.; Steadman, A.; Blasnitz, L.; Zhou, Y.; Kastanek, L.; Meng, B.; Zhang, L. Toll-Like Receptor 7 Stimulates the Expression of Epstein-Barr Virus Latent Membrane Protein 1. PLoS ONE 2012, 7, e43317. [Google Scholar] [CrossRef] [Green Version]
- Martin, H.J.; Lee, J.M.; Walls, D.; Hayward, S.D. Manipulation of the Toll-like Receptor 7 Signaling Pathway by Epstein-Barr Virus. J. Virol. 2007, 81, 9748–9758. [Google Scholar] [CrossRef] [Green Version]


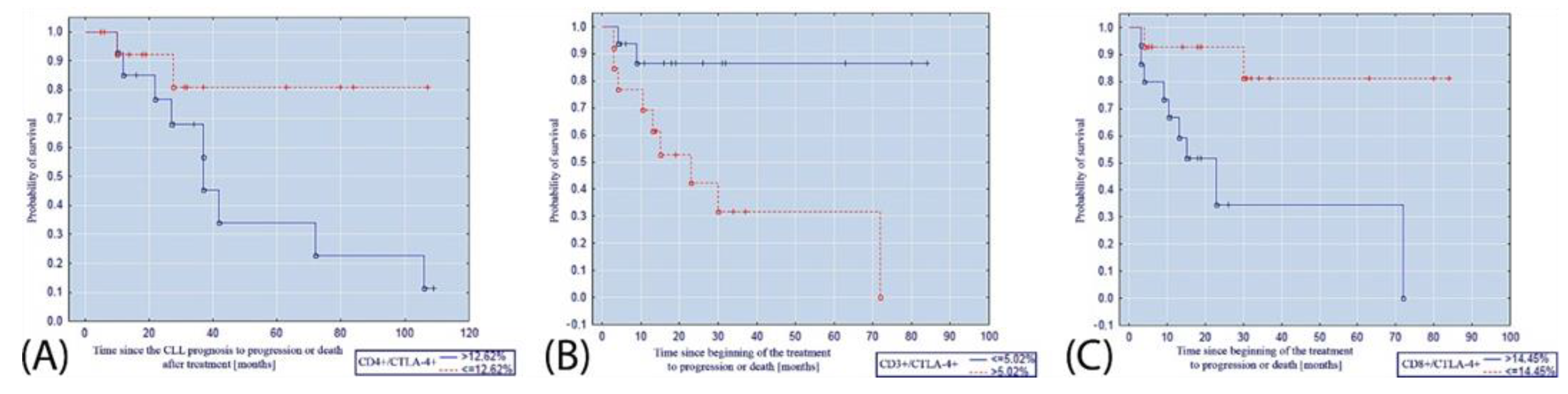


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EBV(+) Patients with CLL | EBV(−) Patients with CLL | Controls | p | |
|---|---|---|---|---|
| N | 59 | 51 | 20 | |
| AGE, YEARS, MEAN (SD) | 63.81 (9.37) | 63.63 (10.76) | 64.50 (7.15) | 0.943 |
| WHITE BLOOD CELL COUNT G/L, MEDIAN [IQR] | 28.30 [20.47, 48.92] | 30.40 [19.85, 46.72] | 7.02 [6.12, 7.85] | <0.001 a |
| LYMPHOCYTE COUNT G/L, MEDIAN [IQR] | 22.44 [15.40, 43.01] | 22.58 [14.93, 40.55] | 2.62 [2.19, 3.05] | <0.001 a |
| HEMOGLOBIN, G/DL, MEAN (SD) | 12.09 (1.42) | 14.12 (1.27) | 14.29 (1.19) | <0.001 b |
| PLATELET COUNT, G/L, MEAN (SD) | 150.02 (58.07) | 180.51 (46.98) | 279.00 (57.05) | <0.001 c |
| BETA-2 MICROGLOBULIN, MG/DL, MEDIAN [IQR] | 3.47 [2.78, 4.23] | 2.37 [1.98, 2.87] | 1.68 [1.29, 1.91] | <0.001 c |
| LACTATE DEHYDROGENASE, U/L MEDIAN [IQR] | 327.00 [290.50, 378.00] | 254.00 [214.50, 334.00] | 155.50 [137.00, 178.25] | <0.001 c |
| RAI STAGE (%) | 0.06 | |||
| 0 | 24 (40.7) | 24 (47.1) | - | |
| I | 11 (18.6) | 15 (29.4) | - | |
| II | 16 (27.1) | 12 (23.5) | - | |
| III | 2 (3.4) | 0 (0.0) | - | |
| IV | 6 (10.2) | 0 (0.0) | - | |
| BINET CLASSIFICATION (%) | 0.018 | |||
| A | 24 (40.7) | 24 (47.1) | - | |
| B | 27 (45.8) | 27 (52.9) | - | |
| C | 8 (13.6) | 0 (0.0) | - | |
| CD19+ ZAP70+ CELLS > 20%, N (%) | 27 (45.8) | 11 (21.6) | - | 0.009 |
| CD19+ CD38+ CELLS > 30%, N (%) | 34 (57.6) | 6 (11.8) | - | <0.001 |
| CD19+ ZAP70+ CELLS, % MEDIAN [IQR] | 17.96 [11.09, 29.04] | 5.80 [3.22, 18.19] | - | <0.001 |
| CD19+ CD38+ CELLS, % MEDIAN [IQR] | 33.53 [12.02, 59.89] | 1.64 [0.78, 6.27] | - | <0.001 |
| Variable | Group | Median | Minimum | Maximum | p |
|---|---|---|---|---|---|
| LYMPHOCYTES CD19+/CD86+ [%] | EBV(+) | 35.37 | 4.43 | 75.6 | <0.0001 0.0002 |
| EBV(−) | 16.73 | 1.5 | 46.99 | ||
| Control | 11.32 | 2.98 | 18.19 | ||
| ABSOLUTE NUMBER OF LYMPHOCYTES CD19+/CD86+ [X103 CELLS/UL] | EBV(+) | 6.4495 | 0.4433 | 32.656 | 0.0354 <0.0001 0.0256 |
| EBV(−) | 2.5725 | 0.2094 | 22.0708 | ||
| Control | 0.0292 | 0.0108 | 0.0403 | ||
| CD86 ANTIGEN EXPRESSION ON LYMPHOCYTES CD19+ [MFI] | EBV(+) | 21.83 | 14.18 | 131.24 | 0.0256 0.0067 |
| EBV(−) | 18.655 | 14.24 | 29.44 | ||
| Control | 15.495 | 13.24 | 18.97 |
| Variable | Group | Median | Minimum | Maximum | p |
|---|---|---|---|---|---|
| LYMPHOCYTES CD3+/CTLA-4+ (AMONG CD3+) [%] | EBV(+) | 5.41 | 2.13 | 14.36 | 0.0395 |
| EBV(−) | 4.715 | 1.95 | 8.06 | ||
| Control | 2.975 | 1.34 | 5.8 | ||
| ABSOLUTE NUMBER OF LYMPHOCYTES CD3+/CTLA-4+ (AMONG CD3+) [X103 CELLS/UL] | EBV(+) | 0.1433 | 0.0356 | 0.423 | 0.0078 0.0314 |
| EBV(−) | 0.1515 | 0.0407 | 0.289 | ||
| Control | 0.0542 | 0.0129 | 0.0787 | ||
| LYMPHOCYTES CD4+/CTLA-4+ (AMONG CD4+) [%] | EBV(+) | 14.755 | 9.56 | 31.14 | 0.0012 0.0001 0.0091 |
| EBV(−) | 8.435 | 5.96 | 14.25 | ||
| Control | 2.85 | 2.17 | 3.19 | ||
| ABSOLUTE NUMBER OF LYMPHOCYTES CD4+/CTLA-4+ (AMONG CD4+) [X103 CELLS/UL] | EBV(+) | 0.1705 | 0.0913 | 0.908 | 0.0001 0.0150 |
| EBV(−) | 0.1367 | 0.0761 | 0.2681 | ||
| Control | 0.0303 | 0.0147 | 0.0406 | ||
| LYMPHOCYTES CD8+/CTLA-4+ (AMONG CD8+) [%] | EBV(+) | 16.855 | 11.38 | 26.69 | 0.0001 0.0001 0.0011 |
| EBV(−) | 8.785 | 5.91 | 13.75 | ||
| Control | 2.835 | 1.58 | 3.89 | ||
| ABSOLUTE NUMBER OF LYMPHOCYTES CD8+/CTLA-4+ (AMONG CD8+) [X103 CELLS/UL] | EBV(+) | 0.1903 | 0.0322 | 1.487 | 0.0002 0.0420 |
| EBV(−) | 0.1166 | 0.0295 | 0.2913 | ||
| Control | 0.0155 | 0.0101 | 0.0378 | ||
| EXPRESSION OF CTLA-4 ON CD8+ [MFI] | EBV(+) | 28.185 | 26.22 | 34.52 | 0.0434 |
| EBV(−) | 28.38 | 25.43 | 29.52 | ||
| Control | 22.895 | 17.57 | 37.65 | ||
| ABSOLUTE NUMBER OF LYMPHOCYTES CD19+/ CTLA-4+(AMONG CD19+) [X103 CELLS/UL] | EBV(+) | 1.1267 | 0.1395 | 8.676 | 0.0005 0.0019 |
| EBV(−) | 0.9997 | 0.148 | 6.3925 | ||
| Control | 0.0056 | 0.0034 | 0.0203 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grywalska, E.; Mielnik, M.; Podgajna, M.; Hymos, A.; Ludian, J.; Rolińska, A.; Gosik, K.; Kwaśniewski, W.; Sosnowska-Pasiarska, B.; Smok-Kalwat, J.; et al. Expression of CTLA-4 and CD86 Antigens and Epstein-Barr Virus Reactivation in Chronic Lymphocytic Leukemia—Any Link with Known Prognostic Factors? Cancers 2022, 14, 672. https://doi.org/10.3390/cancers14030672
Grywalska E, Mielnik M, Podgajna M, Hymos A, Ludian J, Rolińska A, Gosik K, Kwaśniewski W, Sosnowska-Pasiarska B, Smok-Kalwat J, et al. Expression of CTLA-4 and CD86 Antigens and Epstein-Barr Virus Reactivation in Chronic Lymphocytic Leukemia—Any Link with Known Prognostic Factors? Cancers. 2022; 14(3):672. https://doi.org/10.3390/cancers14030672
Chicago/Turabian StyleGrywalska, Ewelina, Michał Mielnik, Martyna Podgajna, Anna Hymos, Jarosław Ludian, Agnieszka Rolińska, Krzysztof Gosik, Wojciech Kwaśniewski, Barbara Sosnowska-Pasiarska, Jolanta Smok-Kalwat, and et al. 2022. "Expression of CTLA-4 and CD86 Antigens and Epstein-Barr Virus Reactivation in Chronic Lymphocytic Leukemia—Any Link with Known Prognostic Factors?" Cancers 14, no. 3: 672. https://doi.org/10.3390/cancers14030672