
Targeting GPCRs and Their Signaling as a Therapeutic Option in Melanoma


Abstract
:Simple Summary
Abstract
1. Introduction
2. Impact of GPCRs on Melanoma Initiation and Progression
2.1. Melanomagenesis
2.2. GPCRs in Melanoma
2.2.1. Endothelin Receptor Type B (EDNRB)
2.2.2. The Melanocortin Receptor (MC1R)
2.2.3. The Wnt/Frizzled Receptor
2.2.4. Glutamate Receptors (GRM1, GRM3 and GRM5)
2.2.5. PAR1
2.2.6. Chemokine Receptors (CXCR4, CCR7, CCR10)
2.2.7. G-Protein-Coupled Estrogen Receptor 1 (GPER1)
3. GPCR Associated Signaling in Melanoma
3.1. Signaling via cAMP
3.2. Signaling via Inositol Triphosphate and Diacylglycerol
3.3. Signaling via Gα12/13
3.4. Signaling via WNT/β-Catenin
3.5. Signaling via Gβ/Gγ Subunits
3.6. β-Arrestin Signaling
4. Perspectives of Targeting GPCRs in Melanoma
4.1. Limitation of Available Tools
4.2. Novel Structures and Drug Design Approaches
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Insel, P.A.; Sriram, K.; Gorr, M.W.; Wiley, S.Z.; Michkov, A.; Salmerón, C.; Chinn, A.M. GPCRomics: An Approach to Discover GPCR Drug Targets. Trends Pharmacol. Sci. 2019, 40, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR Drug Discovery: New Agents, Targets and Indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Usman, S.; Khawer, M.; Rafique, S.; Naz, Z.; Saleem, K. The Current Status of Anti-GPCR Drugs against Different Cancers. J. Pharm. Anal. 2020, 10, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted Agents and Immunotherapies: Optimizing Outcomes in Melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlino, M.S.; Larkin, J.; Long, G.V. Immune Checkpoint Inhibitors in Melanoma. Lancet 2021, 398, 1002–1014. [Google Scholar] [CrossRef]
- Baik, J.H.; Picetti, R.; Saiardi, A.; Thiriet, G.; Dierich, A.; Depaulis, A.; Le Meur, M.; Borrelli, E. Parkinsonian-like Locomotor Impairment in Mice Lacking Dopamine D2 Receptors. Nature 1995, 377, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Hosoda, K.; Hammer, R.E.; Richardson, J.A.; Baynash, A.G.; Cheung, J.C.; Giaid, A.; Yanagisawa, M. Targeted and Natural (Piebald-Lethal) Mutations of Endothelin-B Receptor Gene Produce Megacolon Associated with Spotted Coat Color in Mice. Cell 1994, 79, 1267–1276. [Google Scholar] [CrossRef]
- Wang, Y.; Huso, D.; Cahill, H.; Ryugo, D.; Nathans, J. Progressive Cerebellar, Auditory, and Esophageal Dysfunction Caused by Targeted Disruption of Thefrizzled-4 Gene. J. Neurosci. 2001, 21, 4761–4771. [Google Scholar] [CrossRef] [Green Version]
- Incerti, B.; Cortese, K.; Pizzigoni, A.; Surace, E.M.; Varani, S.; Coppola, M.; Jeffery, G.; Seeliger, M.; Jaissle, G.; Bennett, D.C.; et al. Oa1 Knock-out: New Insights on the Pathogenesis of Ocular Albinism Type 1. Hum. Mol. Genet. 2000, 9, 2781–2788. [Google Scholar] [CrossRef] [Green Version]
- Matteson, P.G.; Desai, J.; Korstanje, R.; Lazar, G.; Borsuk, T.E.; Rollins, J.; Kadambi, S.; Joseph, J.; Rahman, T.; Wink, J.; et al. The Orphan G Protein-Coupled Receptor, Gpr161, Encodes the Vacuolated Lens Locus and Controls Neurulation and Lens Development. Proc. Natl. Acad. Sci. USA 2008, 105, 2088–2093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollock, P.M.; Cohen-Solal, K.; Sood, R.; Namkoong, J.; Martino, J.J.; Koganti, A.; Zhu, H.; Robbins, C.; Makalowska, I.; Shin, S.-S.; et al. Melanoma Mouse Model Implicates Metabotropic Glutamate Signaling in Melanocytic Neoplasia. Nat. Genet. 2003, 34, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Robbins, L.S.; Nadeau, J.H.; Johnson, K.R.; Kelly, M.A.; Roselli-Rehfuss, L.; Baack, E.; Mountjoy, K.G.; Cone, R.D. Pigmentation Phenotypes of Variant Extension Locus Alleles Result from Point Mutations That Alter MSH Receptor Function. Cell 1993, 72, 827–834. [Google Scholar] [CrossRef]
- Twigg, S.R.F.; Hufnagel, R.B.; Miller, K.A.; Zhou, Y.; McGowan, S.J.; Taylor, J.; Craft, J.; Taylor, J.C.; Santoro, S.L.; Huang, T.; et al. A Recurrent Mosaic Mutation in SMO, Encoding the Hedgehog Signal Transducer Smoothened, Is the Major Cause of Curry-Jones Syndrome. Am. J. Hum. Genet. 2016, 98, 1256–1265. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Wall, B.; Chen, S. G-Protein-Coupled Receptors and Melanoma. Pigment Cell Melanoma Res. 2008, 21, 415–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baxter, L.L.; Watkins-Chow, D.E.; Pavan, W.J.; Loftus, S.K. A Curated Gene List for Expanding the Horizons of Pigmentation Biology. Pigment Cell Melanoma Res. 2019, 32, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Aktary, Z.; McMahon, M.; Larue, L. Animal Models of Melanoma. In Melanoma; Fisher, D.E., Bastian, B.C., Eds.; Springer: New York, NY, USA, 2017; pp. 1–31. ISBN 978-1-4614-7322-0. [Google Scholar]
- Delmas, V.; Martinozzi, S.; Bourgeois, Y.; Holzenberger, M.; Larue, L. Cre-Mediated Recombination in the Skin Melanocyte Lineage. Genesis 2003, 36, 73–80. [Google Scholar] [CrossRef]
- Yajima, I.; Belloir, E.; Bourgeois, Y.; Kumasaka, M.; Delmas, V.; Larue, L. Spatiotemporal Gene Control by the Cre-ERT2 System in Melanocytes. Genesis 2006, 44, 34–43. [Google Scholar] [CrossRef]
- Bosenberg, M.; Muthusamy, V.; Curley, D.P.; Wang, Z.; Hobbs, C.; Nelson, B.; Nogueira, C.; Horner II, J.W.; DePinho, R.; Chin, L. Characterization of Melanocyte-Specific Inducible Cre Recombinase Transgenic Mice. Genesis 2006, 44, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Jain, F.; Longakit, A.; Huang, J.L.-Y.; Raamsdonk, C.D.V. Endothelin Signaling Promotes Melanoma Tumorigenesis Driven by Constitutively Active GNAQ. Pigment Cell Melanoma Res. 2020, 33, 834–849. [Google Scholar] [CrossRef]
- Swope, V.B.; Starner, R.J.; Rauck, C.; Abdel-Malek, Z.A. Endothelin-1 and α-Melanocortin Have Redundant Effects on Global Genome Repair in UV-Irradiated Human Melanocytes despite Distinct Signaling Pathways. Pigment Cell Melanoma Res. 2020, 33, 293–304. [Google Scholar] [CrossRef]
- Mitra, D.; Luo, X.; Morgan, A.; Wang, J.; Hoang, M.P.; Lo, J.; Guerrero, C.R.; Lennerz, J.K.; Mihm, M.C.; Wargo, J.A.; et al. An Ultraviolet-Radiation-Independent Pathway to Melanoma Carcinogenesis in the Red Hair/Fair Skin Background. Nature 2012, 491, 449–453. [Google Scholar] [CrossRef]
- D’Orazio, J.A.; Nobuhisa, T.; Cui, R.; Arya, M.; Spry, M.; Wakamatsu, K.; Igras, V.; Kunisada, T.; Granter, S.R.; Nishimura, E.K.; et al. Topical Drug Rescue Strategy and Skin Protection Based on the Role of Mc1r in UV-Induced Tanning. Nature 2006, 443, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Tiwary, S.; Xu, L. FRIZZLED7 Is Required for Tumor Initiation and Metastatic Growth of Melanoma Cells. PLoS ONE 2016, 11, e0147638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Hernandez, I.; Maiques, O.; Kohlhammer, L.; Cantelli, G.; Perdrix-Rosell, A.; Monger, J.; Fanshawe, B.; Bridgeman, V.L.; Karagiannis, S.N.; Penin, R.M.; et al. WNT11-FZD7-DAAM1 Signalling Supports Tumour Initiating Abilities and Melanoma Amoeboid Invasion. Nat. Commun. 2020, 11, 5315. [Google Scholar] [CrossRef] [PubMed]
- Natale, C.A.; Li, J.; Zhang, J.; Dahal, A.; Dentchev, T.; Stanger, B.Z.; Ridky, T.W. Activation of G Protein-Coupled Estrogen Receptor Signaling Inhibits Melanoma and Improves Response to Immune Checkpoint Blockade. Elife 2018, 7, e31770. [Google Scholar] [CrossRef] [PubMed]
- Schiffner, S.; Chen, S.; Becker, J.C.; Bosserhoff, A.-K. Highly Pigmented Tg(Grm1) Mouse Melanoma Develops Non-Pigmented Melanoma Cells in Distant Metastases. Exp. Derm. 2012, 21, 786–788. [Google Scholar] [CrossRef] [PubMed]
- Prickett, T.D.; Wei, X.; Cardenas-Navia, I.; Teer, J.K.; Lin, J.C.; Walia, V.; Gartner, J.; Jiang, J.; Cherukuri, P.F.; Molinolo, A.; et al. Exon Capture Analysis of G Protein-Coupled Receptors Identifies Activating Mutations in GRM3 in Melanoma. Nat. Genet. 2011, 43, 1119–1126. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.Y.; Chang, K.; Pickel, J.M.; Badger, J.D.; Roche, K.W. Expression of the Metabotropic Glutamate Receptor 5 (MGluR5) Induces Melanoma in Transgenic Mice. Proc. Natl. Acad. Sci. USA 2011, 108, 15219–15224. [Google Scholar] [CrossRef] [Green Version]
- Villares, G.J.; Zigler, M.; Wang, H.; Melnikova, V.O.; Wu, H.; Friedman, R.; Leslie, M.C.; Vivas-Mejia, P.E.; Lopez-Berestein, G.; Sood, A.K.; et al. Targeting Melanoma Growth and Metastasis with Systemic Delivery of Liposome-Incorporated Protease-Activated Receptor-1 Small Interfering RNA. Cancer Res. 2008, 68, 9078–9086. [Google Scholar] [CrossRef] [Green Version]
- Murakami, T.; Maki, W.; Cardones, A.R.; Fang, H.; Tun Kyi, A.; Nestle, F.O.; Hwang, S.T. Expression of CXC Chemokine Receptor-4 Enhances the Pulmonary Metastatic Potential of Murine B16 Melanoma Cells. Cancer Res. 2002, 62, 7328–7334. [Google Scholar] [PubMed]
- Ieranò, C.; D’Alterio, C.; Giarra, S.; Napolitano, M.; Rea, G.; Portella, L.; Santagata, A.; Trotta, A.M.; Barbieri, A.; Campani, V.; et al. CXCL12 Loaded-Dermal Filler Captures CXCR4 Expressing Melanoma Circulating Tumor Cells. Cell Death Dis. 2019, 10, 562. [Google Scholar] [CrossRef]
- Wiley, H.E.; Gonzalez, E.B.; Maki, W.; Wu, M.T.; Hwang, S.T. Expression of CC Chemokine Receptor-7 and Regional Lymph Node Metastasis of B16 Murine Melanoma. J. Natl. Cancer Inst. 2001, 93, 1638–1643. [Google Scholar] [CrossRef] [Green Version]
- Murakami, T.; Cardones, A.R.; Finkelstein, S.E.; Restifo, N.P.; Klaunberg, B.A.; Nestle, F.O.; Castillo, S.S.; Dennis, P.A.; Hwang, S.T. Immune Evasion by Murine Melanoma Mediated through CC Chemokine Receptor-10. J. Exp. Med. 2003, 198, 1337. [Google Scholar] [CrossRef] [PubMed]
- Baynash, A.G.; Hosoda, K.; Giaid, A.; Richardson, J.A.; Emoto, N.; Hammer, R.E.; Yanagisawa, M. Interaction of Endothelin-3 with Endothelin-B Receptor Is Essential for Development of Epidermal Melanocytes and Enteric Neurons. Cell 1994, 79, 1277–1285. [Google Scholar] [CrossRef]
- Garcia, R.J.; Ittah, A.; Mirabal, S.; Figueroa, J.; Lopez, L.; Glick, A.B.; Kos, L. Endothelin 3 Induces Skin Pigmentation in a Keratin-Driven Inducible Mouse Model. J. Invest. Derm. 2008, 128, 131–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benaduce, A.P.; Batista, D.; Grilo, G.; Jorge, K.; Cardero, D.; Milikowski, C.; Kos, L. Novel UV-Induced Melanoma Mouse Model Dependent on Endothelin3 Signaling. Pigment Cell Melanoma Res. 2014, 27, 839–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumasaka, M.Y.; Yajima, I.; Hossain, K.; Iida, M.; Tsuzuki, T.; Ohno, T.; Takahashi, M.; Yanagisawa, M.; Kato, M. A Novel Mouse Model for De Novo Melanoma. Cancer Res. 2010, 70, 24–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demunter, A.; De Wolf-Peeters, C.; Degreef, H.; Stas, M.; van den Oord, J.J. Expression of the Endothelin-B Receptor in Pigment Cell Lesions of the Skin. Evidence for Its Role as Tumor Progression Marker in Malignant Melanoma. Virchows Arch. 2001, 438, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Asundi, J.; Reed, C.; Arca, J.; McCutcheon, K.; Ferrando, R.; Clark, S.; Luis, E.; Tien, J.; Firestein, R.; Polakis, P. An Antibody-Drug Conjugate Targeting the Endothelin B Receptor for the Treatment of Melanoma. Clin. Cancer Res. 2011, 17, 965–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahav, R.; Heffner, G.; Patterson, P.H. An Endothelin Receptor B Antagonist Inhibits Growth and Induces Cell Death in Human Melanoma Cells in Vitro and in Vivo. Proc. Natl. Acad. Sci. USA 1999, 96, 11496–11500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahav, R.; Suvà, M.-L.; Rimoldi, D.; Patterson, P.H.; Stamenkovic, I. Endothelin Receptor B Inhibition Triggers Apoptosis and Enhances Angiogenesis in Melanomas. Cancer Res. 2004, 64, 8945–8953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Koschembahr, A.M.; Swope, V.B.; Starner, R.J.; Abdel-Malek, Z.A. Endothelin-1 Protects Human Melanocytes from UV-Induced DNA Damage by Activating JNK and P38 Signalling Pathways. Exp. Derm. 2015, 24, 269–274. [Google Scholar] [CrossRef]
- Freitas, J.T.; Lopez, J.; Llorian, C.; Boroni, M.; Kos, L. The Immunosuppressive Role of Edn3 Overexpression in the Melanoma Microenvironment. Pigment Cell Melanoma Res. 2021, 34, 1084–1093. [Google Scholar] [CrossRef]
- Bagnato, A.; Natali, P.G. Endothelin Receptors as Novel Targets in Tumor Therapy. J. Transl. Med. 2004, 2, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kefford, R.; Beith, J.M.; Van Hazel, G.A.; Millward, M.; Trotter, J.M.; Wyld, D.K.; Kusic, R.; Shreeniwas, R.; Morganti, A.; Ballmer, A.; et al. A Phase II Study of Bosentan, a Dual Endothelin Receptor Antagonist, as Monotherapy in Patients with Stage IV Metastatic Melanoma. Invest. New Drugs 2007, 25, 247–252. [Google Scholar] [CrossRef] [Green Version]
- Kefford, R.F.; Clingan, P.R.; Brady, B.; Ballmer, A.; Morganti, A.; Hersey, P. A Randomized, Double-Blind, Placebo-Controlled Study of High-Dose Bosentan in Patients with Stage IV Metastatic Melanoma Receiving First-Line Dacarbazine Chemotherapy. Mol. Cancer 2010, 9, 69. [Google Scholar] [CrossRef] [Green Version]
- Sandhu, S.; McNeil, C.M.; LoRusso, P.; Patel, M.R.; Kabbarah, O.; Li, C.; Sanabria, S.; Flanagan, W.M.; Yeh, R.-F.; Brunstein, F.; et al. Phase I Study of the Anti-Endothelin B Receptor Antibody-Drug Conjugate DEDN6526A in Patients with Metastatic or Unresectable Cutaneous, Mucosal, or Uveal Melanoma. Invest. New Drugs 2020, 38, 844–854. [Google Scholar] [CrossRef]
- García-Borrón, J.C.; Abdel-Malek, Z.; Jiménez-Cervantes, C. MC1R, the CAMP Pathway and the Response to Solar UV: Extending the Horizon beyond Pigmentation. Pigment Cell Melanoma Res. 2014, 27, 699–720. [Google Scholar] [CrossRef]
- Bertolotto, C.; Abbe, P.; Hemesath, T.J.; Bille, K.; Fisher, D.E.; Ortonne, J.P.; Ballotti, R. Microphthalmia Gene Product as a Signal Transducer in CAMP-Induced Differentiation of Melanocytes. J. Cell Biol. 1998, 142, 827–835. [Google Scholar] [CrossRef] [Green Version]
- Goding, C.R.; Arnheiter, H. MITF—the First 25 Years. Genes Dev. 2019, 33, 983–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deraredj Nadim, W.; Hassanaly, S.; Bénédetti, H.; Kieda, C.; Grillon, C.; Morisset-Lopez, S. The GTPase-Activating Protein-Related Domain of Neurofibromin Interacts with MC1R and Regulates Pigmentation-Mediated Signaling in Human Melanocytes. Biochem. Biophys. Res. Commun. 2021, 534, 758–764. [Google Scholar] [CrossRef] [PubMed]
- Tamate, H.B.; Takeuchi, T. Action of the e Locus of Mice in the Response of Phaeomelanic Hair Follicles to α-Melanocyte-Stimulating Hormone in Vitro. Science 1984, 224, 1241–1242. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.; Healy, E.; Siddiqui, S.; Flanagan, N.; Steijlen, P.M.; Rosdahl, I.; Jacques, J.P.; Rogers, S.; Turner, R.; Jackson, I.J.; et al. Melanocortin 1 Receptor Variants in an Irish Population. J. Invest. Derm. 1998, 111, 119–122. [Google Scholar] [CrossRef] [Green Version]
- Harding, R.M.; Healy, E.; Ray, A.J.; Ellis, N.S.; Flanagan, N.; Todd, C.; Dixon, C.; Sajantila, A.; Jackson, I.J.; Birch-Machin, M.A.; et al. Evidence for Variable Selective Pressures at MC1R. Am. J. Hum. Genet. 2000, 66, 1351–1361. [Google Scholar] [CrossRef] [Green Version]
- Scott, M.C.; Wakamatsu, K.; Ito, S.; Kadekaro, A.L.; Kobayashi, N.; Groden, J.; Kavanagh, R.; Takakuwa, T.; Virador, V.; Hearing, V.J.; et al. Human Melanocortin 1 Receptor Variants, Receptor Function and Melanocyte Response to UV Radiation. J. Cell Sci. 2002, 115, 2349–2355. [Google Scholar] [CrossRef]
- Krude, H.; Biebermann, H.; Luck, W.; Horn, R.; Brabant, G.; Grüters, A. Severe Early-Onset Obesity, Adrenal Insufficiency and Red Hair Pigmentation Caused by POMC Mutations in Humans. Nat. Genet. 1998, 19, 155–157. [Google Scholar] [CrossRef]
- Herraiz, C.; Martínez-Vicente, I.; Maresca, V. The α-Melanocyte-Stimulating Hormone/Melanocortin-1 Receptor Interaction: A Driver of Pleiotropic Effects beyond Pigmentation. Pigment Cell Melanoma Res. 2021, 34, 748–761. [Google Scholar] [CrossRef]
- Swope, V.B.; Abdel-Malek, Z.A. Significance of the Melanocortin 1 and Endothelin B Receptors in Melanocyte Homeostasis and Prevention of Sun-Induced Genotoxicity. Front. Genet. 2016, 7, 146. [Google Scholar] [CrossRef] [Green Version]
- Castejón-Griñán, M.; Herraiz, C.; Olivares, C.; Jiménez-Cervantes, C.; García-Borrón, J.C. CAMP-Independent Non-Pigmentary Actions of Variant Melanocortin 1 Receptor: AKT-Mediated Activation of Protective Responses to Oxidative DNA Damage. Oncogene 2018, 37, 3631–3646. [Google Scholar] [CrossRef]
- Guida, S.; Guida, G.; Goding, C.R. MC1R Functions, Expression, and Implications for Targeted Therapy. J. Investig. Dermatol. 2022, 142, 293–302.e1. [Google Scholar] [CrossRef] [PubMed]
- Bautista, R.-M.F.; Carter, K.M.; Jarrett, S.G.; Napier, D.; Wakamatsu, K.; Ito, S.; D’Orazio, J.A. Cutaneous Pharmacologic CAMP Induction Induces Melanization of the Skin and Improves Recovery from Ultraviolet Injury in Melanocortin 1 Receptor-Intact or Heterozygous Skin. Pigment Cell Melanoma Res. 2020, 33, 30–40. [Google Scholar] [CrossRef]
- Koikov, L.; Starner, R.J.; Swope, V.B.; Upadhyay, P.; Hashimoto, Y.; Freeman, K.T.; Knittel, J.J.; Haskell-Luevano, C.; Abdel-Malek, Z.A. Development of HMC1R Selective Small Agonists for Sunless Tanning and Prevention of Genotoxicity of UV in Melanocytes. J. Invest. Derm. 2021, 141, 1819–1829. [Google Scholar] [CrossRef]
- Veeman, M.T.; Axelrod, J.D.; Moon, R.T. A Second Canon. Functions and Mechanisms of Beta-Catenin-Independent Wnt Signaling. Dev. Cell 2003, 5, 367–377. [Google Scholar] [CrossRef] [Green Version]
- Abou Azar, F.; Lim, G.E. Metabolic Contributions of Wnt Signaling: More Than Controlling Flight. Front. Cell Dev. Biol 2021, 9, 709823. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Larue, L.; Delmas, V. The WNT/Beta-Catenin Pathway in Melanoma. Front. Biosci 2006, 11, 733–742. [Google Scholar] [CrossRef] [Green Version]
- Dorsky, R.I.; Moon, R.T.; Raible, D.W. Control of Neural Crest Cell Fate by the Wnt Signalling Pathway. Nature 1998, 396, 370–373. [Google Scholar] [CrossRef]
- Dunn, K.J.; Williams, B.O.; Li, Y.; Pavan, W.J. Neural Crest-Directed Gene Transfer Demonstrates Wnt1 Role in Melanocyte Expansion and Differentiation during Mouse Development. Proc. Natl. Acad. Sci. USA 2000, 97, 10050–10055. [Google Scholar] [CrossRef] [Green Version]
- Ikeya, M.; Lee, S.M.K.; Johnson, J.E.; McMahon, A.P.; Takada, S. Wnt Signalling Required for Expansion of Neural Crest and CNS Progenitors. Nature 1997, 389, 966–970. [Google Scholar] [CrossRef]
- Dorsky, R.I.; Raible, D.W.; Moon, R.T. Direct Regulation of Nacre, a Zebrafish MITF Homolog Required for Pigment Cell Formation, by the Wnt Pathway. Genes Dev. 2000, 14, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Hari, L.; Brault, V.; Kléber, M.; Lee, H.-Y.; Ille, F.; Leimeroth, R.; Paratore, C.; Suter, U.; Kemler, R.; Sommer, L. Lineage-Specific Requirements of β-Catenin in Neural Crest Development. J. Cell Biol. 2002, 159, 867–880. [Google Scholar] [CrossRef] [Green Version]
- Luciani, F.; Champeval, D.; Herbette, A.; Denat, L.; Aylaj, B.; Martinozzi, S.; Ballotti, R.; Kemler, R.; Goding, C.R.; De Vuyst, F.; et al. Biological and Mathematical Modeling of Melanocyte Development. Development 2011, 138, 3943–3954. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, S.J.; Rambow, F.; Kumasaka, M.; Champeval, D.; Bellacosa, A.; Delmas, V.; Larue, L. Beta-Catenin Inhibits Melanocyte Migration but Induces Melanoma Metastasis. Oncogene 2013, 32, 2230–2238. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, X. Targeting the Wnt/β-Catenin Signaling Pathway in Cancer. J. Hematol. Oncol. 2020, 13, 165. [Google Scholar] [CrossRef]
- Delmas, V.; Beermann, F.; Martinozzi, S.; Carreira, S.; Ackermann, J.; Kumasaka, M.; Denat, L.; Goodall, J.; Luciani, F.; Viros, A.; et al. Beta-Catenin Induces Immortalization of Melanocytes by Suppressing P16INK4a Expression and Cooperates with N-Ras in Melanoma Development. Genes Dev. 2007, 21, 2923–2935. [Google Scholar] [CrossRef] [Green Version]
- Damsky, W.E.; Curley, D.P.; Santhanakrishnan, M.; Rosenbaum, L.E.; Platt, J.T.; Gould Rothberg, B.E.; Taketo, M.M.; Dankort, D.; Rimm, D.L.; McMahon, M.; et al. β-Catenin Signaling Controls Metastasis in Braf-Activated Pten-Deficient Melanomas. Cancer Cell 2011, 20, 741–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nsengimana, J.; Laye, J.; Filia, A.; O’Shea, S.; Muralidhar, S.; Poźniak, J.; Droop, A.; Chan, M.; Walker, C.; Parkinson, L.; et al. β-Catenin-Mediated Immune Evasion Pathway Frequently Operates in Primary Cutaneous Melanomas. J. Clin. Investig. 2018, 128, 2048–2063. [Google Scholar] [CrossRef] [Green Version]
- Shah, K.V.; Chien, A.J.; Yee, C.; Moon, R.T. CTLA-4 Is a Direct Target of Wnt/β-Catenin Signaling and Is Expressed in Human Melanoma Tumors. J. Investig. Dermatol. 2008, 128, 2870–2879. [Google Scholar] [CrossRef] [Green Version]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-Intrinsic β-Catenin Signalling Prevents Anti-Tumour Immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Eddy, K.; Chen, S. Glutamatergic Signaling a Therapeutic Vulnerability in Melanoma. Cancers 2021, 13, 3874. [Google Scholar] [CrossRef]
- Chen, S.; Zhu, H.; Wetzel, W.J.; Philbert, M.A. Spontaneous Melanocytosis in Transgenic Mice. J. Investig. Derm. 1996, 106, 1145–1151. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.-S.; Namkoong, J.; Wall, B.A.; Gleason, R.; Lee, H.J.; Chen, S. Oncogenic Activities of Metabotropic Glutamate Receptor 1 (Grm1) in Melanocyte Transformation. Pigment Cell Melanoma Res. 2008, 21, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, Y.; Harada, T.; Funasaka, Y.; Nakao, K.; Takahara, C.; Abdel-Daim, M.; Sakai, N.; Saito, N.; Nishigori, C.; Aiba, A. Metabotropic Glutamate Receptor Subtype-1 Is Essential for in Vivo Growth of Melanoma. Oncogene 2008, 27, 7162–7170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marín, Y.E.; Namkoong, J.; Cohen-Solal, K.; Shin, S.-S.; Martino, J.J.; Oka, M.; Chen, S. Stimulation of Oncogenic Metabotropic Glutamate Receptor 1 in Melanoma Cells Activates ERK1/2 via PKCepsilon. Cell Signal 2006, 18, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.-S.; Wall, B.A.; Goydos, J.S.; Chen, S. AKT2 Is a Downstream Target of Metabotropic Glutamate Receptor 1 (Grm1). Pigment Cell Melanoma Res. 2010, 23, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Namkoong, J.; Shin, S.-S.; Lee, H.J.; Marín, Y.E.; Wall, B.A.; Goydos, J.S.; Chen, S. Metabotropic Glutamate Receptor 1 and Glutamate Signaling in Human Melanoma. Cancer Res. 2007, 67, 2298–2305. [Google Scholar] [CrossRef] [Green Version]
- Mehnert, J.M.; Silk, A.W.; Lee, J.H.; Dudek, L.; Jeong, B.-S.; Li, J.; Schenkel, J.M.; Sadimin, E.; Kane, M.; Lin, H.; et al. A Phase II Trial of Riluzole, an Antagonist of Metabotropic Glutamate Receptor 1 (GRM1) Signaling, in Patients with Advanced Melanoma. Pigment Cell Melanoma Res. 2018, 31, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Singh, S.J.; Eddy, K.; Filipp, F.V.; Chen, S. Concurrent Targeting of Glutaminolysis and Metabotropic Glutamate Receptor 1 (GRM1) Reduces Glutamate Bioavailability in GRM1+ Melanoma. Cancer Res. 2019, 79, 1799–1809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neto, A.; Ceol, C.J. Melanoma-Associated GRM3 Variants Dysregulate Melanosome Trafficking and CAMP Signaling. Pigment Cell Melanoma Res. 2018, 31, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.-N.; Li, C.; Huang, L.; Chen, L.; Tang, Z.; Han, G.; Liu, Y. Characterization of Group I Metabotropic Glutamate Receptors in Rat and Human Adrenal Glands. Front. Physiol. 2020, 11, 401. [Google Scholar] [CrossRef] [PubMed]
- Tucker, B.; Richards, R.I.; Lardelli, M. Contribution of MGluR and Fmr1 Functional Pathways to Neurite Morphogenesis, Craniofacial Development and Fragile X Syndrome. Hum. Mol. Genet. 2006, 15, 3446–3458. [Google Scholar] [CrossRef] [PubMed]
- Zigler, M.; Kamiya, T.; Brantley, E.C.; Villares, G.J.; Bar-Eli, M. PAR-1 and Thrombin: The Ties That Bind the Microenvironment to Melanoma Metastasis. Cancer Res. 2011, 71, 6561–6566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacquelot, N.; Duong, C.P.M.; Belz, G.T.; Zitvogel, L. Targeting Chemokines and Chemokine Receptors in Melanoma and Other Cancers. Front. Immunol. 2018, 9, 2480. [Google Scholar] [CrossRef]
- Revankar, C.M.; Cimino, D.F.; Sklar, L.A.; Arterburn, J.B.; Prossnitz, E.R. A Transmembrane Intracellular Estrogen Receptor Mediates Rapid Cell Signaling. Science 2005, 307, 1625–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Liu, D. Does GPER Really Function as a G Protein-Coupled Estrogen Receptor in Vivo? Front. Endocrinol (Lausanne) 2020, 11, 148. [Google Scholar] [CrossRef] [PubMed]
- Hamm, H.E. The Many Faces of G Protein Signaling. J. Biol. Chem. 1998, 273, 669–672. [Google Scholar] [CrossRef] [Green Version]
- Martin, E.L.; Rens-Domiano, S.; Schatz, P.J.; Hamm, H.E. Potent Peptide Analogues of a G Protein Receptor-Binding Region Obtained with a Combinatorial Library. J. Biol. Chem. 1996, 271, 361–366. [Google Scholar] [CrossRef] [Green Version]
- Wettschureck, N.; Offermanns, S. Mammalian G Proteins and Their Cell Type Specific Functions. Physiol Rev. 2005, 85, 1159–1204. [Google Scholar] [CrossRef] [Green Version]
- Inoue, A.; Raimondi, F.; Kadji, F.M.N.; Singh, G.; Kishi, T.; Uwamizu, A.; Ono, Y.; Shinjo, Y.; Ishida, S.; Arang, N.; et al. Illuminating G-Protein-Coupling Selectivity of GPCRs. Cell 2019, 177, 1933–1947.e25. [Google Scholar] [CrossRef]
- Sassone-Corsi, P. The Cyclic AMP Pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011148. [Google Scholar] [CrossRef]
- Sutherland, E.W.; Rall, T.W. Fractionation and Characterization of a Cyclic Adenine Ribonucleotide Formed by Tissue Particles. J. Biol. Chem. 1958, 232, 1077–1091. [Google Scholar] [CrossRef]
- Zaccolo, M.; Zerio, A.; Lobo, M.J. Subcellular Organization of the CAMP Signaling Pathway. Pharm. Rev. 2021, 73, 278–309. [Google Scholar] [CrossRef] [PubMed]
- Hanoune, J.; Defer, N. Regulation and Role of Adenylyl Cyclase Isoforms. Annu. Rev. Pharm. Toxicol. 2001, 41, 145–174. [Google Scholar] [CrossRef] [PubMed]
- Dessauer, C.W.; Scully, T.T.; Gilman, A.G. Interactions of Forskolin and ATP with the Cytosolic Domains of Mammalian Adenylyl Cyclase. J. Biol. Chem. 1997, 272, 22272–22277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plagge, A.; Kelsey, G.; Germain-Lee, E.L. Physiological Functions of the Imprinted Gnas Locus and Its Protein Variants Gαs and XLαs in Human and Mouse. J. Endocrinol. 2008, 196, 193–214. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Más, J.; Guillo, L.A.; Zanna, P.; Jiménez-Cervantes, C.; García-Borrón, J.C. Role of G Protein-Coupled Receptor Kinases in the Homologous Desensitization of the Human and Mouse Melanocortin 1 Receptors. Mol. Endocrinol. 2005, 19, 1035–1048. [Google Scholar] [CrossRef] [Green Version]
- Innamorati, G.; Wilkie, T.M.; Kantheti, H.S.; Valenti, M.T.; Dalle Carbonare, L.; Giacomello, L.; Parenti, M.; Melisi, D.; Bassi, C. The Curious Case of Gαs Gain-of-Function in Neoplasia. BMC Cancer 2018, 18, 293. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [CrossRef] [Green Version]
- Frey, U.; Fritz, A.; Rotterdam, S.; Schmid, K.; Potthoff, A.; Altmeyer, P.; Siffert, W.; Brockmeyer, N. GNAS1 T393C Polymorphism and Disease Progression in Patients with Malignant Melanoma. Eur J. Med. Res. 2010, 15, 422–427. [Google Scholar] [CrossRef] [Green Version]
- Frey, U.H.; Alakus, H.; Wohlschlaeger, J.; Schmitz, K.J.; Winde, G.; van Calker, H.G.; Jöckel, K.-H.; Siffert, W.; Schmid, K.W. GNAS1 T393C Polymorphism and Survival in Patients with Sporadic Colorectal Cancer. Clin. Cancer Res. 2005, 11, 5071–5077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frey, U.H.; Eisenhardt, A.; Lümmen, G.; Rübben, H.; Jöckel, K.-H.; Schmid, K.W.; Siffert, W. The T393C Polymorphism of the G Alpha s Gene (GNAS1) Is a Novel Prognostic Marker in Bladder Cancer. Cancer Epidemiol. Biomark. Prev. 2005, 14, 871–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thul, P.J.; Åkesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Ait Blal, H.; Alm, T.; Asplund, A.; Björk, L.; Breckels, L.M.; et al. A Subcellular Map of the Human Proteome. Science 2017, 356, eaal3321. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A Pathology Atlas of the Human Cancer Transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishina, H.; Nimota, K.; Kukimoto, I.; Maehama, T.; Takahashi, K.; Hoshino, S.; Kanaho, Y.; Katada, T. Significance of Thr182 in the Nucleotide-Exchange and GTP-Hydrolysis Reactions of the Alpha Subunit of GTP-Binding Protein Gi2. J. Biochem. 1995, 118, 1083–1089. [Google Scholar] [CrossRef] [PubMed]
- Pace, A.M.; Wong, Y.H.; Bourne, H.R. A Mutant Alpha Subunit of Gi2 Induces Neoplastic Transformation of Rat-1 Cells. Proc. Natl. Acad. Sci. USA 1991, 88, 7031–7035. [Google Scholar] [CrossRef] [Green Version]
- Cooper, D.M.F. Regulation and Organization of Adenylyl Cyclases and CAMP. Biochem. J. 2003, 375, 517–529. [Google Scholar] [CrossRef] [Green Version]
- Halls, M.L.; Cooper, D.M.F. Adenylyl Cyclase Signalling Complexes—Pharmacological Challenges and Opportunities. Pharmacol. Ther. 2017, 172, 171–180. [Google Scholar] [CrossRef]
- Kleinboelting, S.; Diaz, A.; Moniot, S.; van den Heuvel, J.; Weyand, M.; Levin, L.R.; Buck, J.; Steegborn, C. Crystal Structures of Human Soluble Adenylyl Cyclase Reveal Mechanisms of Catalysis and of Its Activation through Bicarbonate. Proc. Natl. Acad. Sci. USA 2014, 111, 3727–3732. [Google Scholar] [CrossRef] [Green Version]
- Magro, C.M.; Neil Crowson, A.; Desman, G.; Zippin, J.H. Soluble Adenylyl Cyclase Antibody Profile as a Diagnostic Adjunct in the Assessment of Pigmented Lesions. Arch. Derm. 2012, 148, 335–344. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Wu, F.; Shi, Y.; Yang, D.; Xu, M.; Lai, Y.; Liu, Y. Identification of Key Candidate Genes Involved in Melanoma Metastasis. Mol. Med. Rep. 2019, 20, 903–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, M.; Dai, J.; Tang, H.; Xu, T.; Yu, S.; Si, L.; Cui, C.; Sheng, X.; Chi, Z.; Mao, L.; et al. MicroRNA-23a-3p Inhibits Mucosal Melanoma Growth and Progression through Targeting Adenylate Cyclase 1 and Attenuating CAMP and MAPK Pathways. Theranostics 2019, 9, 945–960. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, C.I.; Castro-Pérez, E.; Prabhakar, K.; Block, L.; Longley, B.J.; Wisinski, J.A.; Kimple, M.E.; Setaluri, V. EPAC–RAP1 Axis-Mediated Switch in the Response of Primary and Metastatic Melanoma to Cyclic AMP. Mol. Cancer Res. 2017, 15, 1792–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, C.I.; Castro-Pérez, E.; Longley, B.J.; Setaluri, V. Elevated Cyclic AMP Levels Promote BRAFCA/Pten-/- Mouse Melanoma Growth but PCREB Is Negatively Correlated with Human Melanoma Progression. Cancer Lett 2018, 414, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, C.M.; Johnson, L.A.; Piccioni, F.; Townes, A.; Frederick, D.T.; Donahue, M.K.; Narayan, R.; Flaherty, K.T.; Wargo, J.A.; Root, D.E.; et al. A Melanocyte Lineage Program Confers Resistance to MAP Kinase Pathway Inhibition. Nature 2013, 504, 138–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omori, K.; Kotera, J. Overview of PDEs and Their Regulation. Circ. Res. 2007, 100, 309–327. [Google Scholar] [CrossRef] [PubMed]
- Bang, J.; Zippin, J.H. Cyclic Adenosine Monophosphate (CAMP) Signaling in Melanocyte Pigmentation and Melanomagenesis. Pigment Cell Melanoma Res. 2021, 34, 28–43. [Google Scholar] [CrossRef] [PubMed]
- Khaled, M.; Levy, C.; Fisher, D.E. Control of Melanocyte Differentiation by a MITF–PDE4D3 Homeostatic Circuit. Genes Dev. 2010, 24, 2276–2281. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.-C.; Xu, L.; Ding, L.-W.; Sharma, A.; Liu, L.-Z.; Yang, H.; Tan, P.; Vadgama, J.; Karlan, B.Y.; Lester, J.; et al. Genomic and Functional Characterizations of Phosphodiesterase Subtype 4D in Human Cancers. Proc. Natl. Acad. Sci. USA 2013, 110, 6109–6114. [Google Scholar] [CrossRef] [Green Version]
- Marquette, A.; André, J.; Bagot, M.; Bensussan, A.; Dumaz, N. ERK and PDE4 Cooperate to Induce RAF Isoform Switching in Melanoma. Nat. Struct. Mol. Biol 2011, 18, 584–591. [Google Scholar] [CrossRef]
- Dumaz, N.; Hayward, R.; Martin, J.; Ogilvie, L.; Hedley, D.; Curtin, J.A.; Bastian, B.C.; Springer, C.; Marais, R. In Melanoma, RAS Mutations Are Accompanied by Switching Signaling from BRAF to CRAF and Disrupted Cyclic AMP Signaling. Cancer Res. 2006, 66, 9483–9491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delyon, J.; Servy, A.; Laugier, F.; André, J.; Ortonne, N.; Battistella, M.; Mourah, S.; Bensussan, A.; Lebbé, C.; Dumaz, N. PDE4D Promotes FAK-Mediated Cell Invasion in BRAF-Mutated Melanoma. Oncogene 2017, 36, 3252–3262. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.A.; Perkins, J.P.; Krebs, E.G. An Adenosine 3′,5′-Monophosphate-Dependant Protein Kinase from Rabbit Skeletal Muscle. J. Biol. Chem. 1968, 243, 3763–3765. [Google Scholar] [CrossRef]
- Skalhegg, B.S.; Tasken, K. Specificity in the CAMP/PKA Signaling Pathway. Differential Expression, Regulation, and Subcellular Localization of Subunits of PKA. Front. Biosci. 2000, 5, D678–D693. [Google Scholar] [CrossRef] [Green Version]
- Taskén, K.; Skålhegg, B.S.; Taskén, K.A.; Solberg, R.; Knutsen, H.K.; Levy, F.O.; Sandberg, M.; Orstavik, S.; Larsen, T.; Johansen, A.K.; et al. Structure, Function, and Regulation of Human CAMP-Dependent Protein Kinases. Adv. Second Messenger Phosphoprot. Res. 1997, 31, 191–204. [Google Scholar] [CrossRef]
- Kirschner, L.S.; Carney, J.A.; Pack, S.D.; Taymans, S.E.; Giatzakis, C.; Cho, Y.S.; Cho-Chung, Y.S.; Stratakis, C.A. Mutations of the Gene Encoding the Protein Kinase A Type I-Alpha Regulatory Subunit in Patients with the Carney Complex. Nat. Genet. 2000, 26, 89–92. [Google Scholar] [CrossRef]
- Liu, Q.; Tong, D.; Liu, G.; Yi, Y.; Zhang, D.; Zhang, J.; Zhang, Y.; Huang, Z.; Li, Y.; Chen, R.; et al. Carney Complex with PRKAR1A Gene Mutation. Medicine 2017, 96, e8999. [Google Scholar] [CrossRef]
- Cohen, J.N.; Yeh, I.; Mully, T.W.; LeBoit, P.E.; McCalmont, T.H. Genomic and Clinicopathologic Characteristics of PRKAR1A-Inactivated Melanomas: Toward Genetic Distinctions of Animal-Type Melanoma/Pigment Synthesizing Melanoma. Am. J. Surg. Pathol. 2020, 44, 805–816. [Google Scholar] [CrossRef]
- Beebe, S.J.; Salomonsky, P.; Holroyd, C.; Becker, D. Differential Expression of Cyclic AMP-Dependent Protein Kinase Isozymes in Normal Human Melanocytes and Malignant Melanomas. Cell Growth Differ. 1993, 4, 1005. [Google Scholar]
- Hiramoto, K.; Murata, T.; Shimizu, K.; Morita, H.; Inui, M.; Manganiello, V.C.; Tagawa, T.; Arai, N. Role of Phosphodiesterase 2 in Growth and Invasion of Human Malignant Melanoma Cells. Cell Signal. 2014, 26, 1807–1817. [Google Scholar] [CrossRef] [Green Version]
- Lyons, J.; Bastian, B.C.; McCormick, F. MC1R and CAMP Signaling Inhibit Cdc25B Activity and Delay Cell Cycle Progression in Melanoma Cells. Proc. Natl. Acad. Sci. USA 2013, 110, 13845–13850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, M.; Li, Y.; Dillon, T.J.; Stork, P.J.S. Phosphorylation of Rap1 by CAMP-Dependent Protein Kinase (PKA) Creates a Binding Site for KSR to Sustain ERK Activation by CAMP. J. Biol. Chem. 2017, 292, 1449–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostojić, J.; Yoon, Y.-S.; Sonntag, T.; Nguyen, B.; Vaughan, J.M.; Shokhirev, M.; Montminy, M. Transcriptional Co-Activator Regulates Melanocyte Differentiation and Oncogenesis by Integrating CAMP and MAPK/ERK Pathways. Cell Rep. 2021, 35, 109136. [Google Scholar] [CrossRef] [PubMed]
- Buscà, R.; Ballotti, R. Cyclic AMP a Key Messenger in the Regulation of Skin Pigmentation. Pigment. Cell Res. 2000, 13, 60–69. [Google Scholar] [CrossRef]
- Mobley, A.K.; Braeuer, R.R.; Kamiya, T.; Shoshan, E.; Bar-Eli, M. Driving Transcriptional Regulators in Melanoma Metastasis. Cancer Metastasis Rev. 2012, 31, 621–632. [Google Scholar] [CrossRef]
- Dobroff, A.S.; Wang, H.; Melnikova, V.O.; Villares, G.J.; Zigler, M.; Huang, L.; Bar-Eli, M. Silencing CAMP-Response Element-Binding Protein (CREB) Identifies CYR61 as a Tumor Suppressor Gene in Melanoma. J. Biol. Chem. 2009, 284, 26194–26206. [Google Scholar] [CrossRef] [Green Version]
- Xie, S.; Price, J.E.; Luca, M.; Jean, D.; Ronai, Z.; Bar-Eli, M. Dominant-Negative CREB Inhibits Tumor Growth and Metastasis of Human Melanoma Cells. Oncogene 1997, 15, 2069–2075. [Google Scholar] [CrossRef] [Green Version]
- Braeuer, R.R.; Zigler, M.; Villares, G.J.; Dobroff, A.S.; Bar-Eli, M. Transcriptional Control of Melanoma Metastasis: The Importance of the Tumor Microenvironment. Semin. Cancer Biol. 2011, 21, 83–88. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zhou, X.; Yang, J.; Sun, Q.; Liu, Y.; Li, N.; Zhang, Z.; Xu, H. Circ-GLI1 Promotes Metastasis in Melanoma through Interacting with P70S6K2 to Activate Hedgehog/GLI1 and Wnt/β-Catenin Pathways and Upregulate Cyr61. Cell Death Dis. 2020, 11, 596. [Google Scholar] [CrossRef]
- White, J.R.; Thompson, D.T.; Koch, K.E.; Kiriazov, B.S.; Beck, A.C.; van der Heide, D.M.; Grimm, B.G.; Kulak, M.V.; Weigel, R.J. AP-2α-Mediated Activation of E2F and EZH2 Drives Melanoma Metastasis. Cancer Res. 2021, 81, 4455–4470. [Google Scholar] [CrossRef]
- Nemlich, Y.; Baruch, E.N.; Besser, M.J.; Shoshan, E.; Bar-Eli, M.; Anafi, L.; Barshack, I.; Schachter, J.; Ortenberg, R.; Markel, G. ADAR1-Mediated Regulation of Melanoma Invasion. Nat. Commun. 2018, 9, 2154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W.; Jin, L.; Jiang, C.C.; Long, G.V.; Scolyer, R.A.; Wu, Q.; Zhang, X.D.; Mei, Y.; Wu, M. AEBP1 Upregulation Confers Acquired Resistance to BRAF (V600E) Inhibition in Melanoma. Cell Death Dis. 2013, 4, e914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Kong, Q.; Wang, J.; Jiang, Y.; Hua, H. Complex Roles of CAMP–PKA–CREB Signaling in Cancer. Exp. Hematol. Oncol. 2020, 9, 32. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Murata, T.; Shimizu, K.; Nakagawa, T.; Sugiyama, T.; Inui, M.; Hiramoto, K.; Tagawa, T. A Role for Cyclic Nucleotide Phosphodiesterase 4 in Regulation of the Growth of Human Malignant Melanoma Cells. Oncol. Rep. 2007, 17, 1133–1139. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, C.I.; Setaluri, V. EPAC Mediates the Dual Role of CAMP Signaling in Melanoma. Oncoscience 2018, 6, 283–284. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, A.M.; Piazza, G.A.; Tinsley, H.N. The Role of Cyclic Nucleotide Signaling Pathways in Cancer: Targets for Prevention and Treatment. Cancers 2014, 6, 436–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer Cell 2017, 32, 204–220.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Raamsdonk, C.D.; Griewank, K.G.; Crosby, M.B.; Garrido, M.C.; Vemula, S.; Wiesner, T.; Obenauf, A.C.; Wackernagel, W.; Green, G.; Bouvier, N.; et al. Mutations in GNA11 in Uveal Melanoma. N. Engl. J. Med. 2010, 363, 2191–2199. [Google Scholar] [CrossRef] [Green Version]
- Urtatiz, O.; Van Raamsdonk, C.D. Gnaq and Gna11 in the Endothelin Signaling Pathway and Melanoma. Front. Genet. 2016, 7, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Hayre, M.; Vázquez-Prado, J.; Kufareva, I.; Stawiski, E.W.; Handel, T.M.; Seshagiri, S.; Gutkind, J.S. The Emerging Mutational Landscape of G Proteins and G-Protein-Coupled Receptors in Cancer. Nat. Rev. Cancer 2013, 13, 412–424. [Google Scholar] [CrossRef]
- Maziarz, M.; Leyme, A.; Marivin, A.; Luebbers, A.; Patel, P.P.; Chen, Z.; Sprang, S.R.; Garcia-Marcos, M. Atypical Activation of the G Protein Gαq by the Oncogenic Mutation Q209P. J. Biol. Chem. 2018, 293, 19586–19599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Möller, I.; Murali, R.; Müller, H.; Wiesner, T.; Jackett, L.A.; Scholz, S.L.; Cosgarea, I.; van de Nes, J.A.; Sucker, A.; Hillen, U.; et al. Activating Cysteinyl Leukotriene Receptor 2 (CYSLTR2) Mutations in Blue Nevi. Mod. Pathol. 2017, 30, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.L.-Y.; Urtatiz, O.; Van Raamsdonk, C.D. Oncogenic G Protein GNAQ Induces Uveal Melanoma and Intravasation in Mice. Cancer Res. 2015, 75, 3384–3397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urtatiz, O.; Cook, C.; Huang, J.L.-Y.; Yeh, I.; Van Raamsdonk, C.D. GNAQQ209L Expression Initiated in Multipotent Neural Crest Cells Drives Aggressive Melanoma of the Central Nervous System. Pigment Cell Melanoma Res. 2020, 33, 96–111. [Google Scholar] [CrossRef]
- Moore, A.R.; Ran, L.; Guan, Y.; Sher, J.J.; Hitchman, T.D.; Zhang, J.Q.; Hwang, C.; Walzak, E.G.; Shoushtari, A.N.; Monette, S.; et al. GNA11 Q209L Mouse Model Reveals RasGRP3 as an Essential Signaling Node in Uveal Melanoma. Cell Rep. 2018, 22, 2455–2468. [Google Scholar] [CrossRef] [Green Version]
- Annala, S.; Feng, X.; Shridhar, N.; Eryilmaz, F.; Patt, J.; Yang, J.; Pfeil, E.M.; Cervantes-Villagrana, R.D.; Inoue, A.; Häberlein, F.; et al. Direct Targeting of Gαq and Gα11 Oncoproteins in Cancer Cells. Sci. Signal. 2019, 12, eaau5948. [Google Scholar] [CrossRef]
- Kostenis, E.; Pfeil, E.M.; Annala, S. Heterotrimeric Gq Proteins as Therapeutic Targets? J. Biol. Chem. 2020, 295, 5206–5215. [Google Scholar] [CrossRef] [Green Version]
- Ambrosini, G.; Pratilas, C.A.; Qin, L.-X.; Tadi, M.; Surriga, O.; Carvajal, R.D.; Schwartz, G.K. Identification of Unique MEK-Dependent Genes in GNAQ Mutant Uveal Melanoma Involved in Cell Growth, Tumor Cell Invasion and MEK-Resistance. Clin. Cancer Res. 2012, 18, 3552–3561. [Google Scholar] [CrossRef] [Green Version]
- Asundi, J.; Lacap, J.A.; Clark, S.; Nannini, M.; Roth, L.; Polakis, P. MAPK Pathway Inhibition Enhances the Efficacy of an Anti-Endothelin B Receptor Drug Conjugate by Inducing Target Expression in Melanoma. Mol. Cancer 2014, 13, 1599–1610. [Google Scholar] [CrossRef] [Green Version]
- Hitchman, T.D.; Bayshtok, G.; Ceraudo, E.; Moore, A.R.; Lee, C.; Jia, R.; Wang, N.; Pachai, M.R.; Shoushtari, A.N.; Francis, J.H.; et al. Combined Inhibition of Gαq and MEK Enhances Therapeutic Efficacy in Uveal Melanoma. Clin. Cancer Res. 2021, 27, 1476–1490. [Google Scholar] [CrossRef]
- Smrcka, A.V.; Hepler, J.R.; Brown, K.O.; Sternweis, P.C. Regulation of Polyphosphoinositide-Specific Phospholipase C Activity by Purified Gq. Science 1991, 251, 804–807. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.J.; Chae, H.Z.; Rhee, S.G.; Exton, J.H. Activation of the Beta 1 Isozyme of Phospholipase C by Alpha Subunits of the Gq Class of G Proteins. Nature 1991, 350, 516–518. [Google Scholar] [CrossRef] [PubMed]
- Waldo, G.L.; Boyer, J.L.; Morris, A.J.; Harden, T.K. Purification of an AlF4- and G-Protein Beta Gamma-Subunit-Regulated Phospholipase C-Activating Protein. J. Biol. Chem. 1991, 266, 14217–14225. [Google Scholar] [CrossRef]
- Lyon, A.M.; Tesmer, J.J.G. Structural Insights into Phospholipase C-β Function. Mol. Pharm. 2013, 84, 488–500. [Google Scholar] [CrossRef] [Green Version]
- Chua, V.; Lapadula, D.; Randolph, C.; Benovic, J.L.; Wedegaertner, P.B.; Aplin, A.E. Dysregulated GPCR Signaling and Therapeutic Options in Uveal Melanoma. Mol. Cancer Res. 2017, 15, 501–506. [Google Scholar] [CrossRef] [Green Version]
- Johansson, P.; Aoude, L.G.; Wadt, K.; Glasson, W.J.; Warrier, S.K.; Hewitt, A.W.; Kiilgaard, J.F.; Heegaard, S.; Isaacs, T.; Franchina, M.; et al. Deep Sequencing of Uveal Melanoma Identifies a Recurrent Mutation in PLCB4. Oncotarget 2016, 7, 4624–4631. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhao, X.; Wang, D.; He, W.; Zhang, S.; Cao, W.; Huang, Y.; Wang, L.; Zhou, S.; Luo, K. Up-Regulated Expression of Phospholipase C, Β1 Is Associated with Tumor Cell Proliferation and Poor Prognosis in Hepatocellular Carcinoma. Onco Targets 2016, 9, 1697–1706. [Google Scholar] [CrossRef] [Green Version]
- Sengelaub, C.A.; Navrazhina, K.; Ross, J.B.; Halberg, N.; Tavazoie, S.F. PTPRN2 and PLCβ1 Promote Metastatic Breast Cancer Cell Migration through PI(4,5)P2-dependent Actin Remodeling. EMBO J. 2016, 35, 62–76. [Google Scholar] [CrossRef]
- Arita, Y.; O’Driscoll, K.R.; Weinstein, I.B. Growth of Human Melanocyte Cultures Supported by 12-O-Tetradecanoylphorbol-13-Acetate Is Mediated through Protein Kinase C Activation. Cancer Res. 1992, 52, 4514–4521. [Google Scholar]
- Petit, V.; Raymond, J.; Alberti, C.; Pouteaux, M.; Gallagher, S.J.; Nguyen, M.Q.; Aplin, A.E.; Delmas, V.; Larue, L. C57BL/6 Congenic Mouse NRASQ61K Melanoma Cell Lines Are Highly Sensitive to the Combination of Mek and Akt Inhibitors in Vitro and in Vivo. Pigment Cell Melanoma Res. 2019, 32, 829–841. [Google Scholar] [CrossRef]
- Tamura, A.; Halaban, R.; Moellmann, G.; Cowan, J.M.; Lerner, M.R.; Lerner, A.B. Normal Murine Melanocytes in Culture. In Vitro Cell Dev. Biol. 1987, 23, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, T.; Yamauchi, M.; Liang, Z.; Itai, A.; Sakaguchi, M.; Nagano, T.; Kamada, S.; Oka, M. TPA Inhibits Melanoma Growth through Inactivation of STAT3 through Protein Tyrosine Phosphatases. J. Dermatol. Sci. 2017, 86, e94. [Google Scholar] [CrossRef]
- Jørgensen, K.; Skrede, M.; Cruciani, V.; Mikalsen, S.-O.; Slipicevic, A.; Flørenes, V.A. Phorbol Ester Phorbol-12-Myristate-13-Acetate Promotes Anchorage-Independent Growth and Survival of Melanomas through MEK-Independent Activation of ERK1/2. Biochem. Biophys. Res. Commun 2005, 329, 266–274. [Google Scholar] [CrossRef]
- La Porta, C.A.; Porro, D.; Comolli, R. Opposite Effects of TPA on G1/S Transition and on Cell Size in the Low Metastatic B16F1 with Respect to High Metastatic BL6 Murine Melanoma Cells. Cancer Lett. 1998, 132, 159–164. [Google Scholar] [CrossRef]
- Dissanayake, S.K.; Wade, M.; Johnson, C.E.; O’Connell, M.P.; Leotlela, P.D.; French, A.D.; Shah, K.V.; Hewitt, K.J.; Rosenthal, D.T.; Indig, F.E.; et al. The Wnt5A/Protein Kinase C Pathway Mediates Motility in Melanoma Cells via the Inhibition of Metastasis Suppressors and Initiation of an Epithelial to Mesenchymal Transition. J. Biol. Chem. 2007, 282, 17259–17271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Geltinger, C.; Kishikawa, S.; Ohshima, K.; Murata, T.; Nomura, N.; Nakahara, T.; Yokoyama, K.K. Treatment of Mouse Melanoma Cells with Phorbol 12-Myristate 13-Acetate Counteracts Mannosylerythritol Lipid-Induced Growth Arrest and Apoptosis. Cytotechnology 2000, 33, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Foskett, J.K.; White, C.; Cheung, K.-H.; Mak, D.-O.D. Inositol Trisphosphate Receptor Ca2+ Release Channels. Physiol. Rev. 2007, 87, 593–658. [Google Scholar] [CrossRef] [Green Version]
- Cox, J.L.; Lancaster, T.; Carlson, C.G. Changes in the Motility of B16F10 Melanoma Cells Induced by Alterations in Resting Calcium Influx. Melanoma Res. 2002, 12, 211–219. [Google Scholar] [CrossRef]
- Sun, J.; Lu, F.; He, H.; Shen, J.; Messina, J.; Mathew, R.; Wang, D.; Sarnaik, A.A.; Chang, W.-C.; Kim, M.; et al. STIM1- and Orai1-Mediated Ca2+ Oscillation Orchestrates Invadopodium Formation and Melanoma Invasion. J. Cell Biol. 2014, 207, 535–548. [Google Scholar] [CrossRef] [Green Version]
- Umemura, M.; Baljinnyam, E.; Feske, S.; De Lorenzo, M.S.; Xie, L.-H.; Feng, X.; Oda, K.; Makino, A.; Fujita, T.; Yokoyama, U.; et al. Store-Operated Ca2+ Entry (SOCE) Regulates Melanoma Proliferation and Cell Migration. PLoS ONE 2014, 9, e89292. [Google Scholar] [CrossRef]
- Denning, M.F. Specifying Protein Kinase C Functions in Melanoma. Pigment Cell Melanoma Res. 2012, 25, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, N.; Langeberg, L.K.; Gould, C.M.; Newton, A.C.; Scott, J.D. Interaction with AKAP79 Modifies the Cellular Pharmacology of PKC. Mol. Cell 2010, 37, 541–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.-Y.; Wu, H.; Killoran, C.E.; Gilchrest, B.A. The Receptor for Activated C-Kinase-I (RACK-I) Anchors Activated PKC-Beta on Melanosomes. J. Cell Sci. 2004, 117, 3659–3668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schechtman, D.; Mochly-Rosen, D. Adaptor Proteins in Protein Kinase C-Mediated Signal Transduction. Oncogene 2001, 20, 6339–6347. [Google Scholar] [CrossRef] [Green Version]
- Voris, J.P.; Sitailo, L.A.; Rahn, H.R.; Defnet, A.; Gerds, A.T.; Sprague, R.; Yadav, V.; Caroline Le Poole, I.; Denning, M.F. Functional Alterations in Protein Kinase C Beta II Expression in Melanoma. Pigment Cell Melanoma Res. 2010, 23, 216–224. [Google Scholar] [CrossRef]
- Matsuoka, H.; Tsubaki, M.; Yamazoe, Y.; Ogaki, M.; Satou, T.; Itoh, T.; Kusunoki, T.; Nishida, S. Tamoxifen Inhibits Tumor Cell Invasion and Metastasis in Mouse Melanoma through Suppression of PKC/MEK/ERK and PKC/PI3K/Akt Pathways. Exp. Cell Res. 2009, 315, 2022–2032. [Google Scholar] [CrossRef]
- Oka, M.; Kikkawa, U.; Nishigori, C. Protein Kinase C-BetaII Represses Hepatocyte Growth Factor-Induced Invasion by Preventing the Association of Adapter Protein Gab1 and Phosphatidylinositol 3-Kinase in Melanoma Cells. J. Investig. Derm. 2008, 128, 188–195. [Google Scholar] [CrossRef] [Green Version]
- Park, H.Y.; Russakovsky, V.; Ohno, S.; Gilchrest, B.A. The Beta Isoform of Protein Kinase C Stimulates Human Melanogenesis by Activating Tyrosinase in Pigment Cells. J. Biol. Chem. 1993, 268, 11742–11749. [Google Scholar] [CrossRef]
- Chen, X.; Wu, Q.; Depeille, P.; Chen, P.; Thornton, S.; Kalirai, H.; Coupland, S.E.; Roose, J.P.; Bastian, B.C. RasGRP3 Mediates MAPK Pathway Activation in GNAQ Mutant Uveal Melanoma. Cancer Cell 2017, 31, 685–696.e6. [Google Scholar] [CrossRef] [Green Version]
- Lau, E.; Kluger, H.; Varsano, T.; Lee, K.; Scheffler, I.; Rimm, D.L.; Ideker, T.; Ronai, Z.A. PKCε Promotes Oncogenic Functions of ATF2 in the Nucleus While Blocking Its Apoptotic Function at Mitochondria. Cell 2012, 148, 543–555. [Google Scholar] [CrossRef] [Green Version]
- Mhaidat, N.M.; Thorne, R.F.; Zhang, X.D.; Hersey, P. Regulation of Docetaxel-Induced Apoptosis of Human Melanoma Cells by Different Isoforms of Protein Kinase C. Mol. Cancer Res. 2007, 5, 1073–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aiba, Y.; Oh-hora, M.; Kiyonaka, S.; Kimura, Y.; Hijikata, A.; Mori, Y.; Kurosaki, T. Activation of RasGRP3 by Phosphorylation of Thr-133 Is Required for B Cell Receptor-Mediated Ras Activation. Proc. Natl. Acad. Sci. USA 2004, 101, 16612–16617. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.E.; Goulding, R.E.; Ding, Z.; Partovi, A.; Anthony, K.V.; Beaulieu, N.; Tazmini, G.; Cornell, R.B.; Kay, R.J. Differential Membrane Binding and Diacylglycerol Recognition by C1 Domains of RasGRPs. Biochem. J. 2007, 406, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, C.; Stang, S.L.; Zheng, Y.; Beswick, N.S.; Stone, J.C. Integration of DAG Signaling Systems Mediated by PKC-Dependent Phosphorylation of RasGRP3. Blood 2003, 102, 1414–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cozzi, S.-J.; Parsons, P.G.; Ogbourne, S.M.; Pedley, J.; Boyle, G.M. Induction of Senescence in Diterpene Ester-Treated Melanoma Cells via Protein Kinase C-Dependent Hyperactivation of the Mitogen-Activated Protein Kinase Pathway. Cancer Res. 2006, 66, 10083–10091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schönwasser, D.C.; Marais, R.M.; Marshall, C.J.; Parker, P.J. Activation of the Mitogen-Activated Protein Kinase/Extracellular Signal-Regulated Kinase Pathway by Conventional, Novel, and Atypical Protein Kinase C Isotypes. Mol. Cell Biol. 1998, 18, 790–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsubaki, M.; Matsuoka, H.; Yamamoto, C.; Kato, C.; Ogaki, M.; Satou, T.; Itoh, T.; Kusunoki, T.; Tanimori, Y.; Nishida, S. The Protein Kinase C Inhibitor, H7, Inhibits Tumor Cell Invasion and Metastasis in Mouse Melanoma via Suppression of ERK1/2. Clin. Exp. Metastasis 2007, 24, 431–438. [Google Scholar] [CrossRef]
- Johannessen, C.M.; Boehm, J.S.; Kim, S.Y.; Thomas, S.R.; Wardwell, L.; Johnson, L.A.; Emery, C.M.; Stransky, N.; Cogdill, A.P.; Barretina, J.; et al. COT Drives Resistance to RAF Inhibition through MAP Kinase Pathway Reactivation. Nature 2010, 468, 968–972. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Rathod, D.; Patel, K. Protein Kinase C Inhibitor Anchored BRD4 PROTAC PEGylated Nanoliposomes for the Treatment of Vemurafenib-Resistant Melanoma. Exp. Cell Res. 2020, 396, 112275. [Google Scholar] [CrossRef]
- Kwon, H.; Kim, J.; Jho, E.-H. Role of the Hippo Pathway and Mechanisms for Controlling Cellular Localization of YAP/TAZ. FEBS J. 2021, in press. [Google Scholar] [CrossRef]
- Yu, F.-X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; Li, H.; et al. Regulation of the Hippo-YAP Pathway by G-Protein Coupled Receptor Signaling. Cell 2012, 150, 780–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, X.; Degese, M.S.; Iglesias-Bartolome, R.; Vaque, J.P.; Molinolo, A.A.; Rodrigues, M.; Zaidi, M.R.; Ksander, B.R.; Merlino, G.; Sodhi, A.; et al. Hippo-Independent Activation of YAP by the GNAQ Uveal Melanoma Oncogene through a Trio-Regulated Rho GTPase Signaling Circuitry. Cancer Cell 2014, 25, 831–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.-X.; Luo, J.; Mo, J.-S.; Liu, G.; Kim, Y.C.; Meng, Z.; Zhao, L.; Peyman, G.; Ouyang, H.; Jiang, W.; et al. Mutant Gq/11 Promote Uveal Melanoma Tumorigenesis by Activating YAP. Cancer Cell 2014, 25, 822–830. [Google Scholar] [CrossRef] [Green Version]
- Vaqué, J.P.; Dorsam, R.T.; Feng, X.; Iglesias-Bartolome, R.; Forsthoefel, D.J.; Chen, Q.; Debant, A.; Seeger, M.A.; Ksander, B.R.; Teramoto, H.; et al. A Genome-Wide RNAi Screen Reveals a Trio-Regulated Rho GTPase Circuitry Transducing Mitogenic Signals Initiated by G Protein-Coupled Receptors. Mol. Cell 2013, 49, 94–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradis, J.S.; Acosta, M.; Saddawi-Konefka, R.; Kishore, A.; Lubrano, S.; Gomes, F.; Arang, N.; Tiago, M.; Coma, S.; Wu, X.; et al. Synthetic Lethal Screens Reveal Cotargeting FAK and MEK as a Multimodal Precision Therapy for GNAQ-Driven Uveal Melanoma. Clin. Cancer Res. 2021, 27, 3190–3200. [Google Scholar] [CrossRef] [PubMed]
- Shaulian, E.; Karin, M. AP-1 in Cell Proliferation and Survival. Oncogene 2001, 20, 2390–2400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, L.; Anderson, A.; Nguyen, K.; Ojeda, S.S.; Ortiz-Rivera, I.; Nguyen, T.N.; Zhang, T.; Kaoud, T.S.; Gray, N.S.; Dalby, K.N.; et al. JNK2 Is Required for the Tumorigenic Properties of Melanoma Cells. ACS Chem. Biol. 2019, 14, 1426–1435. [Google Scholar] [CrossRef] [PubMed]
- Estrada, Y.; Dong, J.; Ossowski, L. Positive Crosstalk between ERK and P38 in Melanoma Stimulates Migration and in Vivo Proliferation. Pigment Cell Melanoma Res. 2009, 22, 66–76. [Google Scholar] [CrossRef]
- Pathria, G.; Garg, B.; Garg, K.; Wagner, C.; Wagner, S.N. Dual C-Jun N-Terminal Kinase-Cyclin D1 and Extracellular Signal-Related Kinase-c-Jun Disjunction in Human Melanoma. Br. J. Derm. 2016, 175, 1221–1231. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, L.; He, F.; Yang, J.; Ding, Y.; Ge, S.; Fan, X.; Zhou, Y.; Xu, X.; Jia, R. LACTB Suppresses Melanoma Progression by Attenuating PP1A and YAP Interaction. Cancer Lett. 2021, 506, 67–82. [Google Scholar] [CrossRef]
- Nallet-Staub, F.; Marsaud, V.; Li, L.; Gilbert, C.; Dodier, S.; Bataille, V.; Sudol, M.; Herlyn, M.; Mauviel, A. Pro-Invasive Activity of the Hippo Pathway Effectors YAP and TAZ in Cutaneous Melanoma. J. Investig. Derm. 2014, 134, 123–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Yang, L.; Szeto, P.; Abali, G.K.; Zhang, Y.; Kulkarni, A.; Amarasinghe, K.; Li, J.; Vergara, I.A.; Molania, R.; et al. The Hippo Pathway Oncoprotein YAP Promotes Melanoma Cell Invasion and Spontaneous Metastasis. Oncogene 2020, 39, 5267–5281. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Xie, J.; Zhou, X.; Zhang, L.; Cheng, X.; Liang, C. YAP Activation in Melanoma Contributes to Anoikis Resistance and Metastasis. Exp. Biol. Med. 2021, 246, 888–896. [Google Scholar] [CrossRef] [PubMed]
- Lui, J.W.; Moore, S.P.G.; Huang, L.; Ogomori, K.; Li, Y.; Lang, D. YAP Facilitates Melanoma Migration through Regulation of Actin-Related Protein 2/3 Complex Subunit 5 (ARPC5). Pigment Cell Melanoma Res. 2021, 32, 52–65. [Google Scholar] [CrossRef]
- Tan, S.; Zhao, Z.; Qiao, Y.; Zhang, B.; Zhang, T.; Zhang, M.; Qi, J.; Wang, X.; Meng, M.; Zhou, Q. Activation of the Tumor Suppressive Hippo Pathway by Triptonide as a New Strategy to Potently Inhibit Aggressive Melanoma Cell Metastasis. Biochem. Pharmacol. 2021, 185, 114423. [Google Scholar] [CrossRef]
- Flem-Karlsen, K.; McFadden, E.; Omar, N.; Haugen, M.H.; Øy, G.F.; Ryder, T.; Gullestad, H.P.; Hermann, R.; Mælandsmo, G.M.; Flørenes, V.A. Targeting AXL and the DNA Damage Response Pathway as a Novel Therapeutic Strategy in Melanoma. Mol. Cancer 2020, 19, 895–905. [Google Scholar] [CrossRef] [Green Version]
- Müller, J.; Krijgsman, O.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.M.; Foppen, M.H.G.; et al. Low MITF/AXL Ratio Predicts Early Resistance to Multiple Targeted Drugs in Melanoma. Nat. Commun. 2014, 5, 5712. [Google Scholar] [CrossRef]
- Tizpa, E.; Young, H.J.; Bonjoc, K.-J.C.; Chang, C.-W.; Liu, Y.; Foulks, J.M.; Chaudhry, A. Role of AXL in Metastatic Melanoma and Impact of TP-0903 as a Novel Therapeutic Option for Melanoma Brain Metastasis. J. Clin. Oncol. 2020, 38, e22021. [Google Scholar] [CrossRef]
- Kunz, M.; Moeller, S.; Koczan, D.; Lorenz, P.; Wenger, R.H.; Glocker, M.O.; Thiesen, H.-J.; Gross, G.; Ibrahim, S.M. Mechanisms of Hypoxic Gene Regulation of Angiogenesis Factor Cyr61 in Melanoma Cells. J. Biol. Chem. 2003, 278, 45651–45660. [Google Scholar] [CrossRef] [Green Version]
- Borsotti, P.; Ghilardi, C.; Ostano, P.; Silini, A.; Dossi, R.; Pinessi, D.; Foglieni, C.; Scatolini, M.; Lacal, P.M.; Ferrari, R.; et al. Thrombospondin-1 Is Part of a Slug-Independent Motility and Metastatic Program in Cutaneous Melanoma, in Association with VEGFR-1 and FGF-2. Pigment Cell Melanoma Res. 2015, 28, 73–81. [Google Scholar] [CrossRef]
- Jayachandran, A.; Anaka, M.; Prithviraj, P.; Hudson, C.; McKeown, S.J.; Lo, P.-H.; Vella, L.J.; Goding, C.R.; Cebon, J.; Behren, A. Thrombospondin 1 Promotes an Aggressive Phenotype through Epithelial-to-Mesenchymal Transition in Human Melanoma. Oncotarget 2014, 5, 5782–5797. [Google Scholar] [CrossRef] [Green Version]
- Verfaillie, A.; Imrichova, H.; Atak, Z.K.; Dewaele, M.; Rambow, F.; Hulselmans, G.; Christiaens, V.; Svetlichnyy, D.; Luciani, F.; Van den Mooter, L.; et al. Decoding the Regulatory Landscape of Melanoma Reveals TEADS as Regulators of the Invasive Cell State. Nat. Commun. 2015, 6, 6683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, M.J.; Jiang, X.; Kozasa, T.; Roscoe, W.; Singer, W.D.; Gilman, A.G.; Sternweis, P.C.; Bollag, G. Direct Stimulation of the Guanine Nucleotide Exchange Activity of P115 RhoGEF by Galpha13. Science 1998, 280, 2112–2114. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Nakamura, S.; Mano, H.; Kozasa, T. Galpha 12 Activates Rho GTPase through Tyrosine-Phosphorylated Leukemia-Associated RhoGEF. Proc. Natl. Acad. Sci. USA 2003, 100, 733–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Singer, W.D.; Sternweis, P.C.; Sprang, S.R. Structure of the P115RhoGEF RgRGS Domain-Galpha13/I1 Chimera Complex Suggests Convergent Evolution of a GTPase Activator. Nat. Struct. Mol. Biol. 2005, 12, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, S.; Murga, C.; Zohar, M.; Igishi, T.; Gutkind, J.S. A Novel PDZ Domain Containing Guanine Nucleotide Exchange Factor Links Heterotrimeric G Proteins to Rho. J. Biol. Chem. 1999, 274, 5868–5879. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, I.; Cone, R.D.; Im, S.; Nordlund, J.; Abdel-Malek, Z.A. Binding of Melanotropic Hormones to the Melanocortin Receptor MC1R on Human Melanocytes Stimulates Proliferation and Melanogenesis. Endocrinology 1996, 137, 1627–1633. [Google Scholar] [CrossRef]
- Vogt, S.; Grosse, R.; Schultz, G.; Offermanns, S. Receptor-Dependent RhoA Activation in G12/G13-Deficient Cells: Genetic Evidence for an Involvement of Gq/G11. J. Biol. Chem. 2003, 278, 28743–28749. [Google Scholar] [CrossRef] [Green Version]
- Wells, C.D.; Liu, M.-Y.; Jackson, M.; Gutowski, S.; Sternweis, P.M.; Rothstein, J.D.; Kozasa, T.; Sternweis, P.C. Mechanisms for Reversible Regulation between G13 and Rho Exchange Factors. J. Biol. Chem. 2002, 277, 1174–1181. [Google Scholar] [CrossRef] [Green Version]
- Elste, A.P.; Petersen, I. Expression of Proteinase-Activated Receptor 1-4 (PAR 1-4) in Human Cancer. J. Mol. Histol. 2010, 41, 89–99. [Google Scholar] [CrossRef]
- Mo, J.-S.; Yu, F.-X.; Gong, R.; Brown, J.H.; Guan, K.-L. Regulation of the Hippo–YAP Pathway by Protease-Activated Receptors (PARs). Genes Dev. 2012, 26, 2138–2143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, P.; Stemmle, L.N.; Madden, J.F.; Fields, T.A.; Daaka, Y.; Casey, P.J. A Role for the G12 Family of Heterotrimeric G Proteins in Prostate Cancer Invasion. J. Biol. Chem. 2006, 281, 26483–26490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, P.; Moeller, B.J.; Juneja, J.; Booden, M.A.; Der, C.J.; Daaka, Y.; Dewhirst, M.W.; Fields, T.A.; Casey, P.J. The G12 Family of Heterotrimeric G Proteins Promotes Breast Cancer Invasion and Metastasis. Proc. Natl. Acad. Sci. USA 2006, 103, 8173–8178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minami, K.; Ueda, N.; Ishimoto, K.; Tsujiuchi, T. Lysophosphatidic Acid Receptor-2 (LPA2)-Mediated Signaling Enhances Chemoresistance in Melanoma Cells Treated with Anticancer Drugs. Mol. Cell Biochem. 2020, 469, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt Signaling in Cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- de la Fouchardière, A.; Caillot, C.; Jacquemus, J.; Durieux, E.; Houlier, A.; Haddad, V.; Pissaloux, D. β-Catenin Nuclear Expression Discriminates Deep Penetrating Nevi from Other Cutaneous Melanocytic Tumors. Virchows Arch. 2019, 474, 539–550. [Google Scholar] [CrossRef]
- Yeh, I.; Lang, U.E.; Durieux, E.; Tee, M.K.; Jorapur, A.; Shain, A.H.; Haddad, V.; Pissaloux, D.; Chen, X.; Cerroni, L.; et al. Combined Activation of MAP Kinase Pathway and β-Catenin Signaling Cause Deep Penetrating Nevi. Nat. Commun. 2017, 8, 644. [Google Scholar] [CrossRef]
- Rimm, D.L.; Caca, K.; Hu, G.; Harrison, F.B.; Fearon, E.R. Frequent Nuclear/Cytoplasmic Localization of Beta-Catenin without Exon 3 Mutations in Malignant Melanoma. Am. J. Pathol. 1999, 154, 325–329. [Google Scholar] [CrossRef]
- Demunter, A.; Libbrecht, L.; Degreef, H.; De Wolf-Peeters, C.; van den Oord, J.J. Loss of Membranous Expression of β-Catenin Is Associated with Tumor Progression in Cutaneous Melanoma and Rarely Caused by Exon 3 Mutations. Mod. Pathol. 2002, 15, 454–461. [Google Scholar] [CrossRef] [Green Version]
- Takada, R.; Satomi, Y.; Kurata, T.; Ueno, N.; Norioka, S.; Kondoh, H.; Takao, T.; Takada, S. Monounsaturated Fatty Acid Modification of Wnt Protein: Its Role in Wnt Secretion. Dev. Cell 2006, 11, 791–801. [Google Scholar] [CrossRef] [Green Version]
- Willert, K.; Brown, J.D.; Danenberg, E.; Duncan, A.W.; Weissman, I.L.; Reya, T.; Yates, J.R.; Nusse, R. Wnt Proteins Are Lipid-Modified and Can Act as Stem Cell Growth Factors. Nature 2003, 423, 448–452. [Google Scholar] [CrossRef]
- Lee, E.; Salic, A.; Krüger, R.; Heinrich, R.; Kirschner, M.W. The Roles of APC and Axin Derived from Experimental and Theoretical Analysis of the Wnt Pathway. PLoS Biol. 2003, 1, e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salic, A.; Lee, E.; Mayer, L.; Kirschner, M.W. Control of Beta-Catenin Stability: Reconstitution of the Cytoplasmic Steps of the Wnt Pathway in Xenopus Egg Extracts. Mol. Cell 2000, 5, 523–532. [Google Scholar] [CrossRef]
- Valenta, T.; Hausmann, G.; Basler, K. The Many Faces and Functions of β-Catenin. EMBO J. 2012, 31, 2714–2736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aktary, Z.; Bertrand, J.U.; Larue, L. The WNT-Less Wonder: WNT-Independent β-Catenin Signaling. Pigment Cell Melanoma Res. 2016, 29, 524–540. [Google Scholar] [CrossRef] [Green Version]
- Schepsky, A.; Bruser, K.; Gunnarsson, G.J.; Goodall, J.; Hallsson, J.H.; Goding, C.R.; Steingrimsson, E.; Hecht, A. The Microphthalmia-Associated Transcription Factor Mitf Interacts with Beta-Catenin to Determine Target Gene Expression. Mol. Cell. Biol. 2006, 26, 8914–8927. [Google Scholar] [CrossRef] [Green Version]
- Saito, H.; Yasumoto, K.-I.; Takeda, K.; Takahashi, K.; Fukuzaki, A.; Orikasa, S.; Shibahara, S. Melanocyte-Specific Microphthalmia-Associated Transcription Factor Isoform Activates Its Own Gene Promoter through Physical Interaction with Lymphoid-Enhancing Factor 1. J. Biol. Chem. 2002, 277, 28787–28794. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, A.; Fisher, D.E. The Master Role of Microphthalmia-Associated Transcription Factor in Melanocyte and Melanoma Biology. Lab. Investig. 2017, 97, 649–656. [Google Scholar] [CrossRef] [Green Version]
- Ballotti, R.; Cheli, Y.; Bertolotto, C. The Complex Relationship between MITF and the Immune System: A Melanoma ImmunoTherapy (Response) Factor? Mol. Cancer 2020, 19, 170. [Google Scholar] [CrossRef]
- Baljinnyam, E.; Umemura, M.; De Lorenzo, M.S.; Xie, L.-H.; Nowycky, M.; Iwatsubo, M.; Chen, S.; Goydos, J.S.; Iwatsubo, K. Gβγ Subunits Inhibit Epac-Induced Melanoma Cell Migration. BMC Cancer 2011, 11, 256. [Google Scholar] [CrossRef] [Green Version]
- Bonacci, T.M.; Ghosh, M.; Malik, S.; Smrcka, A.V. Regulatory Interactions between the Amino Terminus of G-Protein Betagamma Subunits and the Catalytic Domain of Phospholipase Cbeta2. J. Biol. Chem. 2005, 280, 10174–10181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leopoldt, D.; Hanck, T.; Exner, T.; Maier, U.; Wetzker, R.; Nürnberg, B. Gβγ Stimulates Phosphoinositide 3-Kinase-γ by Direct Interaction with Two Domains of the Catalytic P110 Subunit. J. Biol. Chem. 1998, 273, 7024–7029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeil, E.M.; Brands, J.; Merten, N.; Vögtle, T.; Vescovo, M.; Rick, U.; Albrecht, I.-M.; Heycke, N.; Kawakami, K.; Ono, Y.; et al. Heterotrimeric G Protein Subunit Gαq Is a Master Switch for Gβγ-Mediated Calcium Mobilization by Gi-Coupled GPCRs. Mol. Cell 2020, 80, 940–954.e6. [Google Scholar] [CrossRef] [PubMed]
- Sellers, L.A.; Alderton, F.; Carruthers, A.M.; Schindler, M.; Humphrey, P.P. Receptor Isoforms Mediate Opposing Proliferative Effects through Gbetagamma-Activated P38 or Akt Pathways. Mol. Cell Biol. 2000, 20, 5974–5985. [Google Scholar] [CrossRef]
- Luttrell, L.M.; Ferguson, S.S.; Daaka, Y.; Miller, W.E.; Maudsley, S.; Della Rocca, G.J.; Lin, F.; Kawakatsu, H.; Owada, K.; Luttrell, D.K.; et al. Beta-Arrestin-Dependent Formation of Beta2 Adrenergic Receptor-Src Protein Kinase Complexes. Science 1999, 283, 655–661. [Google Scholar] [CrossRef]
- Miller, W.E.; Maudsley, S.; Ahn, S.; Khan, K.D.; Luttrell, L.M.; Lefkowitz, R.J. Beta-Arrestin1 Interacts with the Catalytic Domain of the Tyrosine Kinase c-SRC. Role of Beta-Arrestin1-Dependent Targeting of c-SRC in Receptor Endocytosis. J. Biol. Chem. 2000, 275, 11312–11319. [Google Scholar] [CrossRef] [Green Version]
- Krupnick, J.G.; Goodman, O.B.; Keen, J.H.; Benovic, J.L. Arrestin/Clathrin Interaction. Localization of the Clathrin Binding Domain of Nonvisual Arrestins to the Carboxy Terminus. J. Biol. Chem. 1997, 272, 15011–15016. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, V.V.; Gurevich, E.V. Arrestins: Critical Players in Trafficking of Many GPCRs. Prog Mol. Biol. Transl. Sci. 2015, 132, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Shenoy, S.K.; Lefkowitz, R.J. Multifaceted Roles of Beta-Arrestins in the Regulation of Seven-Membrane-Spanning Receptor Trafficking and Signalling. Biochem. J. 2003, 375, 503–515. [Google Scholar] [CrossRef]
- Oakley, R.H.; Laporte, S.A.; Holt, J.A.; Caron, M.G.; Barak, L.S. Differential Affinities of Visual Arrestin, ΒArrestin1, and ΒArrestin2 for G Protein-Coupled Receptors Delineate Two Major Classes of Receptors. J. Biol. Chem. 2000, 275, 17201–17210. [Google Scholar] [CrossRef] [Green Version]
- Miller, W.E.; Lefkowitz, R.J. Expanding Roles for Beta-Arrestins as Scaffolds and Adapters in GPCR Signaling and Trafficking. Curr. Opin. Cell Biol. 2001, 13, 139–145. [Google Scholar] [CrossRef]
- Shenoy, S.K.; Lefkowitz, R.J. Seven-Transmembrane Receptor Signaling through Beta-Arrestin. Sci. STKE 2005, 2005, cm10. [Google Scholar] [CrossRef] [PubMed]
- Zhai, P.; Yamamoto, M.; Galeotti, J.; Liu, J.; Masurekar, M.; Thaisz, J.; Irie, K.; Holle, E.; Yu, X.; Kupershmidt, S.; et al. Cardiac-Specific Overexpression of AT1 Receptor Mutant Lacking G Aq/Gαi Causes Hypertrophy and Bradycardia in Transgenic Mice. J. Clin. Investig. 2005, 115, 3045–3056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaulieu, J.-M.; Sotnikova, T.D.; Marion, S.; Lefkowitz, R.J.; Gainetdinov, R.R.; Caron, M.G. An Akt/Beta-Arrestin 2/PP2A Signaling Complex Mediates Dopaminergic Neurotransmission and Behavior. Cell 2005, 122, 261–273. [Google Scholar] [CrossRef] [Green Version]
- McDonald, P.H.; Chow, C.-W.; Miller, W.E.; Laporte, S.A.; Field, M.E.; Lin, F.-T.; Davis, R.J.; Lefkowitz, R.J. β-Arrestin 2: A Receptor-Regulated MAPK Scaffold for the Activation of JNK3. Science 2000, 290, 1574–1577. [Google Scholar] [CrossRef]
- Luttrell, L.M.; Roudabush, F.L.; Choy, E.W.; Miller, W.E.; Field, M.E.; Pierce, K.L.; Lefkowitz, R.J. Activation and Targeting of Extracellular Signal-Regulated Kinases by β-Arrestin Scaffolds. Proc. Natl. Acad. Sci. USA 2001, 98, 2449–2454. [Google Scholar] [CrossRef] [Green Version]
- Perry, S.J.; Baillie, G.S.; Kohout, T.A.; McPhee, I.; Magiera, M.M.; Ang, K.L.; Miller, W.E.; McLean, A.J.; Conti, M.; Houslay, M.D.; et al. Targeting of Cyclic AMP Degradation to Β2-Ad.drenergic Receptors by β-Arrestins. Science 2002, 298, 834–836. [Google Scholar] [CrossRef]
- Abrisqueta, M.; Herraiz, C.; Pérez Oliva, A.B.; Sanchez-Laorden, B.L.; Olivares, C.; Jiménez-Cervantes, C.; García-Borrón, J.C. Differential and Competitive Regulation of Human Melanocortin 1 Receptor Signaling by β-Arrestin Isoforms. J. Cell Sci. 2013, 126, 3724–3737. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Vicente, I.; Abrisqueta, M.; Herraiz, C.; Jiménez-Cervantes, C.; García-Borrón, J.C.; Olivares, C. Functional Characterization of a C-Terminal Splice Variant of the Human Melanocortin 1 Receptor. Exp. Derm. 2020, 29, 610–615. [Google Scholar] [CrossRef]
- Abreu, N.; Acosta-Ruiz, A.; Xiang, G.; Levitz, J. Mechanisms of Differential Desensitization of Metabotropic Glutamate Receptors. Cell Rep. 2021, 35, 109050. [Google Scholar] [CrossRef]
- Ceraudo, E.; Horioka, M.; Mattheisen, J.M.; Hitchman, T.D.; Moore, A.R.; Kazmi, M.A.; Chi, P.; Chen, Y.; Sakmar, T.P.; Huber, T. Direct Evidence That the GPCR CysLTR2 Mutant Causative of Uveal Melanoma Is Constitutively Active with Highly Biased Signaling. J. Biol. Chem. 2021, 296, 100163. [Google Scholar] [CrossRef] [PubMed]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-Targeted Cancer Therapies: Progress, Challenges and Future Directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef] [PubMed]
- Menichetti, R.; Kanekal, K.H.; Bereau, T. Drug–Membrane Permeability across Chemical Space. ACS Cent. Sci. 2019, 5, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Vaghasiya, K.; Ray, E.; Verma, R.K. Lysosomal Targeting Strategies for Design and Delivery of Bioactive for Therapeutic Interventions. J. Drug Target 2018, 26, 208–221. [Google Scholar] [CrossRef]
- Liu, D.; Schilling, B.; Liu, D.; Sucker, A.; Livingstone, E.; Jerby-Arnon, L.; Zimmer, L.; Gutzmer, R.; Satzger, I.; Loquai, C.; et al. Integrative Molecular and Clinical Modeling of Clinical Ou.utcomes to PD1 Blockade in Patients with Metastatic Melanoma. Nat. Med. 2019, 25, 1916–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, A.P.; Smrcka, A.V. Targeting G Protein-Coupled Receptor Signalling by Blocking G Proteins. Nat. Rev. Drug Discov. 2018, 17, 789–803. [Google Scholar] [CrossRef] [PubMed]
- Chidiac, P.; Hebert, T.E.; Valiquette, M.; Dennis, M.; Bouvier, M. Inverse Agonist Activity of Beta-Adrenergic Antagonists. Mol. Pharm. 1994, 45, 490–499. [Google Scholar]
- de Ligt, R.A.F.; Kourounakis, A.P.; IJzerman, A.P. Inverse Agonism at G Protein-Coupled Receptors: (Patho)Physiological Relevance and Implications for Drug Discovery. Br. J. Pharm. 2000, 130, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Pozvek, G.; Hilton, J.M.; Quiza, M.; Houssami, S.; Sexton, P.M. Structure/Function Relationships of Calcitonin Analogues as Agonists, Antagonists, or Inverse Agonists in a Constitutively Activated Receptor Cell System. Mol. Pharm. 1997, 51, 658–665. [Google Scholar] [CrossRef]
- Wu, V.; Yeerna, H.; Nohata, N.; Chiou, J.; Harismendy, O.; Raimondi, F.; Inoue, A.; Russell, R.B.; Tamayo, P.; Gutkind, J.S. Illuminating the Onco-GPCRome: Novel G Protein–Coupled Receptor-Driven Oncocrine Networks and Targets for Cancer Immunotherapy. J. Biol. Chem. 2019, 294, 11062–11086. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.P.; Rowling, E.J.; Miskolczi, Z.; Ferguson, J.; Spoerri, L.; Haass, N.K.; Sloss, O.; McEntegart, S.; Arozarena, I.; von Kriegsheim, A.; et al. Targeting Endothelin Receptor Signalling Overcomes Heterogeneity Driven Therapy Failure. EMBO Mol. Med. 2017, 9, 1011–1029. [Google Scholar] [CrossRef] [PubMed]
- Ben-Baruch, A. Site-Specific Metastasis Formation. Cell Adh. Migr. 2009, 3, 328–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Hoffmann, A.D.; Liu, H.; Liu, X. Organotropism: New Insights into Molecular Mechanisms of Breast Cancer Metastasis. NPJ Precis. Onc. 2018, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the Human Tissue-Specific Expression by Genome-Wide Integration of Transcriptomics and Antibody-Based Proteomics. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forrest, A.R.R.; Kawaji, H.; Rehli, M.; Kenneth Baillie, J.; de Hoon, M.J.L.; Haberle, V.; Lassmann, T.; Kulakovskiy, I.V.; Lizio, M.; Itoh, M.; et al. A Promoter-Level Mammalian Expression Atlas. Nature 2014, 507, 462–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melé, M.; Ferreira, P.G.; Reverter, F.; DeLuca, D.S.; Monlong, J.; Sammeth, M.; Young, T.R.; Goldmann, J.M.; Pervouchine, D.D.; Sullivan, T.J.; et al. Human Genomics. The Human Transcriptome across Tissues and Individuals. Science 2015, 348, 660–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierson, E.; the GTEx Consortium; Koller, D.; Battle, A.; Mostafavi, S. Sharing and Specificity of Co-Expression Networks across 35 Human Tissues. PLoS Comput. Biol. 2015, 11, e1004220. [Google Scholar] [CrossRef] [Green Version]
- Jo, M.; Jung, S.T. Engineering Therapeutic Antibodies Targeting G-Protein–Coupled Receptors. Exp. Mol. Med. 2016, 48, e207. [Google Scholar] [CrossRef]
- Latorraca, N.R.; Venkatakrishnan, A.J.; Dror, R.O. GPCR Dynamics: Structures in Motion. Chem. Rev. 2017, 117, 139–155. [Google Scholar] [CrossRef]
- Pauwels, J.; Fijałkowska, D.; Eyckerman, S.; Gevaert, K. Mass Spectrometry and the Cellular Surfaceome. Mass Spectrom. Rev. 2021, in press. [Google Scholar] [CrossRef]
- Alhosaini, K.; Azhar, A.; Alonazi, A.; Al-Zoghaibi, F. GPCRs: The Most Promiscuous Druggable Receptor of the Mankind. Saudi Pharm J. 2021, 29, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, L.; Cummins, E.; Samudio, I.; Kislinger, T. Cell-Surface Proteomics for the Identification of Novel Therapeutic Targets in Cancer. Expert Rev. Proteom. 2018, 15, 259–275. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Suttapitugsakul, S.; Wu, R. Unraveling the Surface Glycoprotein Interaction Network by Integrating Chemical Crosslinking with MS-Based Proteomics. Chem. Sci. 2021, 12, 2146–2155. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Meng, M.; Yang, D.; Bai, W.; Lu, Y.; Peng, Y.; Song, G.; Wu, Y.; Zhou, Q.; Zhao, S.; et al. High-Throughput Identification of G Protein-Coupled Receptor Modulators through Affinity Mass Spectrometry Screening. Chem. Sci. 2018, 9, 3192–3199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crotty, S.; Pipkin, M.E. In Vivo RNAi Screens: Concepts and Applications. Trends Immunol. 2015, 36, 315–322. [Google Scholar] [CrossRef]
- Driever, W.; Solnica-Krezel, L.; Schier, A.F.; Neuhauss, S.C.; Malicki, J.; Stemple, D.L.; Stainier, D.Y.; Zwartkruis, F.; Abdelilah, S.; Rangini, Z.; et al. A Genetic Screen for Mutations Affecting Embryogenesis in Zebrafish. Development 1996, 123, 37–46. [Google Scholar] [CrossRef]
- Haffter, P.; Granato, M.; Brand, M.; Mullins, M.C.; Hammerschmidt, M.; Kane, D.A.; Odenthal, J.; van Eeden, F.J.; Jiang, Y.J.; Heisenberg, C.P.; et al. The Identification of Genes with Unique and Essential Functions in the Development of the Zebrafish, Danio Rerio. Development 1996, 123, 1–36. [Google Scholar] [CrossRef]
- Keatinge, M.; Tsarouchas, T.M.; Munir, T.; Porter, N.J.; Larraz, J.; Gianni, D.; Tsai, H.-H.; Becker, C.G.; Lyons, D.A.; Becker, T. CRISPR GRNA Phenotypic Screening in Zebrafish Reveals Pro-Regenerative Genes in Spinal Cord Injury. PLoS Genet. 2021, 17, e1009515. [Google Scholar] [CrossRef]
- Trubiroha, A.; Gillotay, P.; Giusti, N.; Gacquer, D.; Libert, F.; Lefort, A.; Haerlingen, B.; De Deken, X.; Opitz, R.; Costagliola, S. A Rapid CRISPR/Cas-Based Mutagenesis Assay in Zebrafish for Identification of Genes Involved in Thyroid Morphogenesis and Function. Sci Rep. 2018, 8, 5647. [Google Scholar] [CrossRef]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The Zebrafish Reference Genome Sequence and Its Relationship to the Human Genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [Green Version]
- Langenhan, T.; Barr, M.M.; Bruchas, M.R.; Ewer, J.; Griffith, L.C.; Maiellaro, I.; Taghert, P.H.; White, B.H.; Monk, K.R. Model Organisms in G Protein–Coupled Receptor Research. Mol. Pharm. 2015, 88, 596–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frantz, W.T.; Ceol, C.J. From Tank to Treatment: Modeling Melanoma in Zebrafish. Cells 2020, 9, 1289. [Google Scholar] [CrossRef] [PubMed]
- Patton, E.E.; Mueller, K.L.; Adams, D.J.; Anandasabapathy, N.; Aplin, A.E.; Bertolotto, C.; Bosenberg, M.; Ceol, C.J.; Burd, C.E.; Chi, P.; et al. Melanoma Models for the next Generation of Therapies. Cancer Cell 2021, 39, 610–631. [Google Scholar] [CrossRef] [PubMed]
- Manzotti, C.; Audisio, R.A.; Pratesi, G. Importance of Orthotopic Implantation for Human Tumors as Model Systems: Relevance to Metastasis and Invasion. Clin. Exp. Metastasis 1993, 11, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Basith, S.; Choi, S. Recent Advances in Structure-Based Drug Design Targeting Class A G Protein-Coupled Receptors Utilizing Crystal Structures and Computational Simulations. J. Med. Chem. 2018, 61, 1–46. [Google Scholar] [CrossRef] [PubMed]
- García-Nafría, J.; Tate, C.G. Cryo-Electron Microscopy: Moving Beyond X-Ray Crystal Structures for Drug Receptors and Drug Development. Annu. Rev. Pharm. Toxicol. 2020, 60, 51–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, T.; Le Trong, I.; Fox, B.A.; Behnke, C.A.; Stenkamp, R.E.; Palczewski, K. X-ray Diffraction Analysis of Three-Dimensional Crystals of Bovine Rhodopsin Obtained from Mixed Micelles. J. Struct. Biol. 2000, 130, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Tate, C.G.; Schertler, G.F.X. Engineering G Protein-Coupled Receptors to Facilitate Their Structure Determination. Curr. Opin. Struct. Biol. 2009, 19, 386–395. [Google Scholar] [CrossRef]
- Lebon, G.; Bennett, K.; Jazayeri, A.; Tate, C.G. Thermostabilisation of an Agonist-Bound Conformation of the Human Adenosine A(2A) Receptor. J. Mol. Biol. 2011, 409, 298–310. [Google Scholar] [CrossRef] [Green Version]
- Rosenbaum, D.M.; Cherezov, V.; Hanson, M.A.; Rasmussen, S.G.F.; Thian, F.S.; Kobilka, T.S.; Choi, H.-J.; Yao, X.-J.; Weis, W.I.; Stevens, R.C.; et al. GPCR Engineering Yields High-Resolution Structural Insights into Beta2-Adrenergic Receptor Function. Science 2007, 318, 1266–1273. [Google Scholar] [CrossRef] [Green Version]
- Tate, C.G. Practical Considerations of Membrane Protein Instability during Purification and Crystallisation. Methods Mol. Biol 2010, 601, 187–203. [Google Scholar] [CrossRef] [PubMed]
- Ishchenko, A.; Stauch, B.; Han, G.W.; Batyuk, A.; Shiriaeva, A.; Li, C.; Zatsepin, N.; Weierstall, U.; Liu, W.; Nango, E.; et al. Toward G Protein-Coupled Receptor Structure-Based Drug Design Using X-Ray Lasers. IUCrJ 2019, 6, 1106–1119. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Lee, W. The XFEL Protein Crystallography: Developments and Perspectives. Int. J. Mol. Sci. 2019, 20, 3421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMullan, G.; Faruqi, A.R.; Henderson, R. Direct Electron Detectors. Methods Enzym. 2016, 579, 1–17. [Google Scholar] [CrossRef]
- Vinothkumar, K.R.; Henderson, R. Single Particle Electron Cryomicroscopy: Trends, Issues and Future Perspective. Q. Rev. Biophys. 2016, 49, e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zivanov, J.; Nakane, T.; Forsberg, B.O.; Kimanius, D.; Hagen, W.J.; Lindahl, E.; Scheres, S.H. New Tools for Automated High-Resolution Cryo-EM Structure Determination in RELION-3. Elife 2018, 7, e42166. [Google Scholar] [CrossRef] [PubMed]
- Congreve, M.; de Graaf, C.; Swain, N.A.; Tate, C.G. Impact of GPCR Structures on Drug Discovery. Cell 2020, 181, 81–91. [Google Scholar] [CrossRef]
- Danev, R.; Belousoff, M.; Liang, Y.-L.; Zhang, X.; Eisenstein, F.; Wootten, D.; Sexton, P.M. Routine Sub-2.5 Å Cryo-EM Structure Determination of GPCRs. Nat. Commun. 2021, 12, 4333. [Google Scholar] [CrossRef]
- Zhang, M.; Gui, M.; Wang, Z.-F.; Gorgulla, C.; Yu, J.J.; Wu, H.; Sun, Z.J.; Klenk, C.; Merklinger, L.; Morstein, L.; et al. Cryo-EM Structure of an Activated GPCR-G Protein Complex in Lipid Nanodiscs. Nat. Struct. Mol. Biol. 2021, 28, 258–267. [Google Scholar] [CrossRef]
- Shimada, I.; Ueda, T.; Kofuku, Y.; Eddy, M.T.; Wüthrich, K. GPCR Drug Discovery: Integrating Solution NMR Data with Crystal and Cryo-EM Structures. Nat. Rev. Drug Discov. 2019, 18, 59–82. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate Prediction of Protein Structures and Interactions Using a Three-Track Neural Network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly Accurate Protein Structure Prediction for the Human Proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Bender, B.J.; Marlow, B.; Meiler, J. Improving Homology Modeling from Low-Sequence Identity Templates in Rosetta: A Case Study in GPCRs. PLoS Comput. Biol. 2020, 16, e1007597. [Google Scholar] [CrossRef]
- Esguerra, M.; Siretskiy, A.; Bello, X.; Sallander, J.; Gutiérrez-de-Terán, H. GPCR-ModSim: A Comprehensive Web Based Solution for Modeling G-Protein Coupled Receptors. Nucleic Acids Res. 2016, 44, W455–W462. [Google Scholar] [CrossRef]
- Worth, C.L.; Kreuchwig, F.; Tiemann, J.K.S.; Kreuchwig, A.; Ritschel, M.; Kleinau, G.; Hildebrand, P.W.; Krause, G. GPCR-SSFE 2.0-a Fragment-Based Molecular Modeling Web Tool for Class A G-Protein Coupled Receptors. Nucleic Acids Res. 2017, 45, W408–W415. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, J.; Jang, R.; Zhang, Y. GPCR-I-TASSER: A Hybrid Approach to G Protein-Coupled Receptor Structure Modeling and the Application to the Human Genome. Structure 2015, 23, 1538–1549. [Google Scholar] [CrossRef] [Green Version]
- Mullard, A. What Does AlphaFold Mean for Drug Discovery? Nat. Rev. Drug Discov. 2021, 20, 725–727. [Google Scholar] [CrossRef]
- Potterton, A.; Heifetz, A.; Townsend-Nicholson, A. Synergistic Use of GPCR Modeling and SDM Experiments to Understand Ligand Binding. In Computational Methods for GPCR Drug Discovery; Heifetz, A., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2018; pp. 335–343. ISBN 978-1-4939-7465-8. [Google Scholar]
- Zhou, Q.; Yang, D.; Wu, M.; Guo, Y.; Guo, W.; Zhong, L.; Cai, X.; Dai, A.; Jang, W.; Shakhnovich, E.I.; et al. Common Activation Mechanism of Class A GPCRs. eLife 2019, 8, e50279. [Google Scholar] [CrossRef]
- Di Roberto, R.B.; Chang, B.; Peisajovich, S.G. The Directed Evolution of Ligand Specificity in a GPCR and the Unequal Contributions of Efficacy and Affinity. Sci Rep. 2017, 7, 16012. [Google Scholar] [CrossRef] [Green Version]
- Hughes, J.; Rees, S.; Kalindjian, S.; Philpott, K. Principles of Early Drug Discovery. Br. J. Pharm. 2011, 162, 1239–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Ahsen, O.; Bömer, U. High-Throughput Screening for Kinase Inhibitors. ChemBioChem 2005, 6, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.J.Y.; Gabriel, F.; Tandale, A.; Nietlispach, D. Structure and Dynamics of GPCRs in Lipid Membranes: Physical Principles and Experimental Approaches. Molecules 2020, 25, 4729. [Google Scholar] [CrossRef] [PubMed]
- Kitaeva, K.V.; Rutland, C.S.; Rizvanov, A.A.; Solovyeva, V.V. Cell Culture Based in Vitro Test Systems for Anticancer Drug Screening. Front. Bioeng. Biotechnol. 2020, 8, 322. [Google Scholar] [CrossRef] [Green Version]
- González, N.; Mantey, S.A.; Pradhan, T.K.; Sancho, V.; Moody, T.W.; Coy, D.H.; Jensen, R.T. Characterization of Putative GRP- and NMB-Receptor Antagonist’s Interaction with Human Receptors. Peptides 2009, 30, 1473–1486. [Google Scholar] [CrossRef] [Green Version]
- Uehara, H.; González, N.; Sancho, V.; Mantey, S.A.; Nuche-Berenguer, B.; Pradhan, T.; Coy, D.H.; Jensen, R.T. Pharmacology and Selectivity of Various Natural and Synthetic Bombesin Related Peptide Agonists for Human and Rat Bombesin Receptors Differs. Peptides 2011, 32, 1685–1699. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Xie, X. Tools for GPCR Drug Discovery. Acta Pharm. Sin. 2012, 33, 372–384. [Google Scholar] [CrossRef] [Green Version]
- Maurel, D.; Comps-Agrar, L.; Brock, C.; Rives, M.-L.; Bourrier, E.; Ayoub, M.A.; Bazin, H.; Tinel, N.; Durroux, T.; Prézeau, L.; et al. Cell-Surface Protein-Protein Interaction Analysis with Time-Resolved FRET and Snap-Tag Technologies: Application to GPCR Oligomerization. Nat. Methods 2008, 5, 561–567. [Google Scholar] [CrossRef]
- Valencia, C.; Dujet, C.; Margathe, J.-F.; Iturrioz, X.; Roux, T.; Trinquet, E.; Villa, P.; Hibert, M.; Dupuis, E.; Llorens-Cortes, C.; et al. A Time-Resolved FRET Cell-Based Binding Assay for the Apelin Receptor. ChemMedChem 2017, 12, 925–931. [Google Scholar] [CrossRef] [Green Version]
- Titus, S.; Neumann, S.; Zheng, W.; Southall, N.; Michael, S.; Klumpp, C.; Yasgar, A.; Shinn, P.; Thomas, C.J.; Inglese, J.; et al. Quantitative High-Throughput Screening Using a Live-Cell CAMP Assay Identifies Small-Molecule Agonists of the TSH Receptor. J. Biomol. Screen 2008, 13, 120–127. [Google Scholar] [CrossRef] [Green Version]
- Trinquet, E.; Bouhelal, R.; Dietz, M. Monitoring Gq-Coupled Receptor Response through Inositol Phosphate Quantification with the IP-One Assay. Expert Opin. Drug Discov. 2011, 6, 981–994. [Google Scholar] [CrossRef] [PubMed]
- Katsuya, K.; Hori, Y.; Oikawa, D.; Yamamoto, T.; Umetani, K.; Urashima, T.; Kinoshita, T.; Ayukawa, K.; Tokunaga, F.; Tamaru, M. High-Throughput Screening for Linear Ubiquitin Chain Assembly Complex (LUBAC) Selective Inhibitors Using Homogenous Time-Resolved Fluorescence (HTRF)-Based Assay System. SLAS Discov. 2018, 23, 1018–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lotta, L.A.; Mokrosiński, J.; Mendes de Oliveira, E.; Li, C.; Sharp, S.J.; Luan, J.; Brouwers, B.; Ayinampudi, V.; Bowker, N.; Kerrison, N.; et al. Human Gain-of-Function MC4R Variants Show Signaling Bias and Protect against Obesity. Cell 2019, 177, 597–607.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zindel, D.; Vol, C.; Lecha, O.; Bequignon, I.; Bilgic, M.; Vereecke, M.; Charrier-Savournin, F.; Romier, M.; Trinquet, E.; Pin, J.-P.; et al. HTRF® Total and Phospho-YAP (Ser127) Cellular Assays. In The Hippo Pathway: Methods and Protocols; Hergovich, A., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2019; pp. 153–166. ISBN 978-1-4939-8910-2. [Google Scholar]
- Liu, L.; Jockers, R. Structure-Based Virtual Screening Accelerates GPCR Drug Discovery. Trends Pharmacol. Sci. 2020, 41, 382–384. [Google Scholar] [CrossRef] [PubMed]
- Shoichet, B.K.; Kobilka, B.K. Structure-Based Drug Screening for G Protein-Coupled Receptors. Trends Pharm. Sci 2012, 33, 268–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, R.M.; Kang, H.J.; McCorvy, J.D.; Glatfelter, G.C.; Jones, A.J.; Che, T.; Slocum, S.; Huang, X.-P.; Savych, O.; Moroz, Y.S.; et al. Virtual Discovery of Melatonin Receptor Ligands to Modulate Circadian Rhythms. Nature 2020, 579, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Ebalunode, J.O.; Zheng, W.; Tropsha, A. Application of QSAR and Shape Pharmacophore Modeling Approaches for Targeted Chemical Library Design. In Chemical Library Design; Zhou, J.Z., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2011; pp. 111–133. ISBN 978-1-60761-931-4. [Google Scholar]
- Green, H.; Koes, D.R.; Durrant, J.D. DeepFrag: A Deep Convolutional Neural Network for Fragment-Based Lead Optimization. Chem. Sci. 2021, 12, 8036–8047. [Google Scholar] [CrossRef] [PubMed]
- Neves, B.J.; Braga, R.C.; Melo-Filho, C.C.; Moreira-Filho, J.T.; Muratov, E.N.; Andrade, C.H. QSAR-Based Virtual Screening: Advances and Applications in Drug Discovery. Front. Pharm. 2018, 9, 1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, T.D.Y.; Terry, D.B.; Smith, L.H. In Vitro and In Vivo Assessment of ADME and PK Properties During Lead Selection and Lead Optimization—Guidelines, Benchmarks and Rules of Thumb. In Assay Guidance Manual; Markossian, S., Grossman, A., Brimacombe, K., Arkin, M., Auld, D., Austin, C.P., Baell, J., Chung, T.D.Y., Coussens, N.P., Dahlin, J.L., et al., Eds.; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004. [Google Scholar]
- Guan, L.; Yang, H.; Cai, Y.; Sun, L.; Di, P.; Li, W.; Liu, G.; Tang, Y. ADMET-Score—A Comprehensive Scoring Function for Evaluation of Chemical Drug-Likeness. Med. Chem. Commun. 2018, 10, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Akil, H.; Quintana, M.; Raymond, J.H.; Billoux, T.; Benboubker, V.; Besse, S.; Auzeloux, P.; Delmas, V.; Petit, V.; Larue, L.; et al. Efficacy of Targeted Radionuclide Therapy Using [131I]ICF01012 in 3D Pigmented BRAF- and NRAS-Mutant Melanoma Models and In Vivo NRAS-Mutant Melanoma. Cancers 2021, 13, 1421. [Google Scholar] [CrossRef]
- Norain, A.; Dadachova, E. Targeted Radionuclide Therapy of Melanoma. Semin. Nucl. Med. 2016, 46, 250–259. [Google Scholar] [CrossRef] [PubMed]
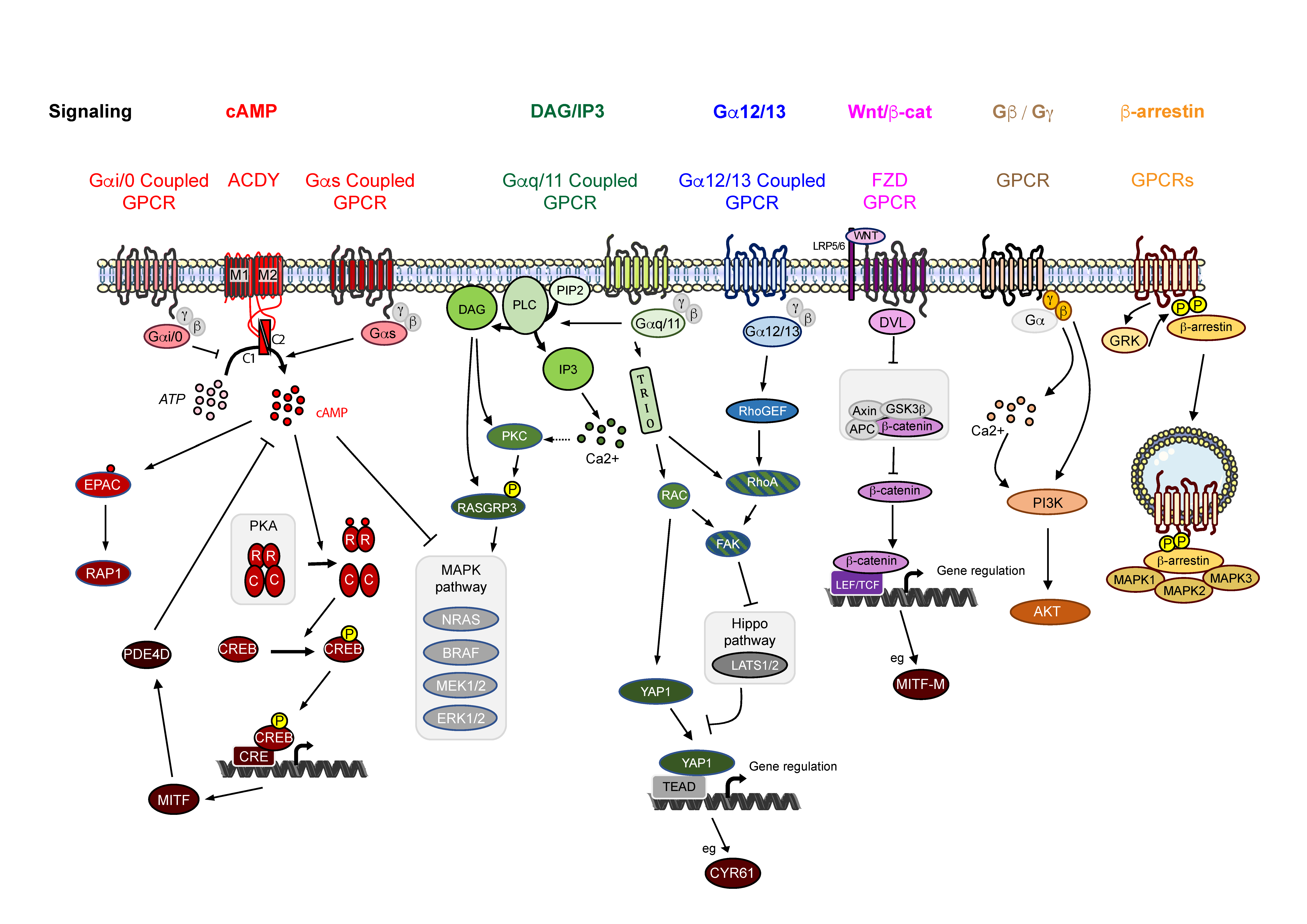

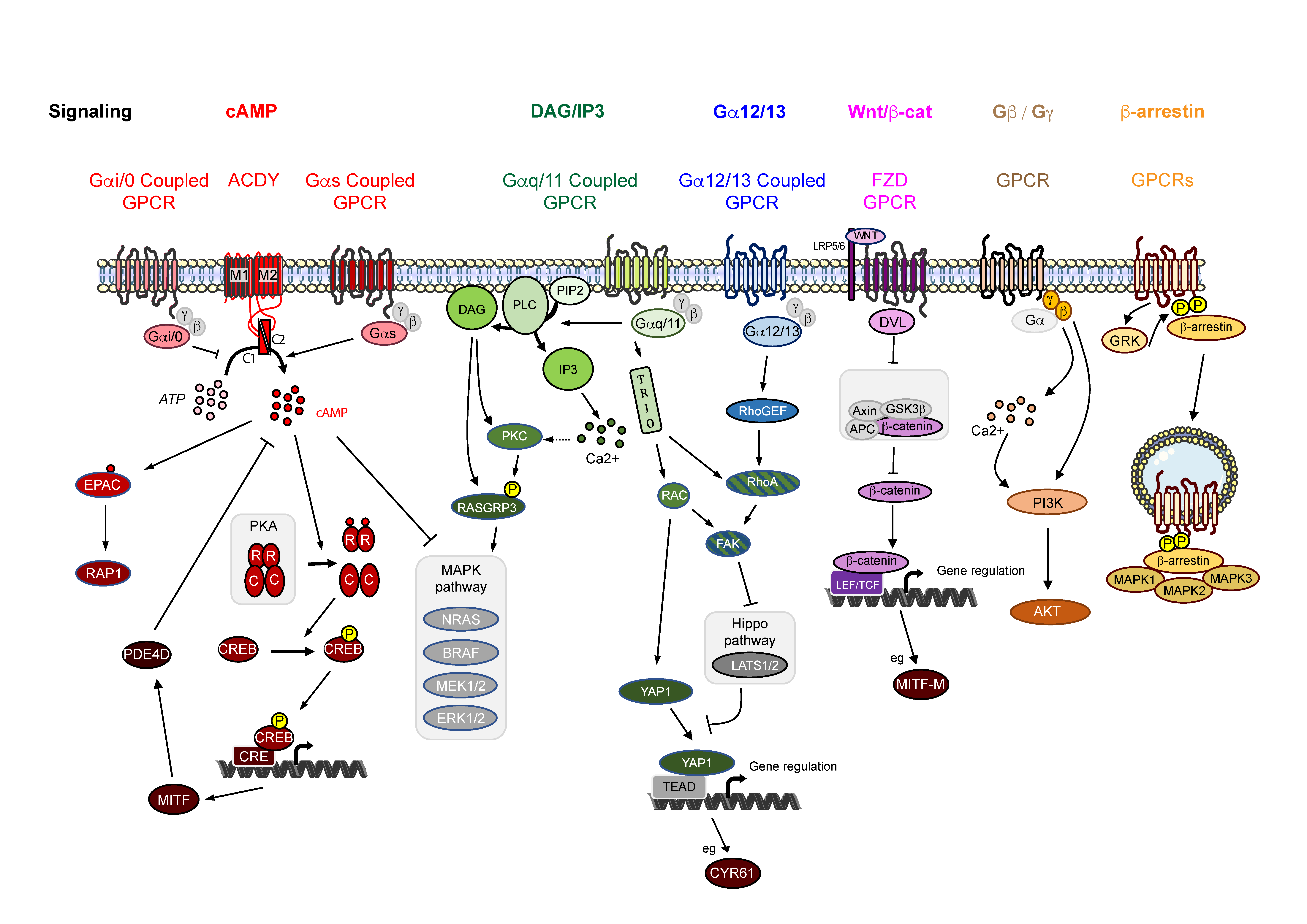{kind=link}
| Gene Cyto.loc | Receptor Name | Signaling | Mouse Model | Human Disease | Function(s) | References |
|---|---|---|---|---|---|---|
| DRD2 11q23.2 | Dopamine D2 | Gαs | Hyperpigmentation eg. Drd2tm1Ebo/Drd2tm1Ebo | αMSH synthesis | [7] | |
| EDNRB 13q22.3 | Endothelin B | Gαq | Hypopigmentation eg. Ednrbs-l/Ednrbs-l | WS4A (#277580) ABCD syndrome (#600501) | Proliferation Survival DNA repair | [8] |
| FZD4 11q14.2 | Frizzled 4 | β-catenin | Hypopigmentation eg. Fzd4tm1Nat/Fzd4tm1Nat | [9] | ||
| GPR143 Xp22.2 | Protein G143 | Gαq | Eye hypopigmentation eg. Gpr143tm1Inc/Y | OA1 (#300808, #300500) | Melanogenesis | [10] |
| GPR161 1q24.2 | Protein G141 | Gαs | White belly spot eg. Gpr161vl/Gpr161vl | Shh negative regulator | [11] | |
| GRM1 6q24.3 | Glutamate receptor metabotropic 1 | Gαq | Hyperpigmentation eg. Tg(Dct-Grm1)ESzc | Proliferation Survival | [12] | |
| MC1R 16q24.3 | Melanocortin 1 receptor | Gαs | Yellow coat Mc1re/Mc1re | Light skin, red/blond hair | Switch eu/pheomelanin DNA repair | [13] |
| SMO 7q32.1 | Smoothened | Gli | Striped depigmentation Curry-Jones syndrome (#601707) | [14] |
| Gene Cyto.loc | Receptor Name | Signaling | Animal Model | Melanomagenesis | Function(s) | References |
|---|---|---|---|---|---|---|
| EDNRB 13q22.3 | Endothelin B | Gαq | Mitf-cre/+; Rosa-fs-GNAQQ209L/+; EdnrbF/F | initiation progression | Proliferation Invasion DNA repair | [21] [22] |
| MC1R 16q24.3 | Melanocortin 1 | Gαs | Tyr::CreERT2/°; BrafV600E; MC1Re/e | initiation | Proliferation DNA repair | [23] [24] |
| FZD7 2q33.1 | Frizzled 7 | β-catenin | Xen., A375P, WM1361 shRNA-FZD7 | progression | Proliferation | [25] [26] |
| GPER1 7p22.3 | G-protein-coupled estrogen receptor | Gαs | Xen., NHEM with BRAFV600E; p53R248W; CDK4R24C; hTERT | initiation | Differentiation | [27] |
| GRM1 6q24.3 | Glutamate receptor metabotropic 1 | Gαq | Dct::Grm1 | initiation progression | Proliferation Invasion | [12] [28] |
| GRM3 7q21.11/12 | Glutamate receptor metabotropic 3 | Gαq | Xen., A375 GRM3 S610L, E767K, or E870K; | initiation progression | Proliferation Migration | [29] |
| GRM5 11q14.2/3 | Glutamate receptor metabotropic 5 | Gαq | Tyrp1::Grm5 | initiation progression | Proliferation Invasion | [30] |
| PAR1/F2R 5q13.3 | Coagulation factor II Thrombin | Gαq | Xen. A375 shRNA-PAR1 | progression: | Proliferation | [31] |
| CXCR4 2q22.1 | CXC motif chemokine 4 | Gαi/0 | Allograft B16 Ectopic expression Blocking peptide T22 | progression | Attraction Growth | [32] [33] |
| CCR7 17q21.2 | CC motif chemokine 7 | Gαi/0 | Allograft B16 Ectopic expression Neutral. anti-CCL21 ab | progression | Attraction Growth | [34] |
| CCR10 17q21.2 | CC motif chemokine 10 | Gαi/0 | Allograft B16 Ectopic expression | progression | Immune invasion Apop. resistance | [35] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raymond, J.H.; Aktary, Z.; Larue, L.; Delmas, V. Targeting GPCRs and Their Signaling as a Therapeutic Option in Melanoma. Cancers 2022, 14, 706. https://doi.org/10.3390/cancers14030706
Raymond JH, Aktary Z, Larue L, Delmas V. Targeting GPCRs and Their Signaling as a Therapeutic Option in Melanoma. Cancers. 2022; 14(3):706. https://doi.org/10.3390/cancers14030706
Chicago/Turabian StyleRaymond, Jérémy H., Zackie Aktary, Lionel Larue, and Véronique Delmas. 2022. "Targeting GPCRs and Their Signaling as a Therapeutic Option in Melanoma" Cancers 14, no. 3: 706. https://doi.org/10.3390/cancers14030706