
MALAT1 Fusions and Basal Cells Contribute to Primary Resistance against Androgen Receptor Inhibition in TRAMP Mice

 , , , and
, , , and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Experiments
2.2. Organoid Culture, Treatment, and Viability Assays
2.3. Immunohistochemistry
2.4. Western Blotting
2.5. RNA Sequencing and Gene Ontology Enrichment Analysis
2.6. qRT-PCR and PCR
2.7. TRAMPC1 Cell Culture, Lentiviral Knockdown, Treatment, and Viability Assays
2.8. MALAT1 Knockdown in TRAMPC1 Cells
2.9. Verification of MALAT1 Knockdown and Alterations in Target Gene Expression by RT-PCR
2.10. Validation of MALAT1-Fusions by PCR
2.11. Statistical Analysis
3. Results
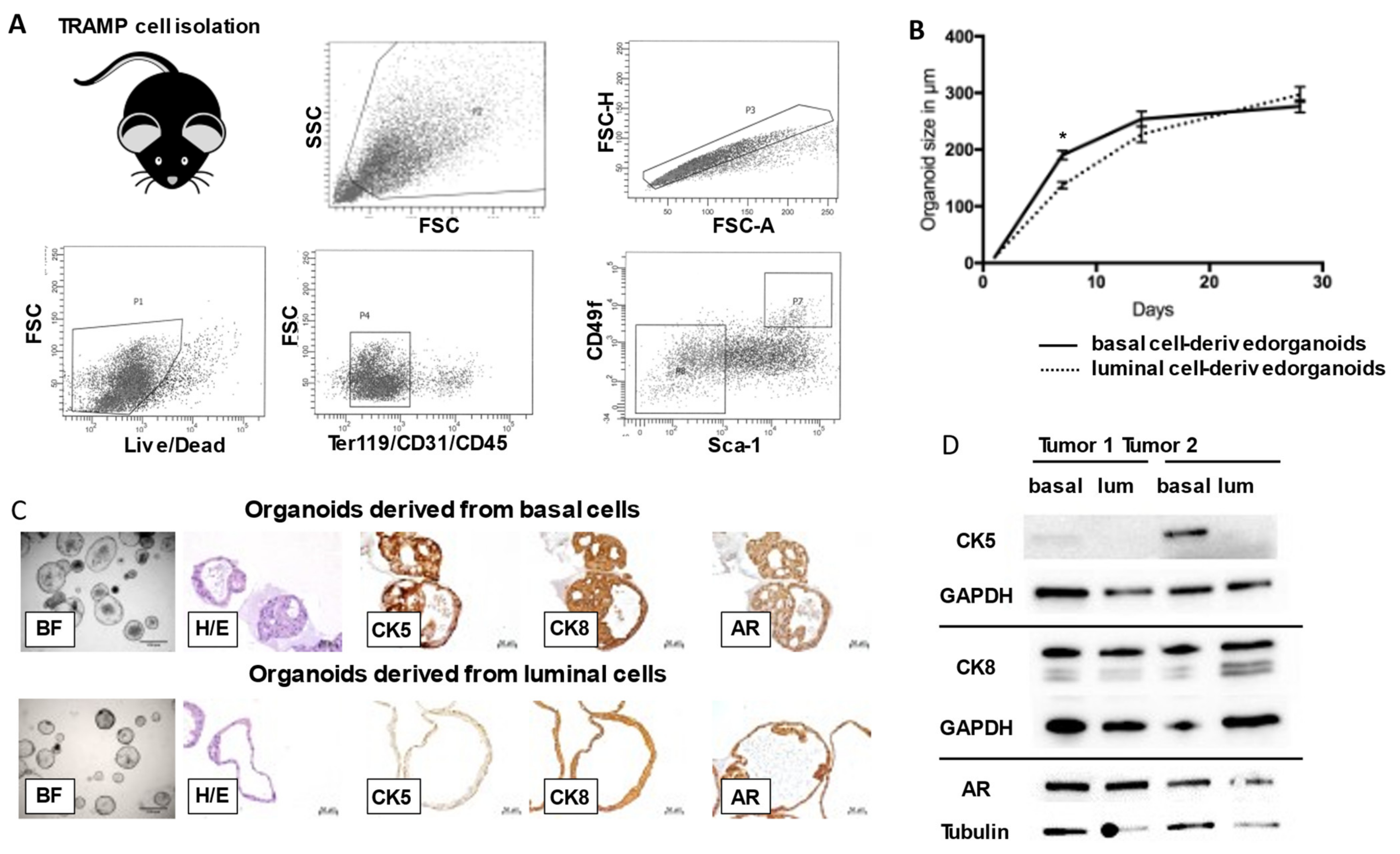
3.1. Basal and Luminal Cell-Derived Organoids from TRAMP Tumors Exhibit Heterogenous Cellular Architecture and Potency
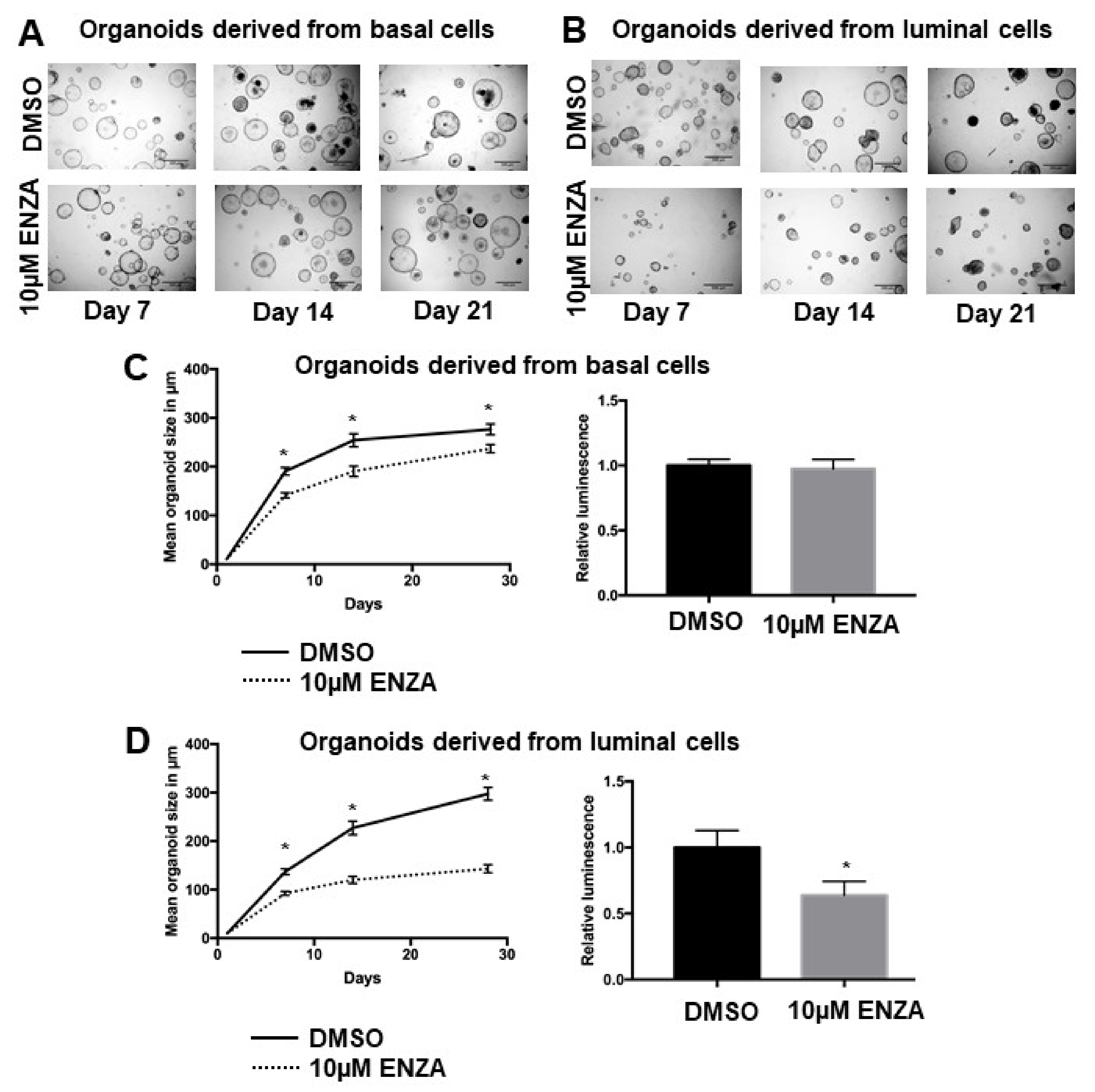
3.2. Basal Cell-Derived Organoids Display Resistance towards Androgen Receptor Inhibition
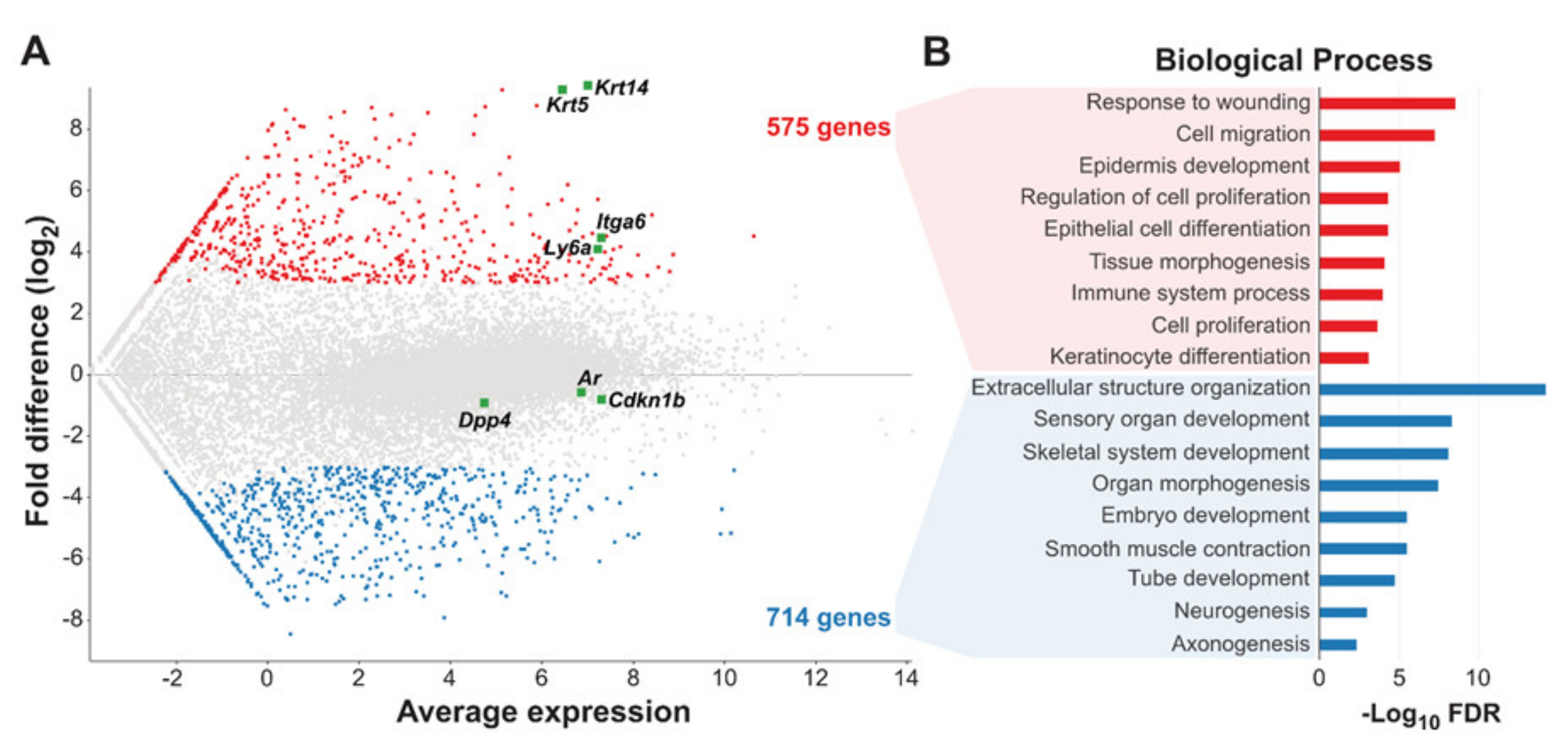
3.3. Gene Expression Profiling Reveals Distinct Epithelial and Neuroendocrine Signatures for Basal and Luminal TRAMP Tumor Cells
3.4. MALAT1-Fusions Are Abundant and Regulate Resistance towards Androgen Receptor Inhibition in TRAMP Tumor Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ording, A.G.; Horváth-Puhó, E.; Lash, T.L.; Ehrenstein, V.; Borre, M.; Vyberg, M.; Sørensen, H.T. Does comorbidity interact with prostate cancer to increase mortality? A Danish cohort study of 45 326 prostate cancer patients diagnosed during 1995–2011. Acta Oncol. 2015, 55, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Ording, A.G.; Horváth-Puhó, E.; Lash, T.L.; Ehrenstein, V.; Borre, M.; Vyberg, M.; Sørensen, H.T. Prostate cancer, comorbidity, and the risk of venous thromboembolism: A cohort study of 44,035 Danish prostate cancer patients, 1995–2011. Cancer 2015, 121, 3692–3699. [Google Scholar] [CrossRef]
- Polesel, J.; Gini, A.; Maso, L.D.; Stocco, C.; Birri, S.; Taborelli, M.; Serraino, D.; Zucchetto, A. The Impact of Diabetes and Other Metabolic Disorders on Prostate Cancer Prognosis. J. Diabetes Complicat. 2016, 30, 591–596. [Google Scholar] [CrossRef]
- Wang, Z.; Mitrofanova, A.; Bergren, S.K.; Abate-Shen, C.; Cardiff, R.D.; Califano, A.; Shen, M.M. Lineage analysis of basal epithelial cells reveals their unexpected plasticity and supports a cell-of-origin model for prostate cancer heterogeneity. Nat. Cell Biol. 2013, 15, 274–283. [Google Scholar] [CrossRef] [Green Version]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef] [Green Version]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [Green Version]
- Aparicio, A.M.; Shen, L.; Tapia, E.M.L.N.; Lu, J.-F.; Chen, H.-C.; Zhang, J.; Wu, G.; Wang, X.; Troncoso, P.; Corn, P.G.; et al. Combined Tumor Suppressor Defects Characterize Clinically Defined Aggressive Variant Prostate Cancers. Clin. Cancer Res. 2015, 22, 1520–1530. [Google Scholar] [CrossRef] [Green Version]
- Park, J.W.; Lee, J.K.; Sheu, K.M.; Wang, L.; Balanis, N.G.; Nguyen, K.; Smith, B.A.; Cheng, C.; Tsai, B.L.; Cheng, D.; et al. Reprogramming normal human epithelial tissues to a common, lethal neuroendocrine cancer lineage. Science 2018, 362, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Gingrich, J.R.; Greenberg, N.M. A Transgenic Mouse Prostate Cancer Model. Toxicol. Pathol. 1996, 24, 502–504. [Google Scholar] [CrossRef]
- Greenberg, N.M.; DeMayo, F.; Finegold, M.J.; Medina, D.; Tilley, W.D.; Aspinall, J.O.; Cunha, G.R.; Donjacour, A.A.; Matusik, R.J.; Rosen, J.M. Prostate cancer in a transgenic mouse. Proc. Natl. Acad. Sci. USA 1995, 92, 3439–3443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenberg, N.M.; DeMayo, F.J.; Sheppard, P.C.; Barrios, R.; Lebovitz, R.; Finegold, M.; Angelopoulou, R.; Dodd, J.G.; Duckworth, M.L.; Rosen, J.M.; et al. The rat probasin gene promoter directs hormonally and developmentally regulated expression of a heterologous gene specifically to the prostate in transgenic mice. Mol. Endocrinol. 1994, 8, 230–239. [Google Scholar]
- Gingrich, J.R.; Barrios, R.J.; Morton, R.A.; Boyce, B.F.; DeMayo, F.; Finegold, M.J.; Angelopoulou, R.; Rosen, J.M.; Greenberg, N.M. Metastatic prostate cancer in a transgenic mouse. Cancer Res. 1996, 56, 4096–4102. [Google Scholar] [PubMed]
- Gingrich, J.R.; Barrios, R.J.; Kattan, M.; Nahm, H.S.; Finegold, M.J.; Greenberg, N.M. Androgen-independent prostate cancer progression in the TRAMP model. Cancer Res. 1997, 57, 4687–4691. [Google Scholar]
- Johnson, M.A.; Iversen, P.; Schwier, P.; Corn, A.L.; Sandusky, G.; Graff, J.; Neubauer, B.L. Castration triggers growth of previously static androgen-independent lesions in the transgenic adenocarcinoma of the mouse prostate (TRAMP) model. Prostate 2005, 62, 322–338. [Google Scholar] [CrossRef] [PubMed]
- Alanee, S.; Moore, A.; Nutt, M.; Holland, B.; Dynda, D.; El-Zawahry, A.; McVary, K.T. Contemporary Incidence and Mortality Rates of Neuroendocrine Prostate Cancer. Anticancer Res. 2015, 35, 4145–4150. [Google Scholar]
- Fisher, K.W.; Montironi, R.; Beltran, A.L.; Moch, H.; Wang, L.; Scarpelli, M.; Williamson, S.R.; Koch, M.O.; Cheng, L. Molecular foundations for personalized therapy in prostate cancer. Curr. Drug Targets 2015, 16, 103–114. [Google Scholar] [CrossRef]
- Tang, Y.; Wang, L.; Goloubeva, O.; Khan, M.A.; Lee, D.; Hussain, A. The relationship of neuroendocrine carcinomas to anti-tumor therapies in TRAMP mice. Prostate 2009, 69, 1763–1773. [Google Scholar] [CrossRef]
- Chiaverotti, T.; Couto, S.S.; Donjacour, A.; Mao, J.-H.; Nagase, H.; Cardiff, R.D.; Cunha, G.R.; Balmain, A. Dissociation of Epithelial and Neuroendocrine Carcinoma Lineages in the Transgenic Adenocarcinoma of Mouse Prostate Model of Prostate Cancer. Am. J. Pathol. 2008, 172, 236–246. [Google Scholar] [CrossRef] [Green Version]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef] [PubMed]
- Nicorici, D.; Şatalan, M.; Edgren, H.; Kangaspeska, S.; Murumägi, A.; Kallioniemi, O.; Virtanen, S.; Kilkku, O. FusionCatcher—A tool for finding somatic fusion genes in paired-end RNA-sequencing data. bioRxiv 2014. [Google Scholar] [CrossRef]
- Marhold, M.; Tomasich, E.; El-Gazzar, A.; Heller, G.; Spittler, A.; Horvat, R.; Krainer, M.; Horak, P. HIF1α Regulates mTOR Signaling and Viability of Prostate Cancer Stem Cells. Mol. Cancer Res. 2015, 13, 556–564. [Google Scholar] [CrossRef] [Green Version]
- Chua, C.W.; Shibata, M.; Lei, M.; Toivanen, R.; Barlow, L.J.; Bergren, S.K.; Badani, K.K.; McKiernan, J.M.; Benson, M.C.; Hibshoosh, H.; et al. Single luminal epithelial progenitors can generate prostate organoids in culture. Nat. Cell Biol. 2014, 16, 951–961. [Google Scholar] [CrossRef] [Green Version]
- Berman-Booty, L.D.; Knudsen, K.E. Models of neuroendocrine prostate cancer. Endocr.-Relat. Cancer 2015, 22, R33–R49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puca, L.; Vlachostergios, P.; Beltran, H. Neuroendocrine Differentiation in Prostate Cancer: Emerging Biology, Models, and Therapies. Cold Spring Harb. Perspect. Med. 2019, 9, a030593. [Google Scholar] [CrossRef]
- Garabedian, E.M.; Humphrey, P.A.; Gordon, J.I. A transgenic mouse model of metastatic prostate cancer originating from neuroendocrine cells. Proc. Natl. Acad. Sci. USA 1998, 95, 15382–15387. [Google Scholar] [CrossRef] [Green Version]
- Zou, M.; Toivanen, R.; Mitrofanova, A.; Floch, N.; Hayati, S.; Sun, Y.; Le Magnen, C.; Chester, D.; Mostaghel, E.A.; Califano, A.; et al. Transdifferentiation as a Mechanism of Treatment Resistance in a Mouse Model of Castration-Resistant Prostate Cancer. Cancer Discov. 2017, 7, 736–749. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, A.S.; Stoyanova, T.; Witte, O.N. Primitive origins of prostate cancer: In vivo evidence for prostate-regenerating cells and prostate cancer-initiating cells. Mol. Oncol. 2010, 4, 385–396. [Google Scholar] [CrossRef]
- Lawson, D.A.; Zong, Y.; Memarzadeh, S.; Xin, L.; Huang, J.; Witte, O.N. Basal epithelial stem cells are efficient targets for prostate cancer initiation. Proc. Natl. Acad. Sci. USA 2010, 107, 2610–2615. [Google Scholar] [CrossRef] [Green Version]
- Stoyanova, T.; Cooper, A.R.; Drake, J.M.; Liu, X.; Armstrong, A.J.; Pienta, K.J.; Zhang, H.; Kohn, D.B.; Huang, J.; Witte, O.N.; et al. Prostate cancer originating in basal cells progresses to adenocarcinoma propagated by luminal-like cells. Proc. Natl. Acad. Sci. USA 2013, 110, 20111–20116. [Google Scholar] [CrossRef] [Green Version]
- Mirosevich, J.; Bentel, J.; Zeps, N.; Redmond, S.; D’Antuono, M.; Dawkins, H.; Miyamoto, S.; Irahara, M.; Ushigoe, K.; Kuwahara, A.; et al. Androgen receptor expression of proliferating basal and luminal cells in adult murine ventral prostate. J. Endocrinol. 1999, 162, 341–350. [Google Scholar] [CrossRef]
- Karthaus, W.R.; Hofree, M.; Choi, D.; Linton, E.L.; Turkekul, M.; Bejnood, A.; Carver, B.; Gopalan, A.; Abida, W.; Laudone, V.; et al. Regenerative potential of prostate luminal cells revealed by single-cell analysis. Science 2020, 368, 497–505. [Google Scholar] [CrossRef]
- Crowley, L.; Cambuli, F.; Aparicio, L.; Shibata, M.; Robinson, B.D.; Xuan, S.; Li, W.; Hibshoosh, H.; Loda, M.; Rabadan, R.; et al. A single-cell atlas of the mouse and human prostate reveals heterogeneity and conservation of epithelial progenitors. eLife 2020, 9, e59465. [Google Scholar] [CrossRef]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal Feedback Regulation of PI3K and Androgen Receptor Signaling in PTEN-Deficient Prostate Cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef] [Green Version]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.-W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent Fusion of TMPRSS2 and ETS Transcription Factor Genes in Prostate Cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Sowalsky, A.G.; Xia, Z.; Wang, L.; Zhao, H.; Chen, S.; Bubley, G.J.; Balk, S.P.; Li, W. Whole Transcriptome Sequencing Reveals Extensive Unspliced mRNA in Metastatic Castration-Resistant Prostate Cancer. Mol. Cancer Res. 2015, 13, 98–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, S.; Liu, Y.; Xu, W.; Sun, Y.; Lu, J.; Wang, F.; Wei, M.; Shen, J.; Hou, J.; Gao, X.; et al. Long Noncoding RNA MALAT-1 is a New Potential Therapeutic Target for Castration Resistant Prostate Cancer. J. Urol. 2013, 190, 2278–2287. [Google Scholar] [CrossRef]
- Dai, X.; Liang, Z.; Liu, L.; Guo, K.; Xu, S.; Wang, H. Silencing of MALAT1 inhibits migration and invasion by sponging miR-1-3p in prostate cancer cells. Mol. Med. Rep. 2019, 20, 3499–3508. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Liu, L.; Liang, Z.; Guo, K.; Xu, S.; Wang, H. Silencing of lncRNA MALAT1 inhibits cell cycle progression via androgen receptor signaling in prostate cancer cells. Pathol.-Res. Pract. 2019, 215, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Wang, F.; Shen, J.; Sun, Y.; Xu, W.; Lu, J.; Wei, M.; Xu, C.; Wu, C.; Zhang, Z.; et al. Long non-coding RNA metastasis associated in lung adenocarcinoma transcript 1 derived miniRNA as a novel plasma-based biomarker for diagnosing prostate cancer. Eur. J. Cancer 2013, 49, 2949–2959. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Ren, S.; Chen, R.; Lu, J.; Shi, X.; Zhu, Y.; Zhang, W.; Jing, T.; Zhang, C.; Shen, J.; et al. Development and prospective multicenter evaluation of the long noncoding RNA MALAT-1 as a diagnostic urinary biomarker for prostate cancer. Oncotarget 2014, 5, 11091–11102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, G.; Guo, L.; Xing, Y.; Sun, W.; Yuan, M. Identification of prognostic biomarkers of prostate cancer with long non-coding RNA-mediated competitive endogenous RNA network. Exp. Ther. Med. 2019, 17, 3035–3040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Mao, R.; Ren, G.; Liu, X.; Zhang, Y.; Wang, J.; Wang, Y.; Li, M.; Qiu, Q.; Wang, L.; et al. Whole Exome Sequencing Identifies Putative Predictors of Recurrent Prostate Cancer with High Accuracy. OMICS 2019, 23, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Goyal, B.; Yadav, S.R.M.; Awasthee, N.; Gupta, S.; Kunnumakkara, A.B.; Gupta, S.C. Diagnostic, prognostic, and therapeutic significance of long non-coding RNA MALAT1 in cancer. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188502. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.M.; Chang, H.Y. Long Noncoding RNAs in Cancer Pathways. Cancer Cell 2016, 29, 452–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Sun, Y.; Li, L.; Niu, Y.; Lin, W.; Lin, C.; Antonarakis, E.S.; Luo, J.; Yeh, S.; Chang, C. Preclinical Study using Malat1 Small Interfering RNA or Androgen Receptor Splicing Variant 7 Degradation Enhancer ASC-J9((R)) to Suppress Enzalutamide-resistant Prostate Cancer Progression. Eur. Urol. 2017, 72, 835–844. [Google Scholar] [CrossRef] [Green Version]
- Stone, L. Prostate cancer: Escaping enzalutamide: Malat1 contributes to resistance. Nat. Rev. Urol. 2017, 14, 450. [Google Scholar] [CrossRef]
- Chou, F.-J.; Lin, C.; Tian, H.; Lin, W.; You, B.; Lu, J.; Sahasrabudhe, D.; Huang, C.-P.; Yang, V.; Yeh, S.; et al. Preclinical studies using cisplatin/carboplatin to restore the Enzalutamide sensitivity via degrading the androgen receptor splicing variant 7 (ARv7) to further suppress Enzalutamide resistant prostate cancer. Cell Death Dis. 2020, 11, 942. [Google Scholar] [CrossRef]
- Lin, C.; Chou, F.-J.; Lu, J.; Lin, W.; Truong, M.; Tian, H.; Sun, Y.; Luo, J.; Yang, R.; Niu, Y.; et al. Preclinical studies show using enzalutamide is less effective in docetaxel-pretreated than in docetaxel-naïve prostate cancer cells. Aging 2020, 12, 17694–17712. [Google Scholar] [CrossRef]
- Nanni, S.; Aiello, A.; Salis, C.; Re, A.; Cencioni, C.; Bacci, L.; Pierconti, F.; Pinto, F.; Ripoli, C.; Ostano, P.; et al. Metabolic Reprogramming by Malat1 Depletion in Prostate Cancer (Stage 2). Cancers 2020, 13, 15. [Google Scholar] [CrossRef] [PubMed]
- Hao, T.; Wang, Z.; Yang, J.; Zhang, Y.; Shang, Y.; Sun, J. MALAT1 knockdown inhibits prostate cancer progression by regulating miR-140/BIRC6 axis. Biomed Pharmacother. 2020, 123, 109666. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Xu, W.; Du, X.; Hou, J. MALAT1 silencing suppresses prostate cancer progression by upregulating miR-1 and downregulating KRAS. OncoTargets Ther. 2018, 11, 3461–3473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Ding, L.; Wang, L.; Zhao, Y.; Sun, Z.; Karnes, R.J.; Zhang, J.; Huang, H. LncRNA MALAT1 enhances oncogenic activities of EZH2 in castration-resistant prostate cancer. Oncotarget 2015, 6, 41045–41055. [Google Scholar] [CrossRef]
- Aiello, A.; Bacci, L.; Re, A.; Ripoli, C.; Pierconti, F.; Pinto, F.; Masetti, R.; Grassi, C.; Gaetano, C.; Bassi, P.F.; et al. MALAT1 and HOTAIR Long Non-Coding RNAs Play Opposite Role in Estrogen-Mediated Transcriptional Regulation in Prostate Cancer Cells. Sci. Rep. 2016, 6, 38414. [Google Scholar] [CrossRef]
- Rambout, X.; Maquat, L.E. NCBP3: A Multifaceted Adaptive Regulator of Gene Expression. Trends Biochem. Sci. 2020, 46, 87–96. [Google Scholar] [CrossRef]
- Scheffer, G.L.; Wijngaard, P.L.J.; Flens, M.J.; Izquierdo, M.A.; Slovak, M.L.; Pinedo, H.M.; Meijer, C.J.L.M.; Clevers, H.C.; Scheper, R.J. The drug resistance-related protein LRP is the human major vault protein. Nat. Med. 1995, 1, 578–582. [Google Scholar] [CrossRef]
- Van Brussel, J.P.; van Steenbrugge, G.J.; van Krimpen, C.; Bogdanowicz, J.F.; van der Kwast, T.H.; Schröder, F.H.; Mickisch, G.H. Expression of multidrug resistance related proteins and proliferative activity is increased in advanced clinical prostate cancer. J. Urol. 2001, 165, 130–135. [Google Scholar] [CrossRef]
- Ramberg, H.; Richardsen, E.; de Souza, G.A.; Rakaee, M.; Stensland, M.E.; Braadland, P.R.; Nygård, S.; Ögren, O.; Guldvik, I.J.; Berge, V.; et al. Proteomic analyses identify major vault protein as a prognostic biomarker for fatal prostate cancer. Carcinogenesis 2021, 42, 685–693. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| V3SM11247-246169059 | AAAAGGCTCGTTCACCTGT |
| V3SM11247-246186748 | TGCGATTTCCTCGGGCTGA |
| V3SM11247-246310696 | AACCCTACTGACGAATCTG |
| Fwd | Rev | |
|---|---|---|
| MALAT1 | TGCTGCATTAAGCCTGGAGT | ACGAAACATTGGCACACTGC |
| AR | AATGAGTACCGCATGCACAA | CCCATCCACTGGAATAATGC |
| ACTIN | ATGAGCTGCCTGACGGCCAGGTCATC | TGGTACCACCAGACAGCACTGTGTTG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marhold, M.; Udovica, S.; Topakian, T.; Horak, P.; Horvat, R.; Tomasich, E.; Heller, G.; Krainer, M. MALAT1 Fusions and Basal Cells Contribute to Primary Resistance against Androgen Receptor Inhibition in TRAMP Mice. Cancers 2022, 14, 749. https://doi.org/10.3390/cancers14030749
Marhold M, Udovica S, Topakian T, Horak P, Horvat R, Tomasich E, Heller G, Krainer M. MALAT1 Fusions and Basal Cells Contribute to Primary Resistance against Androgen Receptor Inhibition in TRAMP Mice. Cancers. 2022; 14(3):749. https://doi.org/10.3390/cancers14030749
Chicago/Turabian StyleMarhold, Maximilian, Simon Udovica, Thais Topakian, Peter Horak, Reinhard Horvat, Erwin Tomasich, Gerwin Heller, and Michael Krainer. 2022. "MALAT1 Fusions and Basal Cells Contribute to Primary Resistance against Androgen Receptor Inhibition in TRAMP Mice" Cancers 14, no. 3: 749. https://doi.org/10.3390/cancers14030749
APA StyleMarhold, M., Udovica, S., Topakian, T., Horak, P., Horvat, R., Tomasich, E., Heller, G., & Krainer, M. (2022). MALAT1 Fusions and Basal Cells Contribute to Primary Resistance against Androgen Receptor Inhibition in TRAMP Mice. Cancers, 14(3), 749. https://doi.org/10.3390/cancers14030749