
Targeting Mitochondrial COX-2 Enhances Chemosensitivity via Drp1-Dependent Remodeling of Mitochondrial Dynamics in Hepatocellular Carcinoma

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Bioinformatics Analysis
2.2. HCC Tissue Specimens
2.3. In Vivo Subcutaneous Xenograft Models
2.4. Immunohistochemical (IHC) Analysis
2.5. Cell Culture
2.6. Establishment of Stable Cell Lines and RNA Interference
2.7. Antibodies and Reagents
2.8. Western Blotting Analysis
2.9. Mitochondrial and Cytoplasmic Fractions and Mitochondrial Protein Immunoprecipitation (Mito-IP)
2.10. Immunofluorescence (IF) Assay
2.11. Proximity Ligation Assay (PLA)
2.12. I-TASSER Analysis
2.13. Cell Viability, EdU Incorporation Assay, and Apoptosis Analysis
2.14. Statistical Analyses
3. Results
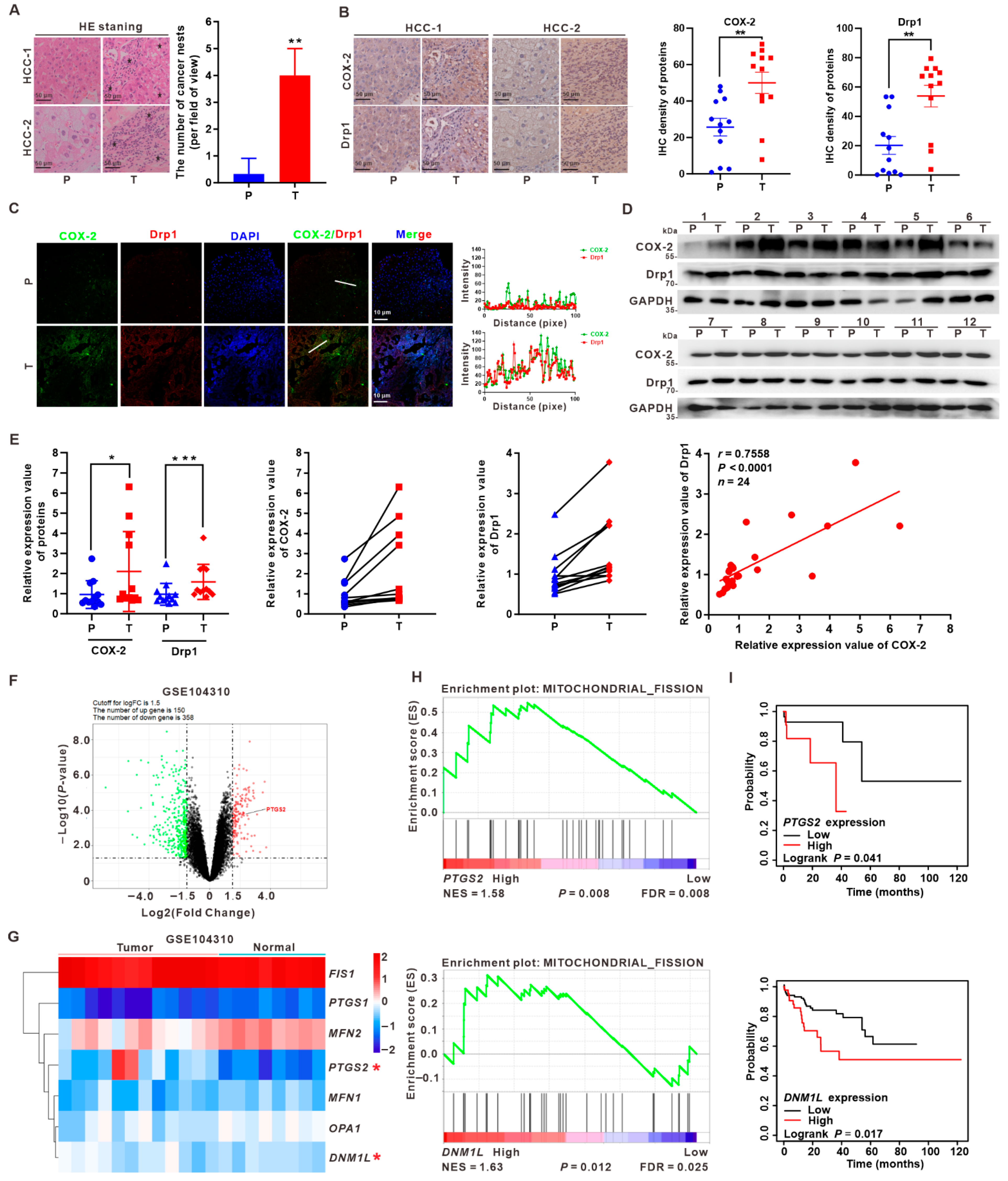
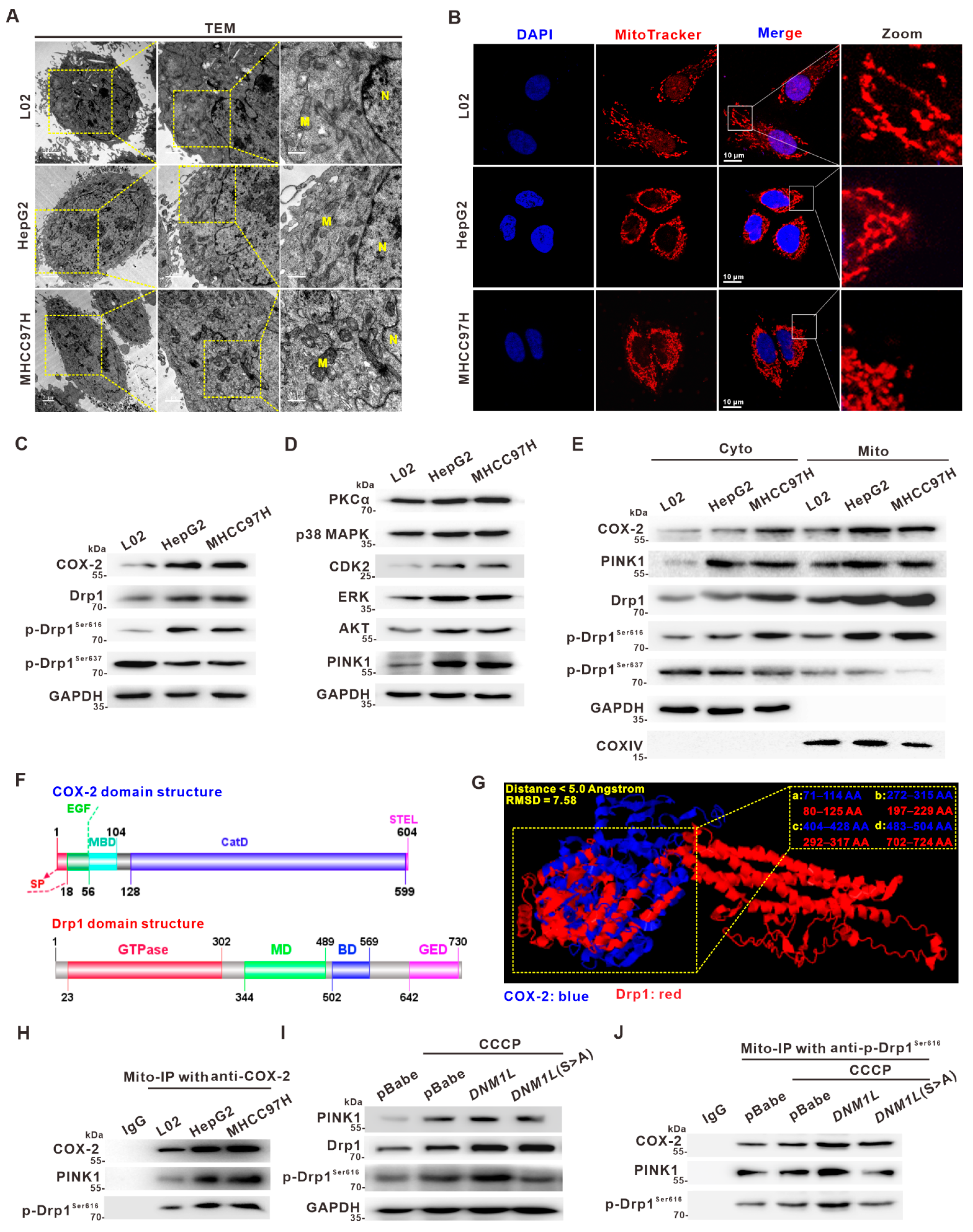
3.1. Upregulation of COX-2 and Drp1 Is Associated with the Poorer Prognosis of HCC Patients
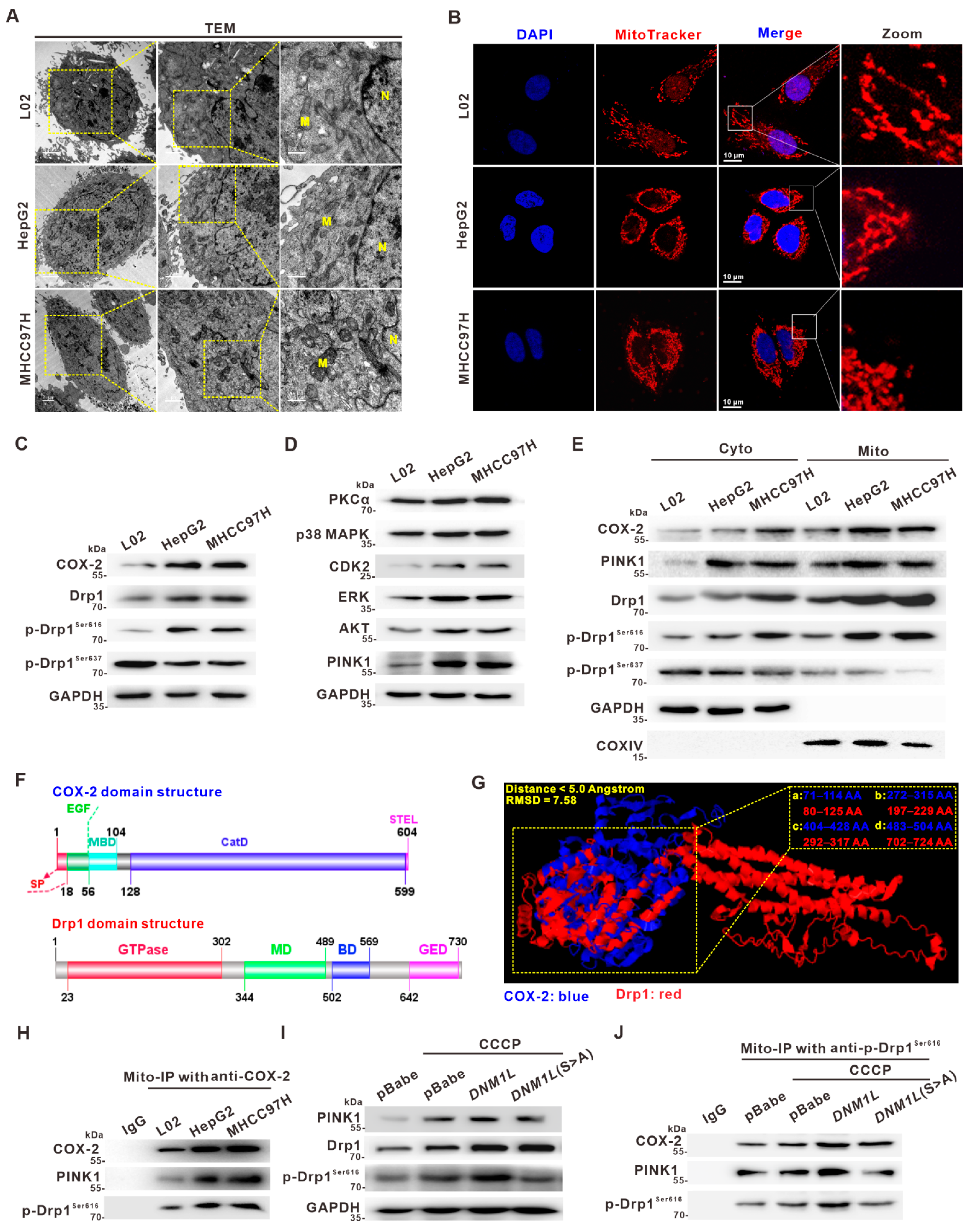
3.2. Activation of Drp1 Enhances Mitochondrial Fission and Its Molecular Association with COX-2 in HCC Cells
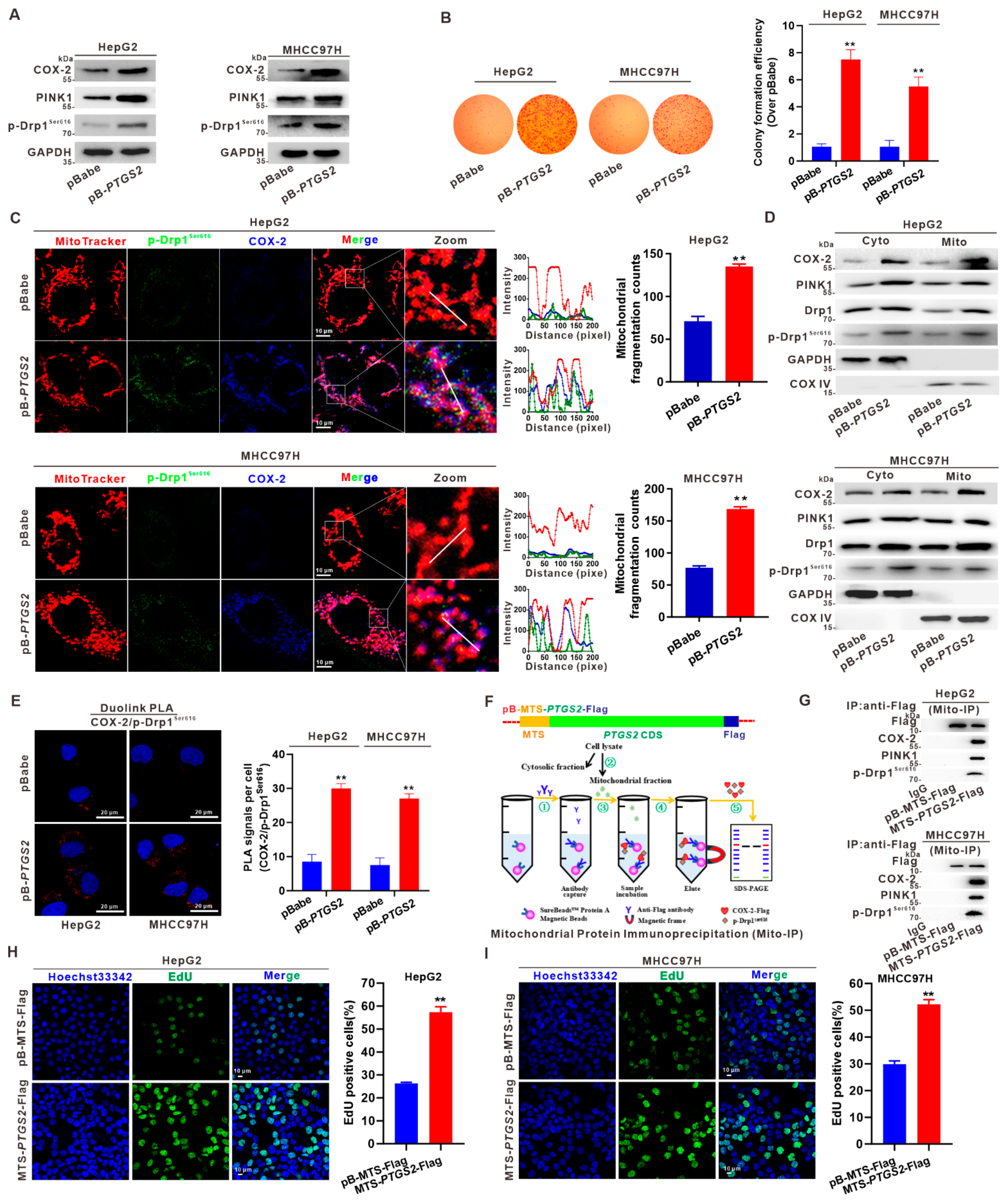
3.3. Mito-COX-2 Modulates Mitochondrial Fission by Stabilizing the Activity of p-Drp1Ser616 in HCC Cells
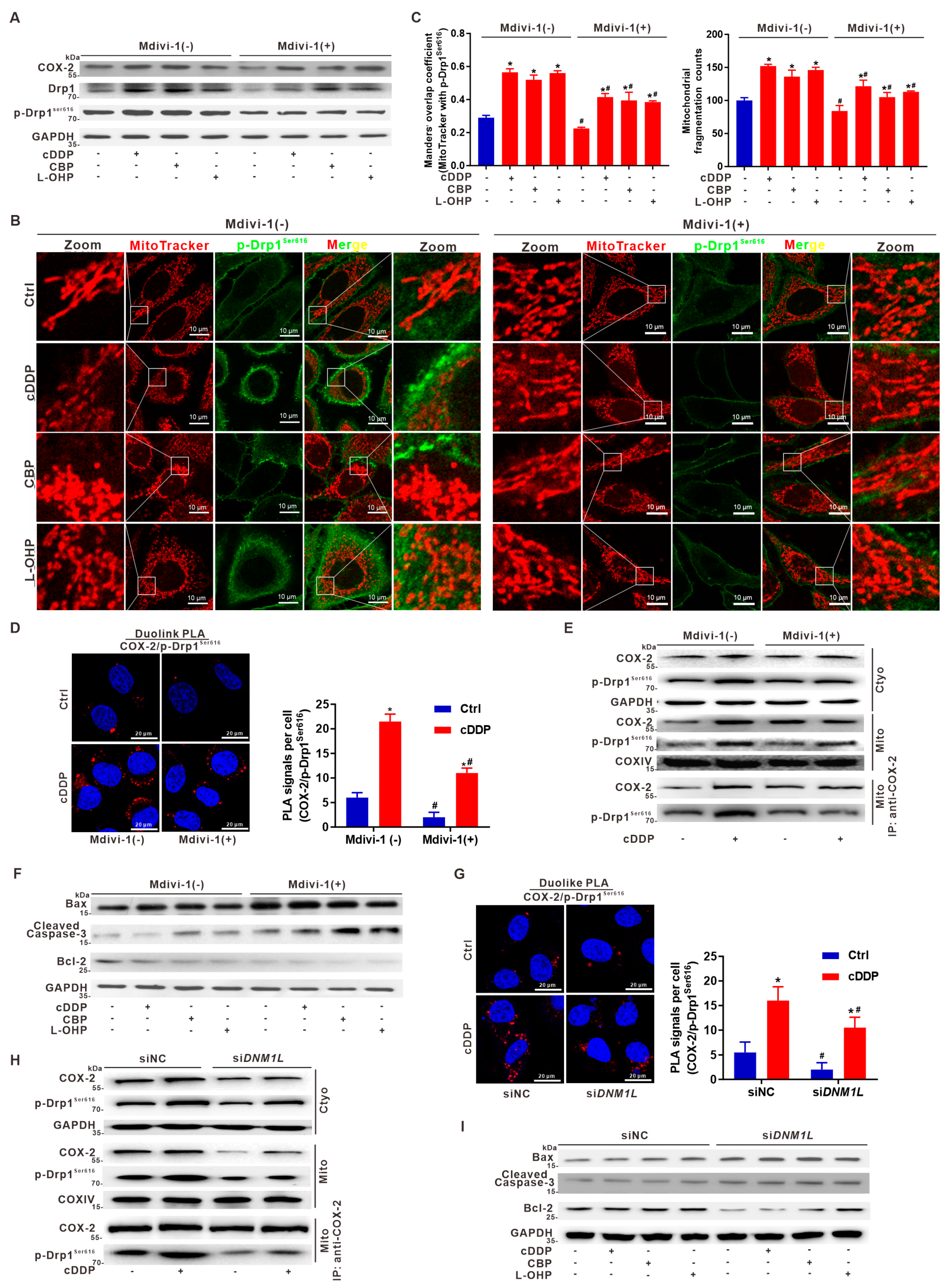
3.4. Suppression of Mito-COX-2 Translocation Decreases Its Interaction with p-Drp1Ser616 and Modulates Mitochondrial Fission in HCC Cells
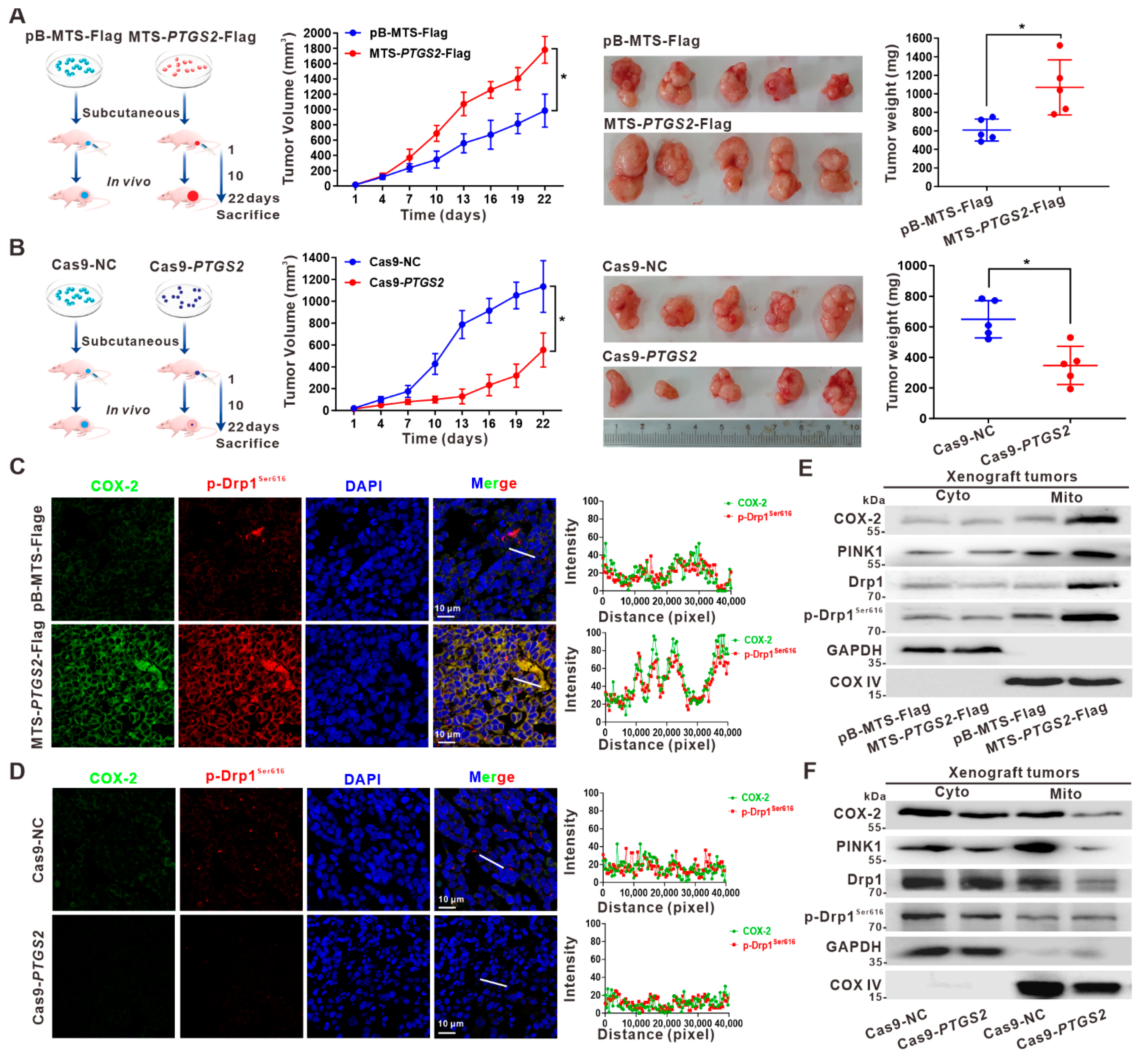
3.5. Suppression of HCC Xenograft Growth via the Inhibition of p-Drp1Ser616 by Targeted Intervention on Mito-COX-2 Translocation In Vivo
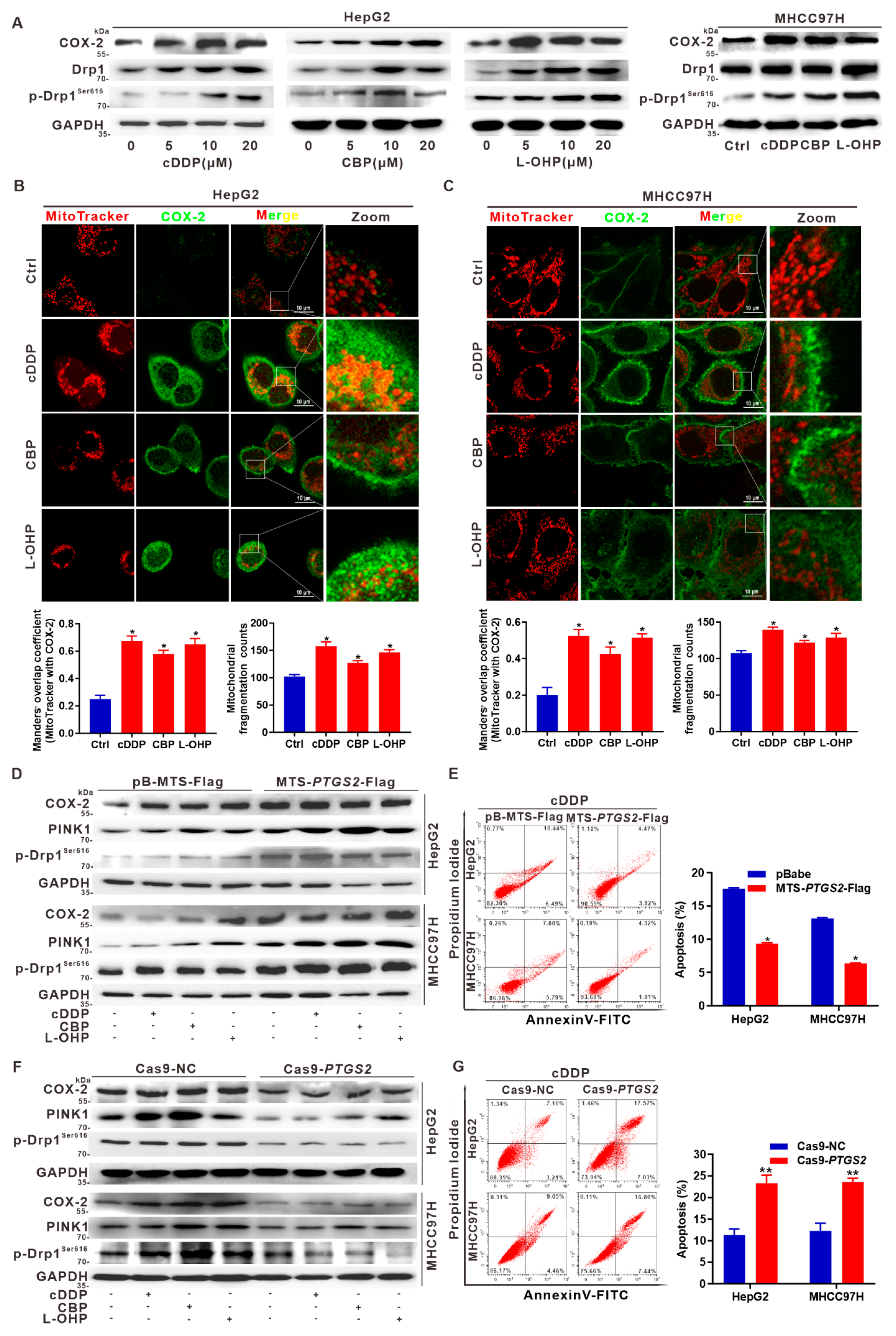
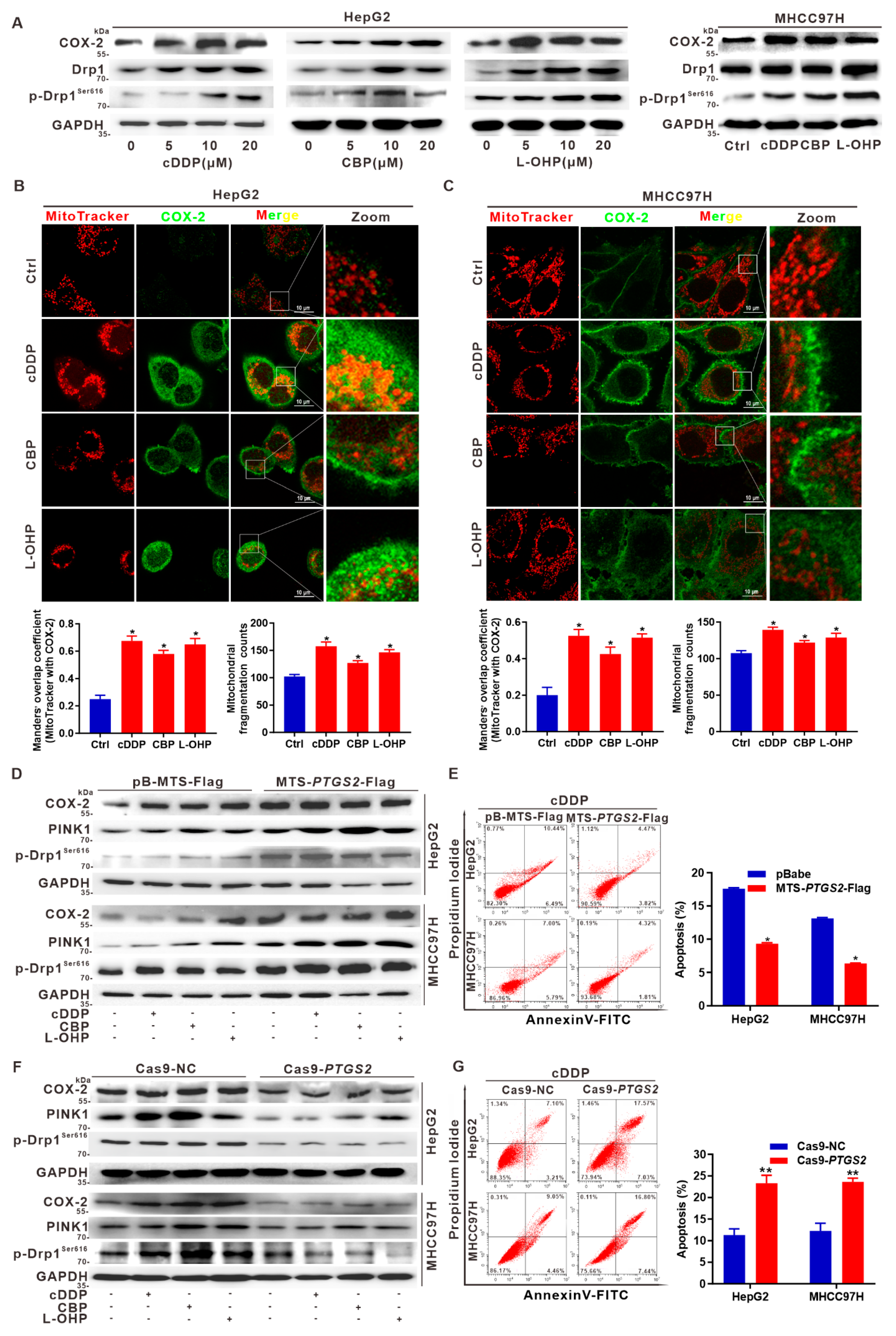
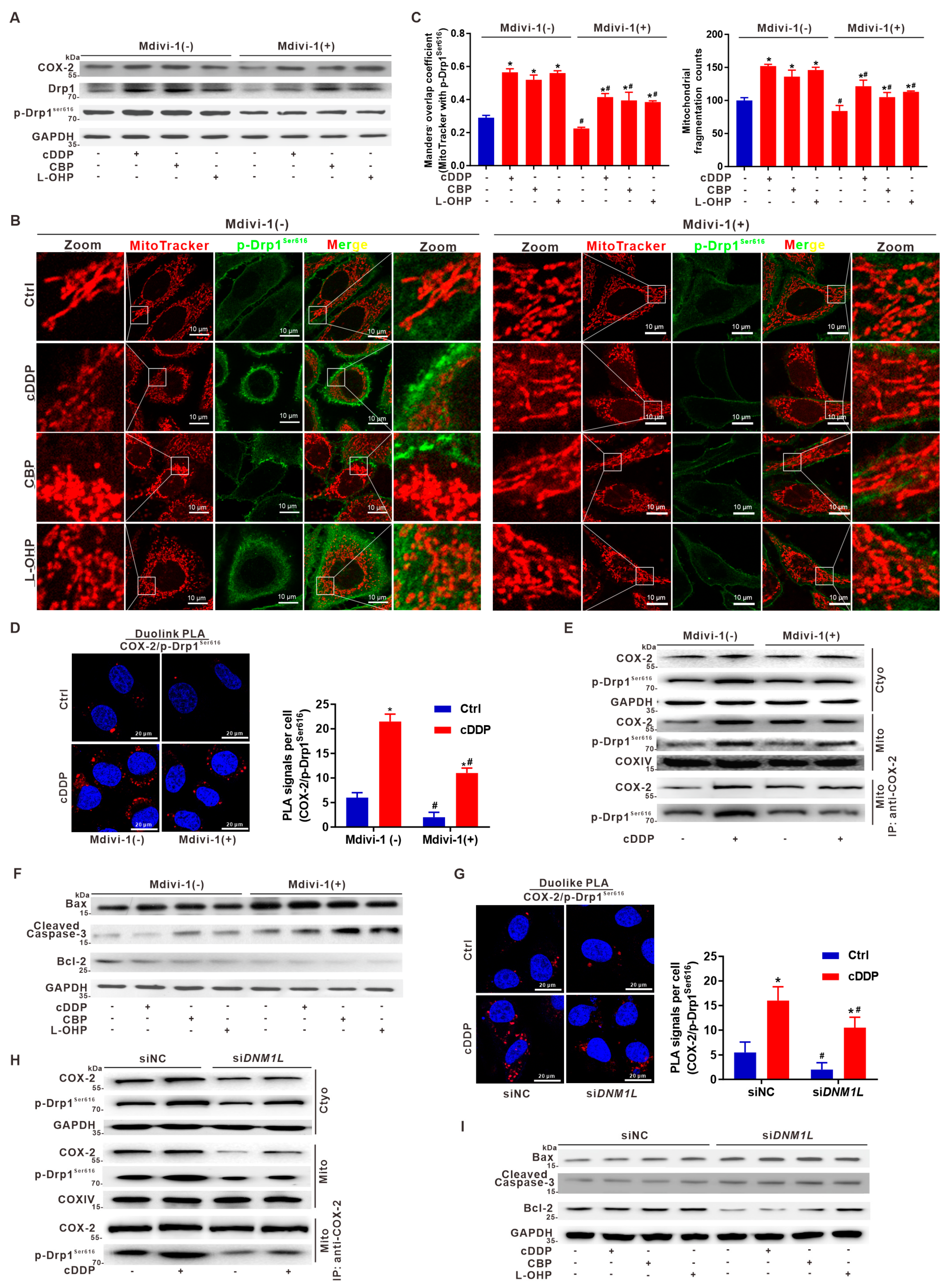
3.6. Targeted Intervention on Mito-COX-2 Enhances Chemosensitivity by Inhibiting p-Drp1Ser616-Driven Mitochondrial Fission in Platinum Drug-Treated HCC Cells
3.7. Suppression of Drp1 Promotes Apoptosis via Inhibition of Mito-COX-2/p-Drp1Ser616 Interaction in Platinum Drug-Treated HCC Cells
3.8. Deacetylation of Mito-COX-2 via SIRT3 Activation Mediates Higher Sensitivity of HCC to Cisplatin by Inhibiting Mito-COX-2/p-Drp1Ser616 Interactions In Vitro and In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Llovet, J.M.; Castet, F.; Heikenwalder, M.; Maini, M.K.; Mazzaferro, V.; Pinato, D.J.; Pikarsky, E.; Zhu, A.X.; Finn, R.S. Immunotherapies for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2021. [Google Scholar] [CrossRef]
- Goh, G.B.; Chang, P.E.; Tan, C.K. Changing epidemiology of hepatocellular carcinoma in Asia. Best Pract. Res. Clin. Gastroenterol. 2015, 29, 919–928. [Google Scholar] [CrossRef]
- Chu, Y.J.; Yang, H.I.; Wu, H.C.; Lee, M.H.; Liu, J.; Wang, L.Y.; Lu, S.N.; Jen, C.L.; You, S.L.; Santella, R.M.; et al. Aflatoxin B1 exposure increases the risk of hepatocellular carcinoma associated with hepatitis C virus infection or alcohol consumption. Eur. J. Cancer 2018, 94, 37–46. [Google Scholar] [CrossRef]
- Chen, J.H.; Wang, Y.Y.; Lv, W.B.; Gan, Y.; Chang, W.; Tian, N.N.; Huang, X.H.; Liu, L.; Yu, X.F.; Chen, S.D. Effects of interactions between environmental factors and KIF1B genetic variants on the risk of hepatocellular carcinoma in a Chinese cohort. World J. Gastroenterol. 2016, 22, 4183–4190. [Google Scholar] [CrossRef]
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.C.; Llovet, J.M. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology 2015, 149, 1226–1239.e4. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Wang, H. Precision diagnosis and treatment of liver cancer in China. Cancer Lett. 2018, 412, 283–288. [Google Scholar] [CrossRef]
- Kraus, F.; Roy, K.; Pucadyil, T.J.; Ryan, M.T. Function and regulation of the divisome for mitochondrial fission. Nature 2021, 590, 57–66. [Google Scholar] [CrossRef]
- Tabish, T.A.; Narayan, R.J. Mitochondria-targeted graphene for advanced cancer therapeutics. Acta Biomater. 2021, 129, 43–56. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, H.; Li, J.; Jiang, X.; Zhang, Y.; Wu, Q.; Shen, L.; Shi, J.; Gao, N. ROCK1 activation-mediated mitochondrial translocation of Drp1 and cofilin are required for arnidiol-induced mitochondrial fission and apoptosis. J. Exp. Clin. Cancer Res. 2020, 39, 37. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Lee, J.; Kim, J.Y.; Wang, L.; Tian, Y.; Chan, S.T.; Cho, C.; Machida, K.; Chen, D.; Ou, J.J. Mitophagy Controls the Activities of Tumor Suppressor p53 to Regulate Hepatic Cancer Stem Cells. Mol. Cell 2017, 68, 281–292.e5. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Li, W.; Chen, H.; Jiang, L.; Zhu, R.; Feng, D. FUNDC1 is a novel mitochondrial-associated-membrane (MAM) protein required for hypoxia-induced mitochondrial fission and mitophagy. Autophagy 2016, 12, 1675–1676. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Liu, H.; Liu, J.; Long, J. Post-translational modifications on mitochondrial metabolic enzymes in cancer. Free Radic. Biol. Med. 2022, 179, 11–23. [Google Scholar] [CrossRef]
- Zhang, S.; Che, L.; He, C.; Huang, J.; Guo, N.; Shi, J.; Lin, Y.; Lin, Z. Drp1 and RB interaction to mediate mitochondria-dependent necroptosis induced by cadmium in hepatocytes. Cell Death Dis. 2019, 10, 523. [Google Scholar] [CrossRef] [Green Version]
- Otera, H.; Mihara, K. Molecular mechanisms and physiologic functions of mitochondrial dynamics. J. Biochem. 2011, 149, 241–251. [Google Scholar] [CrossRef] [Green Version]
- Cereghetti, G.M.; Costa, V.; Scorrano, L. Inhibition of Drp1-dependent mitochondrial fragmentation and apoptosis by a polypeptide antagonist of calcineurin. Cell Death Differ. 2010, 17, 1785–1794. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Ryu, H.J.; Kim, M.J.; Kang, T.C. LIM kinase-2 induces programmed necrotic neuronal death via dysfunction of DRP1-mediated mitochondrial fission. Cell Death Differ. 2014, 21, 1036–1049. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Cao, H.; Zhan, L.; Yin, C.; Wang, G.; Liang, P.; Li, J.; Wang, Z.; Liu, B.; Huang, Q.; et al. Mitochondrial fission promotes cell migration by Ca2+ /CaMKII/ERK/FAK pathway in hepatocellular carcinoma. Liver Int. 2018, 38, 1263–1272. [Google Scholar] [CrossRef]
- Huang, Q.; Zhan, L.; Cao, H.; Li, J.; Lyu, Y.; Guo, X.; Zhang, J.; Ji, L.; Ren, T.; An, J.; et al. Increased mitochondrial fission promotes autophagy and hepatocellular carcinoma cell survival through the ROS-modulated coordinated regulation of the NFKB and TP53 pathways. Autophagy 2016, 12, 999–1014. [Google Scholar] [CrossRef]
- Kashatus, J.A.; Nascimento, A.; Myers, L.J.; Sher, A.; Byrne, F.L.; Hoehn, K.L.; Counter, C.M.; Kashatus, D.F. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol. Cell 2015, 57, 537–551. [Google Scholar] [CrossRef] [Green Version]
- Smith, W.L.; DeWitt, D.L.; Garavito, R.M. Cyclooxygenases: Structural, cellular, and molecular biology. Annu. Rev. Biochem. 2000, 69, 145–182. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Lin, Y.; Hu, Y.; Shan, W.; Liu, S.; Xu, Y.; Zhang, H.; Cai, S.; Yu, X.; Cai, Z.; et al. mTOR inhibition improves the immunomodulatory properties of human bone marrow mesenchymal stem cells by inducing COX-2 and PGE2. Stem Cell Res. 2017, 8, 292. [Google Scholar] [CrossRef] [Green Version]
- Norouzi, M.; Norouzi, S.; Amini, M.; Amanzadeh, A.; Irian, S.; Salimi, M. Apoptotic effects of two COX-2 inhibitors on breast adenocarcinoma cells through COX-2 independent pathway. J. Cell Biochem. 2015, 116, 81–90. [Google Scholar] [CrossRef]
- Niu, Z.; Zhang, W.; Gu, X.; Zhang, X.; Qi, Y.; Zhang, Y. Mitophagy inhibits proliferation by decreasing cyclooxygenase-2 (COX-2) in arsenic trioxide-treated HepG2 cells. Environ. Toxicol. Pharm. 2016, 45, 212–221. [Google Scholar] [CrossRef]
- Zhou, T.J.; Zhang, S.L.; He, C.Y.; Zhuang, Q.Y.; Han, P.Y.; Jiang, S.W.; Yao, H.; Huang, Y.J.; Ling, W.H.; Lin, Y.C.; et al. Downregulation of mitochondrial cyclooxygenase-2 inhibits the stemness of nasopharyngeal carcinoma by decreasing the activity of dynamin-related protein 1. Theranostics 2017, 7, 1389–1406. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Lin, Y.; Han, P.Y.; Jiang, S.; Che, L.; He, C.Y.; Lin, Y.C.; Lin, Z.N. HBx combined with AFB1 triggers hepatic steatosis via COX-2-mediated necrosome formation and mitochondrial dynamics disorder. J. Cell Mol. Med. 2019, 23, 5920–5933. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.Y.; Zhan, D.L.; Chen, Y.Y.; Wang, W.H.; He, C.Y.; Lin, Y.; Lin, Y.C.; Lin, Z.N. Aflatoxin B1 enhances pyroptosis of hepatocytes and activation of Kupffer cells to promote liver inflammatory injury via dephosphorylation of cyclooxygenase-2: An in vitro, ex vivo and in vivo study. Arch. Toxicol. 2019, 93, 3305–3320. [Google Scholar] [CrossRef]
- Jin, L.; Galonek, H.; Israelian, K.; Choy, W.; Morrison, M.; Xia, Y.; Wang, X.; Xu, Y.; Yang, Y.; Smith, J.J.; et al. Biochemical characterization, localization, and tissue distribution of the longer form of mouse SIRT3. Protein Sci. 2009, 18, 514–525. [Google Scholar] [CrossRef]
- Song, C.L.; Tang, H.; Ran, L.K.; Ko, B.C.; Zhang, Z.Z.; Chen, X.; Ren, J.H.; Tao, N.N.; Li, W.Y.; Huang, A.L.; et al. Sirtuin 3 inhibits hepatocellular carcinoma growth through the glycogen synthase kinase-3beta/BCL2-associated X protein-dependent apoptotic pathway. Oncogene 2016, 35, 631–641. [Google Scholar] [CrossRef]
- Cheng, Y.; Ren, X.; Gowda, A.S.; Shan, Y.; Zhang, L.; Yuan, Y.S.; Patel, R.; Wu, H.; Huber-Keener, K.; Yang, J.W.; et al. Interaction of Sirt3 with OGG1 contributes to repair of mitochondrial DNA and protects from apoptotic cell death under oxidative stress. Cell Death Dis. 2013, 4, e731. [Google Scholar] [CrossRef] [Green Version]
- Kuo, M.T.; Huang, Y.F.; Chou, C.Y.; Chen, H.H.W. Targeting the Copper Transport System to Improve Treatment Efficacies of Platinum-Containing Drugs in Cancer Chemotherapy. Pharmaceuticals 2021, 14, 549. [Google Scholar] [CrossRef]
- Kohno, K.; Wang, K.Y.; Takahashi, M.; Kurita, T.; Yoshida, Y.; Hirakawa, M.; Harada, Y.; Kuma, A.; Izumi, H.; Matsumoto, S. Mitochondrial Transcription Factor A and Mitochondrial Genome as Molecular Targets for Cisplatin-Based Cancer Chemotherapy. Int. J. Mol. Sci. 2015, 16, 19836–19850. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, Q.; Zhou, T.; He, C.; Zhang, S.; Qiu, Y.; Luo, B.; Zhao, R.; Liu, H.; Lin, Y.; Lin, Z. Protein phosphatase 2A-B55delta enhances chemotherapy sensitivity of human hepatocellular carcinoma under the regulation of microRNA-133b. J. Exp. Clin. Cancer Res. 2016, 35, 67. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.F.; Liu, T.Q.; Zhi, X.T.; Zou, J.; Zhong, J.T.; Li, T.; Mo, X.L.; Zhou, W.; Guo, W.W.; Liu, X.; et al. COX-2/PGE2 Axis Regulates HIF2alpha Activity to Promote Hepatocellular Carcinoma Hypoxic Response and Reduce the Sensitivity of Sorafenib Treatment. Clin. Cancer Res. 2018, 24, 3204–3216. [Google Scholar] [CrossRef] [Green Version]
- Carrico, C.; Meyer, J.G.; He, W.; Gibson, B.W.; Verdin, E. The Mitochondrial Acylome Emerges: Proteomics, Regulation by Sirtuins, and Metabolic and Disease Implications. Cell Metab. 2018, 27, 497–512. [Google Scholar] [CrossRef] [Green Version]
- Bagul, P.K.; Katare, P.B.; Bugga, P.; Dinda, A.K.; Banerjee, S.K. SIRT-3 Modulation by Resveratrol Improves Mitochondrial Oxidative Phosphorylation in Diabetic Heart through Deacetylation of TFAM. Cells 2018, 7, 235. [Google Scholar] [CrossRef] [Green Version]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef]
- Woo, S.M.; Min, K.J.; Kwon, T.K. Melatonin-mediated Bim up-regulation and cyclooxygenase-2 (COX-2) down-regulation enhances tunicamycin-induced apoptosis in MDA-MB-231 cells. J. Pineal Res. 2015, 58, 310–320. [Google Scholar] [CrossRef]
- Semaan, J.; Pinon, A.; Rioux, B.; Hassan, L.; Limami, Y.; Pouget, C.; Fagnere, C.; Sol, V.; Diab-Assaf, M.; Simon, A.; et al. Resistance to 3-HTMC-Induced Apoptosis Through Activation of PI3K/Akt, MEK/ERK, and p38/COX-2/PGE2 Pathways in Human HT-29 and HCT116 Colorectal Cancer Cells. J. Cell Biochem. 2016, 117, 2875–2885. [Google Scholar] [CrossRef]
- Xu, G.; Wang, Y.; Li, W.; Cao, Y.; Xu, J.; Hu, Z.; Hao, Y.; Hu, L.; Sun, Y. COX-2 Forms Regulatory Loop with YAP to Promote Proliferation and Tumorigenesis of Hepatocellular Carcinoma Cells. Neoplasia 2018, 20, 324–334. [Google Scholar] [CrossRef]
- Janakiraman, H.; House, R.P.; Talwar, S.; Courtney, S.M.; Hazard, E.S.; Hardiman, G.; Mehrotra, S.; Howe, P.H.; Gangaraju, V.; Palanisamy, V. Repression of caspase-3 and RNA-binding protein HuR cleavage by cyclooxygenase-2 promotes drug resistance in oral squamous cell carcinoma. Oncogene 2017, 36, 3137–3148. [Google Scholar] [CrossRef] [Green Version]
- Zha, L.; Fan, L.; Sun, G.; Wang, H.; Ma, T.; Zhong, F.; Wei, W. Melatonin sensitizes human hepatoma cells to endoplasmic reticulum stress-induced apoptosis. J. Pineal Res. 2012, 52, 322–331. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [Green Version]
- Moore, A.S.; Wong, Y.C.; Simpson, C.L.; Holzbaur, E.L. Dynamic actin cycling through mitochondrial subpopulations locally regulates the fission-fusion balance within mitochondrial networks. Nat. Commun. 2016, 7, 12886. [Google Scholar] [CrossRef]
- Cho, H.M.; Ryu, J.R.; Jo, Y.; Seo, T.W.; Choi, Y.N.; Kim, J.H.; Chung, J.M.; Cho, B.; Kang, H.C.; Yu, S.W.; et al. Drp1-Zip1 Interaction Regulates Mitochondrial Quality Surveillance System. Mol. Cell 2019, 73, 364–376.e8. [Google Scholar] [CrossRef] [Green Version]
- Bohovych, I.; Chan, S.S.; Khalimonchuk, O. Mitochondrial protein quality control: The mechanisms guarding mitochondrial health. Antioxid. Redox Signal. 2015, 22, 977–994. [Google Scholar] [CrossRef] [Green Version]
- Ni, Z.; He, J.; Wu, Y.; Hu, C.; Dai, X.; Yan, X.; Li, B.; Li, X.; Xiong, H.; Li, Y.; et al. AKT-mediated phosphorylation of ATG4B impairs mitochondrial activity and enhances the Warburg effect in hepatocellular carcinoma cells. Autophagy 2018, 14, 685–701. [Google Scholar] [CrossRef] [Green Version]
- Chung, K.P.; Huang, Y.L.; Chen, Y.J.; Juan, Y.H.; Hsu, C.L.; Nakahira, K.; Huang, Y.T.; Lin, M.W.; Wu, S.G.; Shih, J.Y.; et al. Multi-kinase framework promotes proliferation and invasion of lung adenocarcinoma through activation of dynamin-related protein 1. Mol. Oncol. 2021, 15, 560–578. [Google Scholar] [CrossRef]
- Tang, F.; Zhang, R.; Wang, J. Cyclooxygenase-2-Mediated Up-Regulation of Mitochondrial Transcription Factor a Mitigates the Radio-Sensitivity of Cancer Cells. Int. J. Mol. Sci. 2019, 20, 1218. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.Y.; Su, Y.F.; Hsieh, M.T.; Lin, S.; Meng, R.; London, D.; Lin, C.; Tang, H.Y.; Hwang, J.; Davis, F.B.; et al. Nuclear monomeric integrin alphav in cancer cells is a coactivator regulated by thyroid hormone. FASEB J. 2013, 27, 3209–3216. [Google Scholar] [CrossRef]
- Chin, Y.T.; Wei, P.L.; Ho, Y.; Nana, A.W.; Changou, C.A.; Chen, Y.R.; Yang, Y.S.; Hsieh, M.T.; Hercbergs, A.; Davis, P.J.; et al. Thyroxine inhibits resveratrol-caused apoptosis by PD-L1 in ovarian cancer cells. Endocr. Relat. Cancer 2018, 25, 533–545. [Google Scholar] [CrossRef] [Green Version]
- Alexanian, A.; Sorokin, A. Cyclooxygenase 2: Protein-protein interactions and posttranslational modifications. Physiol. Genom. 2017, 49, 667–681. [Google Scholar] [CrossRef]
- Kotiadis, V.N.; Duchen, M.R.; Osellame, L.D. Mitochondrial quality control and communications with the nucleus are important in maintaining mitochondrial function and cell health. Biochim. Biophys. Acta 2014, 1840, 1254–1265. [Google Scholar] [CrossRef] [Green Version]
- Jadiya, P.; Tomar, D. Mitochondrial Protein Quality Control Mechanisms. Genes 2020, 11, 563. [Google Scholar] [CrossRef]
- Zong, W.X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Aventaggiato, M.; Vernucci, E.; Barreca, F.; Russo, M.A.; Tafani, M. Sirtuins’ control of autophagy and mitophagy in cancer. Pharm. Ther. 2021, 221, 107748. [Google Scholar] [CrossRef]
- Liao, G.; Zhao, Z.; Yang, H.; Li, X. Honokiol ameliorates radiation-induced brain injury via the activation of SIRT3. J. Int. Med. Res. 2020, 48, 1–11. [Google Scholar] [CrossRef]
- Tyagi, A.; Mirita, C.; Taher, N.; Shah, I.; Moeller, E.; Tyagi, A.; Chong, T.; Pugazhenthi, S. Metabolic syndrome exacerbates amyloid pathology in a comorbid Alzheimer’s mouse model. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165849. [Google Scholar] [CrossRef]
- Rodrigues, T.; Ferraz, L.S. Therapeutic potential of targeting mitochondrial dynamics in cancer. Biochem. Pharm. 2020, 182, 114282. [Google Scholar] [CrossRef]
- Tian, H.; Zhang, B.; Li, L.; Wang, G.; Li, H.; Zheng, J. Manipulation of Mitochondrial Plasticity Changes the Metabolic Competition Between “Foe” and “Friend” During Tumor Malignant Transformation. Front. Oncol. 2020, 10, 1692. [Google Scholar] [CrossRef]
- Han, Y.; Kim, B.; Cho, U.; Park, I.S.; Kim, S.I.; Dhanasekaran, D.N.; Tsang, B.K.; Song, Y.S. Mitochondrial fission causes cisplatin resistance under hypoxic conditions via ROS in ovarian cancer cells. Oncogene 2019, 38, 7089–7105. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Che, L.; Wu, J.-S.; Du, Z.-B.; He, Y.-Q.; Yang, L.; Lin, J.-X.; Lei, Z.; Chen, X.-X.; Guo, D.-B.; Li, W.-G.; et al. Targeting Mitochondrial COX-2 Enhances Chemosensitivity via Drp1-Dependent Remodeling of Mitochondrial Dynamics in Hepatocellular Carcinoma. Cancers 2022, 14, 821. https://doi.org/10.3390/cancers14030821
Che L, Wu J-S, Du Z-B, He Y-Q, Yang L, Lin J-X, Lei Z, Chen X-X, Guo D-B, Li W-G, et al. Targeting Mitochondrial COX-2 Enhances Chemosensitivity via Drp1-Dependent Remodeling of Mitochondrial Dynamics in Hepatocellular Carcinoma. Cancers. 2022; 14(3):821. https://doi.org/10.3390/cancers14030821
Chicago/Turabian StyleChe, Lin, Jia-Shen Wu, Ze-Bang Du, Yu-Qiao He, Lei Yang, Jin-Xian Lin, Zhao Lei, Xiao-Xuan Chen, Dong-Bei Guo, Wen-Gang Li, and et al. 2022. "Targeting Mitochondrial COX-2 Enhances Chemosensitivity via Drp1-Dependent Remodeling of Mitochondrial Dynamics in Hepatocellular Carcinoma" Cancers 14, no. 3: 821. https://doi.org/10.3390/cancers14030821
APA StyleChe, L., Wu, J.-S., Du, Z.-B., He, Y.-Q., Yang, L., Lin, J.-X., Lei, Z., Chen, X.-X., Guo, D.-B., Li, W.-G., Lin, Y.-C., & Lin, Z.-N. (2022). Targeting Mitochondrial COX-2 Enhances Chemosensitivity via Drp1-Dependent Remodeling of Mitochondrial Dynamics in Hepatocellular Carcinoma. Cancers, 14(3), 821. https://doi.org/10.3390/cancers14030821