
Machine Learning-Based Analysis of Glioma Grades Reveals Co-Enrichment

 , , , , , ,
, , , , , ,
Abstract
Simple Summary
Abstract
1. Introduction
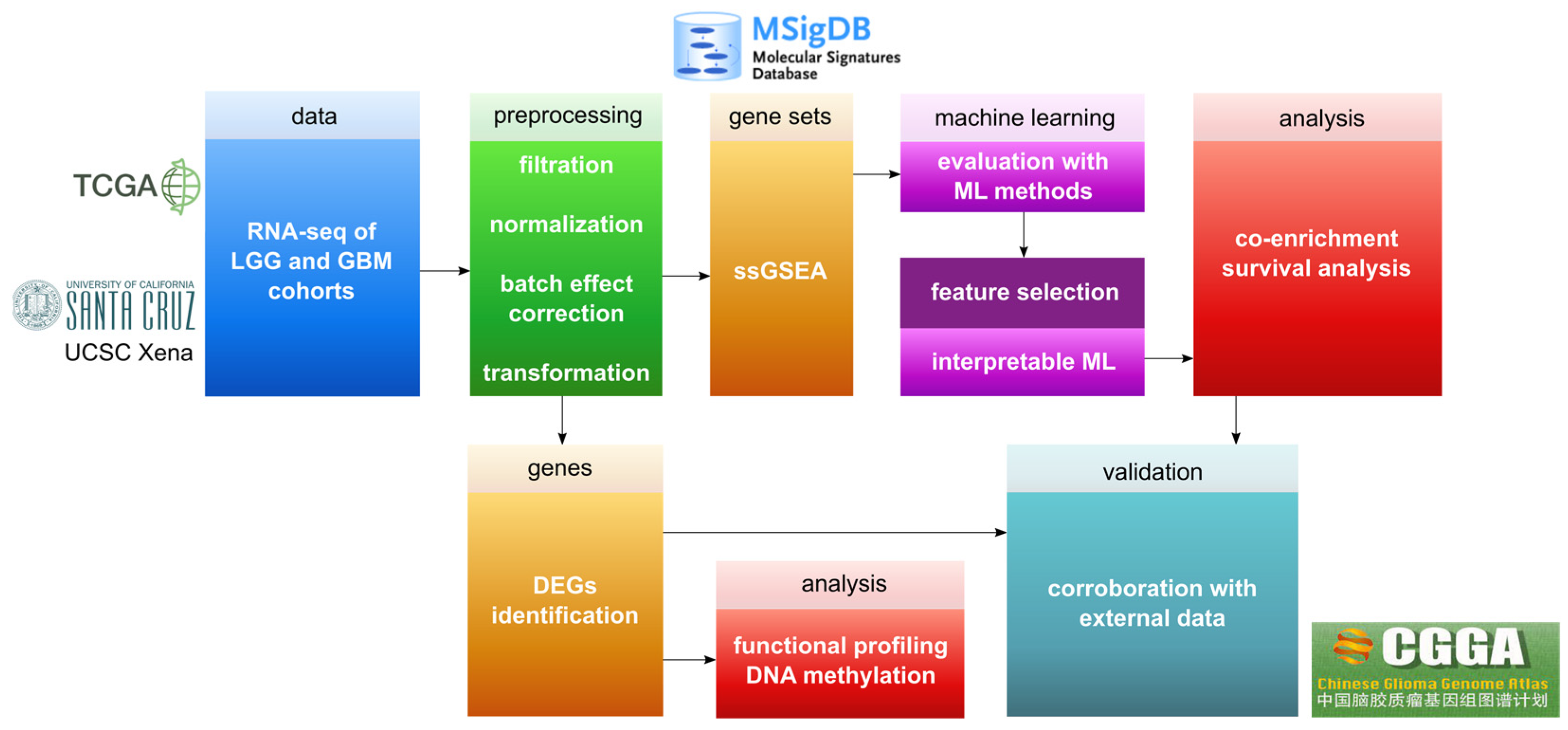
2. Materials and Methods
2.1. Preprocessing of Gene Expression Datasets
2.2. DNA Methylation Data
2.3. ssGSEA Analysis
2.4. ML Evaluation
2.5. Interpretable ML
2.6. Rule-Based Networks of Co-Enrichment
3. Results
3.1. Data Correction
3.2. DEGs Evaluation
3.3. ML for ssGSEA
3.4. Glioma Co-Enrichment
3.5. Survival Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, R.; Smith-Cohn, M.; Cohen, A.L.; Colman, H. Glioma subclassifications and their clinical significance. Neurotherapeutics 2017, 14, 284–297. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Fidler-Benaoudia, M.; Keegan, T.H.; Hipp, H.S.; Jemal, A.; Siegel, R.L. Cancer statistics for adolescents and young adults, 2020. CA A Cancer J. Clin. 2020, 70, 443–459. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Miyake, M.; Takahashi, M.; Hamamoto, R. Observing deep radiomics for the classification of glioma grades. Sci. Rep. 2021, 11, 10942. [Google Scholar] [CrossRef]
- Network, C.G.A.R. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar]
- Tu, J.; Fang, Y.; Han, D.; Tan, X.; Jiang, H.; Gong, X.; Wang, X.; Hong, W.; Wei, W. Activation of nuclear factor-κB in the angiogenesis of glioma: Insights into the associated molecular mechanisms and targeted therapies. Cell Prolif. 2021, 54, e12929. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. NF-κB in immunobiology. Cell Res. 2011, 21, 223–244. [Google Scholar] [CrossRef]
- Cohen, A.L.; Colman, H. Glioma biology and molecular markers. Curr. Underst. Treat. Gliomas 2015, 163, 15–30. [Google Scholar] [CrossRef]
- Network, C.G.A.R. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061. [Google Scholar]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Rasnic, R.; Brandes, N.; Zuk, O.; Linial, M. Substantial batch effects in TCGA exome sequences undermine pan-cancer analysis of germline variants. BMC Cancer 2019, 19, 783. [Google Scholar] [CrossRef] [PubMed]
- Ibing, S.; Michels, B.E.; Mosdzien, M.; Meyer, H.R.; Feuerbach, L.; Körner, C. On the impact of batch effect correction in TCGA isomiR expression data. NAR Cancer 2021, 3, zcab007. [Google Scholar] [CrossRef] [PubMed]
- Leek, J.T.; Scharpf, R.B.; Bravo, H.C.; Simcha, D.; Langmead, B.; Johnson, W.E.; Geman, D.; Baggerly, K.; Irizarry, R.A. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet. 2010, 11, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Collado-Torres, L.; Nellore, A.; Kammers, K.; Ellis, S.E.; Taub, M.A.; Hansen, K.D.; Jaffe, A.E.; Langmead, B.; Leek, J.T. Reproducible RNA-seq analysis using recount2. Nat. Biotechnol. 2017, 35, 319–321. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]
- Deo, R.C. Machine learning in medicine. Circulation 2015, 132, 1920–1930. [Google Scholar] [CrossRef]
- Alimadadi, A.; Aryal, S.; Manandhar, I.; Munroe, P.B.; Joe, B.; Cheng, X. Artificial intelligence and machine learning to fight COVID-19. Physiol. Genom. 2020, 52, 200–202. [Google Scholar] [CrossRef]
- Serra, A.; Galdi, P.; Tagliaferri, R. Machine learning for bioinformatics and neuroimaging. Wiley Interdiscip. Rev. Data Min. Knowl. Discov. 2018, 8, e1248. [Google Scholar] [CrossRef]
- Qiu, J.; Wu, Q.; Ding, G.; Xu, Y.; Feng, S. A survey of machine learning for big data processing. EURASIP J. Adv. Signal Process. 2016, 2016, 67. [Google Scholar] [CrossRef]
- Kourou, K.; Exarchos, T.P.; Exarchos, K.P.; Karamouzis, M.V.; Fotiadis, D.I. Machine learning applications in cancer prognosis and prediction. Comput. Struct. Biotechnol. J. 2015, 13, 8–17. [Google Scholar] [CrossRef]
- Murdoch, W.J.; Singh, C.; Kumbier, K.; Abbasi-Asl, R.; Yu, B. Definitions, methods, and applications in interpretable machine learning. Proc. Natl. Acad. Sci. USA 2019, 116, 22071–22080. [Google Scholar] [CrossRef] [PubMed]
- Soneson, C.; Gerster, S.; Delorenzi, M. Batch effect confounding leads to strong bias in performance estimates obtained by cross-validation. PLoS ONE 2014, 9, e100335. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The molecular signatures database hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Zhang, K.-N.; Wang, Q.; Li, G.; Zeng, F.; Zhang, Y.; Wu, F.; Chai, R.; Wang, Z.; Zhang, C. Chinese Glioma Genome Atlas (CGGA): A comprehensive resource with functional genomic data from Chinese glioma patients. Genom. Proteom. Bioinform. 2021, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a shared vision for cancer genomic data. N. Engl. J. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef]
- Vivian, J.; Rao, A.A.; Nothaft, F.A.; Ketchum, C.; Armstrong, J.; Novak, A.; Pfeil, J.; Narkizian, J.; Deran, A.D.; Musselman-Brown, A. Toil enables reproducible, open source, big biomedical data analyses. Nat. Biotechnol. 2017, 35, 314–316. [Google Scholar] [CrossRef]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef]
- Povey, S.; Lovering, R.; Bruford, E.; Wright, M.; Lush, M.; Wain, H. The HUGO gene nomenclature committee (HGNC). Hum. Genet. 2001, 109, 678–680. [Google Scholar] [CrossRef] [PubMed]
- Barbie, D.A.; Tamayo, P.; Boehm, J.S.; Kim, S.Y.; Moody, S.E.; Dunn, I.F.; Schinzel, A.C.; Sandy, P.; Meylan, E.; Scholl, C. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009, 462, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Dramiński, M.; Koronacki, J. rmcfs: An R package for Monte Carlo feature selection and interdependency discovery. J. Stat. Softw. 2018, 85, 1–28. [Google Scholar] [CrossRef]
- Hornik, K.; Buchta, C.; Zeileis, A. Open-source machine learning: R meets Weka. Comput. Stat. 2009, 24, 225–232. [Google Scholar] [CrossRef]
- Platt, J. Sequential Minimal Optimization: A Fast Algorithm for Training Support Vector Machines; Microsoft Research: Redmond, WA, USA, 1998; pp. 1–21. [Google Scholar]
- Aha, D.W.; Kibler, D.; Albert, M.K. Instance-based learning algorithms. Mach. Learn. 1991, 6, 37–66. [Google Scholar] [CrossRef]
- Breiman, L. Bagging predictors. Mach. Learn. 1996, 24, 123–140. [Google Scholar] [CrossRef]
- Quinlan, J.R. C4. 5: Programs for Machine Learning; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Cohen, W.W. Fast effective rule induction. In Machine Learning Proceedings 1995; Elsevier: Amsterdam, The Netherlands, 1995; pp. 115–123. [Google Scholar]
- Garbulowski, M.; Diamanti, K.; Smolińska, K.; Baltzer, N.; Stoll, P.; Bornelöv, S.; Øhrn, A.; Feuk, L.; Komorowski, J.R. ROSETTA: An interpretable machine learning framework. BMC Bioinform. 2021, 22, 110. [Google Scholar] [CrossRef]
- Pawlak, Z. Rough sets. Int. J. Comput. Inf. Sci. 1982, 11, 341–356. [Google Scholar] [CrossRef]
- Pita-Juárez, Y.; Altschuler, G.; Kariotis, S.; Wei, W.; Koler, K.; Green, C.; Tanzi, R.; Hide, W. The pathway Coexpression network: Revealing pathway relationships. PLoS Comput. Biol. 2018, 14, e1006042. [Google Scholar] [CrossRef]
- Chen, D.; Zhang, H.; Lu, P.; Liu, X.; Cao, H. Synergy evaluation by a pathway–pathway interaction network: A new way to predict drug combination. Mol. BioSystems 2016, 12, 614–623. [Google Scholar] [CrossRef]
- Dutkowski, J.; Kramer, M.; Surma, M.A.; Balakrishnan, R.; Cherry, J.M.; Krogan, N.J.; Ideker, T. A gene ontology inferred from molecular networks. Nat. Biotechnol. 2013, 31, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Smolinska, K.; Garbulowski, M.; Diamanti, K.; Davoy, X.; Anyango, S.O.O.; Barrenäs, F.; Bornelöv, S.; Komorowski, J. VisuNet: An interactive tool for rule network visualization of rule-based learning models. Diva 2021, 2, 141–166. [Google Scholar]
- Meyer, P.E.; Meyer, M.P.E. Package ‘infotheo’. In R Package Version; Citeseer: Princet, NJ, USA, 2009; Volume 1. [Google Scholar]
- Li, F.; Yi, Y.; Miao, Y.; Long, W.; Long, T.; Chen, S.; Cheng, W.; Zou, C.; Zheng, Y.; Wu, X. N6-methyladenosine modulates nonsense-mediated mRNA decay in human glioblastoma. Cancer Res. 2019, 79, 5785–5798. [Google Scholar] [CrossRef]
- Matzuk, M.M.; Lamb, D.J. The biology of infertility: Research advances and clinical challenges. Nat. Med. 2008, 14, 1197–1213. [Google Scholar] [CrossRef] [PubMed]
- Myllykangas, S.; Himberg, J.; Böhling, T.; Nagy, B.; Hollmén, J.; Knuutila, S. DNA copy number amplification profiling of human neoplasms. Oncogene 2006, 25, 7324–7332. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Kessler, P.; Williams, B.R. Transcript profiling of Wilms tumors reveals connections to kidney morphogenesis and expression patterns associated with anaplasia. Oncogene 2005, 24, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, R.; Nemoto, T.; Takahashi, H.; Ohta, T.; Kawai, A.; Seki, K.; Yoshida, T.; Toyama, Y.; Ichikawa, H.; Hasegawa, T. Gene expression analysis of soft tissue sarcomas: Characterization and reclassification of malignant fibrous histiocytoma. Mod. Pathol. 2007, 20, 749–759. [Google Scholar] [CrossRef]
- Xia, H.; Qi, Y.; Ng, S.S.; Chen, X.; Chen, S.; Fang, M.; Li, D.; Zhao, Y.; Ge, R.; Li, G. MicroRNA-15b regulates cell cycle progression by targeting cyclins in glioma cells. Biochem. Biophys. Res. Commun. 2009, 380, 205–210. [Google Scholar] [CrossRef]
- Liu, E.; Wu, J.; Cao, W.; Zhang, J.; Liu, W.; Jiang, X.; Zhang, X. Curcumin induces G2/M cell cycle arrest in a p53-dependent manner and upregulates ING4 expression in human glioma. J. Neuro-Oncol. 2007, 85, 263–270. [Google Scholar] [CrossRef]
- Doan, P.; Musa, A.; Candeias, N.R.; Emmert-Streib, F.; Yli-Harja, O.; Kandhavelu, M. Alkylaminophenol induces G1/S phase cell cycle arrest in glioblastoma cells through p53 and cyclin-dependent kinase signaling pathway. Front. Pharmacol. 2019, 10, 330. [Google Scholar] [CrossRef]
- Willems, E.; Dedobbeleer, M.; Digregorio, M.; Lombard, A.; Goffart, N.; Lumapat, P.N.; Lambert, J.; Van den Ackerveken, P.; Szpakowska, M.; Chevigné, A. Aurora A plays a dual role in migration and survival of human glioblastoma cells according to the CXCL12 concentration. Oncogene 2019, 38, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Lehman, N.L.; O’Donnell, J.P.; Whiteley, L.J.; Stapp, R.T.; Lehman, T.D.; Roszka, K.M.; Schultz, L.R.; Williams, C.J.; Mikkelsen, T.; Brown, S.L. Aurora A is differentially expressed in gliomas, is associated with patient survival in glioblastoma and is a potential chemotherapeutic target in gliomas. Cell Cycle 2012, 11, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Warner, S.L.; Munoz, R.M.; Stafford, P.; Koller, E.; Hurley, L.H.; Von Hoff, D.D.; Han, H. Comparing Aurora A and Aurora B as molecular targets for growth inhibition of pancreatic cancer cells. Mol. Cancer Ther. 2006, 5, 2450–2458. [Google Scholar] [CrossRef][Green Version]
- Liu, W.; Palovcak, A.; Li, F.; Zafar, A.; Yuan, F.; Zhang, Y. Fanconi anemia pathway as a prospective target for cancer intervention. Cell Biosci. 2020, 10, 39. [Google Scholar] [CrossRef] [PubMed]
- Squatrito, M.; Brennan, C.W.; Helmy, K.; Huse, J.T.; Petrini, J.H.; Holland, E.C. Loss of ATM/Chk2/p53 pathway components accelerates tumor development and contributes to radiation resistance in gliomas. Cancer Cell 2010, 18, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dube, C.; Gibert, M.; Cruickshanks, N.; Wang, B.; Coughlan, M.; Yang, Y.; Setiady, I.; Deveau, C.; Saoud, K. The p53 pathway in glioblastoma. Cancers 2018, 10, 297. [Google Scholar] [CrossRef] [PubMed]
- Dunn, I.F.; Heese, O.; Black, P.M. Growth factors in glioma angiogenesis: FGFs, PDGF, EGF, and TGFs. J. Neuro-Oncol. 2000, 50, 121–137. [Google Scholar] [CrossRef] [PubMed]
- Rajan, V.; Menon, K. Involvement of microtubules in lipoprotein degradation and utilization for steroidogenesis in cultured rat luteal cells. Endocrinology 1985, 117, 2408–2416. [Google Scholar] [CrossRef]
- Ahmad, F.; Sun, Q.; Patel, D.; Stommel, J.M. Cholesterol metabolism: A potential therapeutic target in glioblastoma. Cancers 2019, 11, 146. [Google Scholar] [CrossRef]
- Li, D.; Li, S.; Xue, A.Z.; Callahan, L.A.S.; Liu, Y. Expression of SREBP2 and cholesterol metabolism related genes in TCGA glioma cohorts. Medicine 2020, 99, e18815. [Google Scholar] [CrossRef]
- Cavuoto, P.; Fenech, M.F. A review of methionine dependency and the role of methionine restriction in cancer growth control and life-span extension. Cancer Treat. Rev. 2012, 38, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Calinescu, A.-A.; Castro, M.G. Microtubule targeting agents in glioma. Transl. Cancer Res. 2016, 5, S54. [Google Scholar] [CrossRef] [PubMed]
- Therneau, T.M.; Lumley, T. Package ‘survival’. R Top Doc 2015, 128, 28–33. [Google Scholar]
- Kassambara, A.; Kosinski, M.; Biecek, P.; Fabian, S. Package ‘Survminer’, CRAN: CRAN Repository. 2017. Available online: https://cran.microsoft.com/snapshot/2017-04-21/web/packages/survminer/survminer.pdf (accessed on 31 December 2021).
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Han, W.; Guan, W. Valproic Acid: A Promising Therapeutic Agent in Glioma Treatment. Front. Oncol. 2021, 11, 687362. [Google Scholar] [CrossRef]
- Fang, G.; Wang, W.; Paunic, V.; Heydari, H.; Costanzo, M.; Liu, X.; Liu, X.; VanderSluis, B.; Oately, B.; Steinbach, M. Discovering genetic interactions bridging pathways in genome-wide association studies. Nat. Commun. 2019, 10, 4274. [Google Scholar] [CrossRef]
- Pisano, C.; Tucci, M.; Di Stefano, R.F.; Turco, F.; Scagliotti, G.V.; Di Maio, M.; Buttigliero, C. Interactions between androgen receptor signaling and other molecular pathways in prostate cancer progression: Current and future clinical implications. Crit. Rev. Oncol./Hematol. 2021, 157, 103185. [Google Scholar] [CrossRef]
- Jeong, W.-J.; Ro, E.J.; Choi, K.-Y. Interaction between Wnt/β-catenin and RAS-ERK pathways and an anti-cancer strategy via degradations of β-catenin and RAS by targeting the Wnt/β-catenin pathway. NPJ Precis. Oncol. 2018, 2, 5. [Google Scholar] [CrossRef]
- Liu, K.-Q.; Liu, Z.-P.; Hao, J.-K.; Chen, L.; Zhao, X.-M. Identifying dysregulated pathways in cancers from pathway interaction networks. BMC Bioinform. 2012, 13, 126. [Google Scholar] [CrossRef]
- Reimand, J.; Isserlin, R.; Voisin, V.; Kucera, M.; Tannus-Lopes, C.; Rostamianfar, A.; Wadi, L.; Meyer, M.; Wong, J.; Xu, C. Pathway enrichment analysis and visualization of omics data using g: Profiler, GSEA, Cytoscape and EnrichmentMap. Nat. Protoc. 2019, 14, 482–517. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.-M.; Shafi, A.; Nguyen, T.; Draghici, S. Identifying significantly impacted pathways: A comprehensive review and assessment. Genome Biol. 2019, 20, 203. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Wang, W.; Tan, M.; Zhu, L.; Zhang, Y.; Fessler, E.; Vermeulen, L.; Wang, X. DeepCC: A novel deep learning-based framework for cancer molecular subtype classification. Oncogenesis 2019, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Hussain, F.; Tan, C.L.; Dash, M. Discretization: An enabling technique. Data Min. Knowl. Discov. 2002, 6, 393–423. [Google Scholar] [CrossRef]
- Kopper, P.; Pölsterl, S.; Wachinger, C.; Bischl, B.; Bender, A.; Rügamer, D. Semi-structured deep piecewise exponential models. In Proceedings of the Survival Prediction-Algorithms, Challenges and Applications, Palo Alto, CA, USA, 22–24 March 2021; pp. 40–53. [Google Scholar]
- Kopper, P.; Wiegrebe, S.; Bischl, B.; Bender, A.; Rügamer, D. DeepPAMM: Deep Piecewise Exponential Additive Mixed Models for Complex Hazard Structures in Survival Analysis. In Proceedings of the Advances in Knowledge Discovery and Data Mining (PAKDD ’22), Jeju, Korea, 23–26 May 2022. [Google Scholar]
- Rudin, C. Stop explaining black box machine learning models for high stakes decisions and use interpretable models instead. Nat. Mach. Intell. 2019, 1, 206–215. [Google Scholar] [CrossRef]
- Au, Q.; Herbinger, J.; Stachl, C.; Bischl, B.; Casalicchio, G. Grouped feature importance and combined features effect plot. Arxiv Prepr. 2021, arXiv:2104.11688. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TCGA | CGGA | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| GM1 | GM2 | Batch 1 | Batch 2 | ||||||
| GII | GIII | LGG | GBM | GII | GIII | GBM | GII | GIII | GBM |
| 231 | 168 | 108 | 151 | 188 | 255 | 249 | 103 | 79 | 139 |
| CGGA Batch 1 | CGGA Batch 2 | ||||||
|---|---|---|---|---|---|---|---|
| GII vs. GIII | LGG vs. GBM | GII vs. GIII | LGG vs. GBM | ||||
| p < 0.001 | p < 0.05 | p < 0.001 | p < 0.05 | p < 0.001 | p < 0.05 | p < 0.001 | p < 0.05 |
| 62% | 88% | 27% | 44% | 85% | 96% | 52% | 71% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garbulowski, M.; Smolinska, K.; Çabuk, U.; Yones, S.A.; Celli, L.; Yaz, E.N.; Barrenäs, F.; Diamanti, K.; Wadelius, C.; Komorowski, J. Machine Learning-Based Analysis of Glioma Grades Reveals Co-Enrichment. Cancers 2022, 14, 1014. https://doi.org/10.3390/cancers14041014
Garbulowski M, Smolinska K, Çabuk U, Yones SA, Celli L, Yaz EN, Barrenäs F, Diamanti K, Wadelius C, Komorowski J. Machine Learning-Based Analysis of Glioma Grades Reveals Co-Enrichment. Cancers. 2022; 14(4):1014. https://doi.org/10.3390/cancers14041014
Chicago/Turabian StyleGarbulowski, Mateusz, Karolina Smolinska, Uğur Çabuk, Sara A. Yones, Ludovica Celli, Esma Nur Yaz, Fredrik Barrenäs, Klev Diamanti, Claes Wadelius, and Jan Komorowski. 2022. "Machine Learning-Based Analysis of Glioma Grades Reveals Co-Enrichment" Cancers 14, no. 4: 1014. https://doi.org/10.3390/cancers14041014
APA StyleGarbulowski, M., Smolinska, K., Çabuk, U., Yones, S. A., Celli, L., Yaz, E. N., Barrenäs, F., Diamanti, K., Wadelius, C., & Komorowski, J. (2022). Machine Learning-Based Analysis of Glioma Grades Reveals Co-Enrichment. Cancers, 14(4), 1014. https://doi.org/10.3390/cancers14041014