
Combination of Tipifarnib and Sunitinib Overcomes Renal Cell Carcinoma Resistance to Tyrosine Kinase Inhibitors via Tumor-Derived Exosome and T Cell Modulation

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Materials
2.3. Cell Culture
2.4. Exosome Isolation and Characterization
2.5. Cell Viability and the Cytotoxicity
2.6. Transfection, Immunofluorescence, and Western Blot Analyses
2.7. Colony Forming Units (CFU) Assay
2.8. Analysis of the Correlation between Exosome Biogenesis and Secretion Markers and RCC
2.9. Annexin V-FITC/PI Double-Staining Assay
2.10. Real Time-PCR
2.11. Statistical Analysis
3. Results
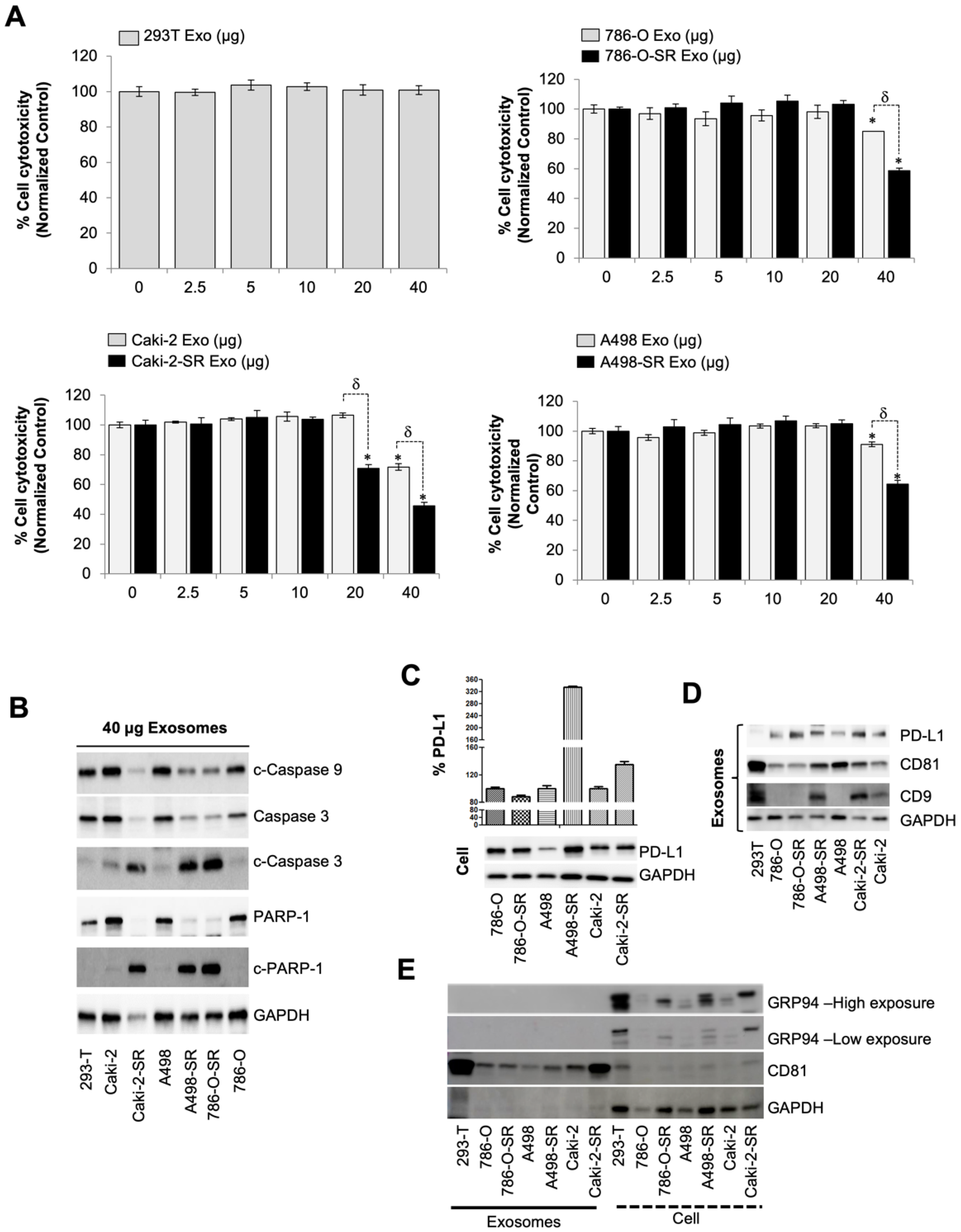
3.1. SR RCC Exosome’s Role in Suppressing the Immune System
3.2. The Level of PD-L1-Containing Exosomes from SS and SR RCC Cells
3.3. Patient PD-L1 Expression and T Cell Modulation after Tumor-Derived Exosome Exposure
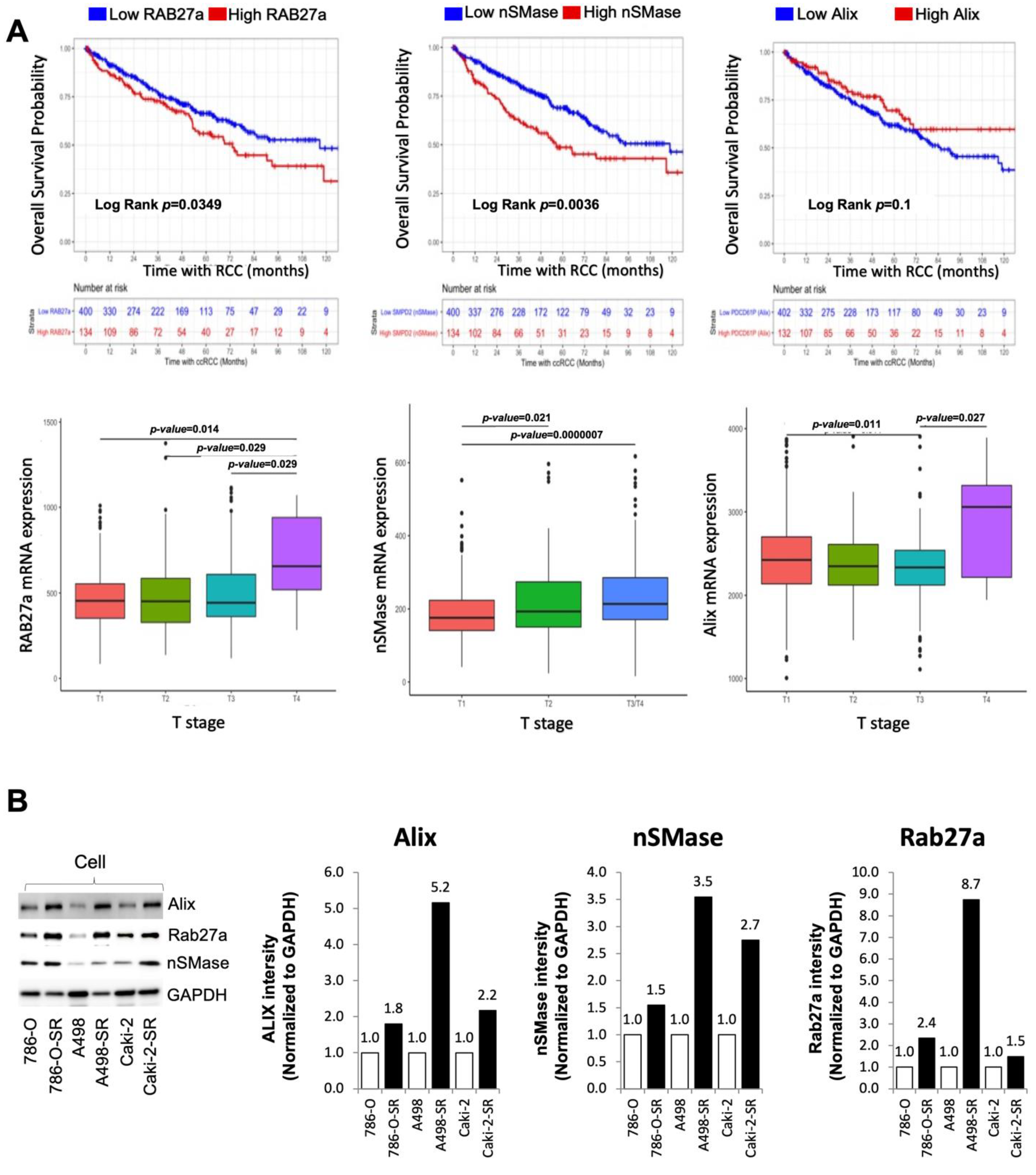
3.4. Clinical Significance of Alix, nSMase, and RAB27a Expression in RCC
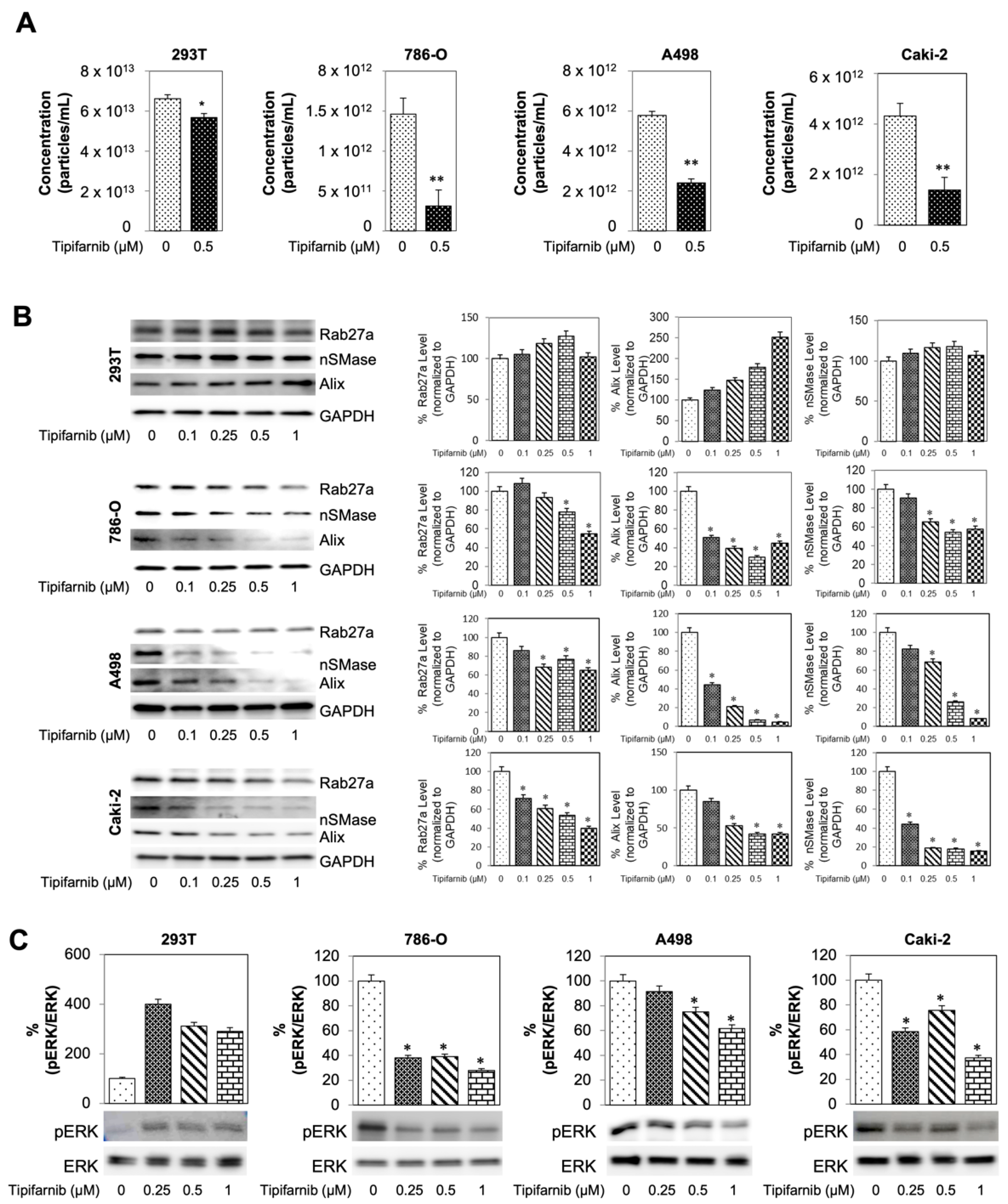
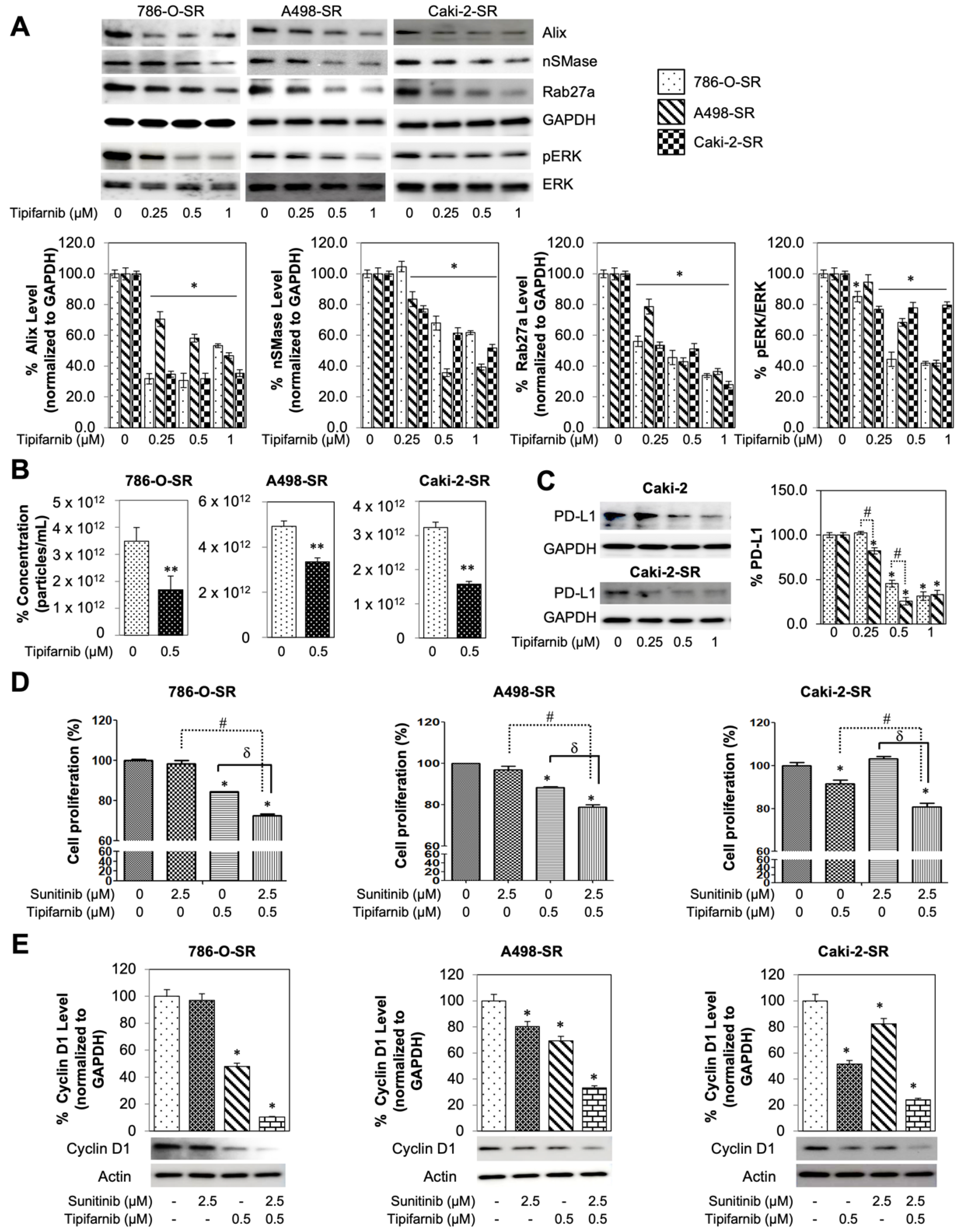
3.5. Tipifarnib Reduces Exosome Load in SS RCC Cell Lines
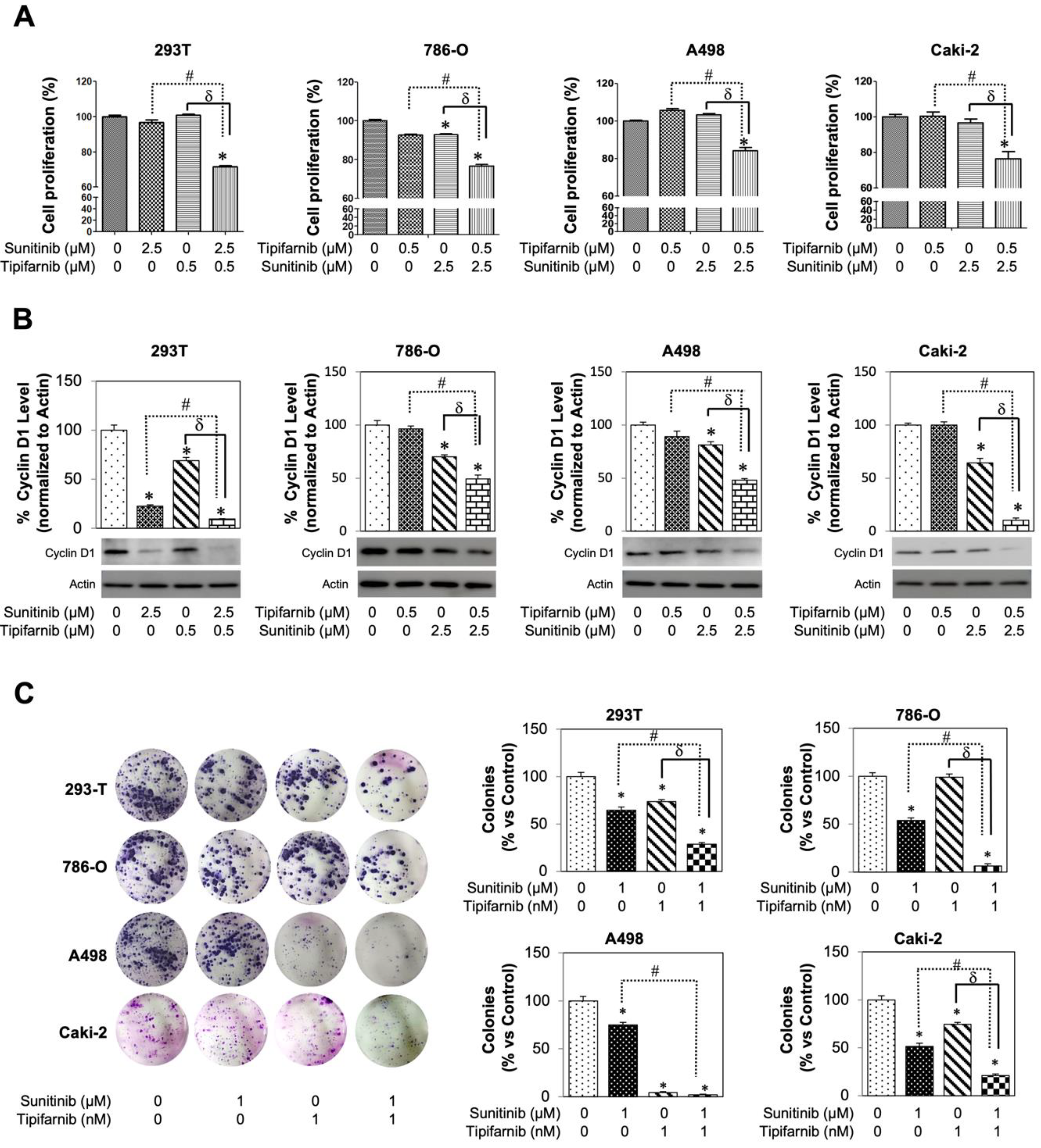
3.6. Tipifarnib Suppresses Cell Proliferation in SS RCC Cell Lines
3.7. In-Vitro Utilization of Tipifarnib for Sunitinib-Resistant RCC Cell Lines
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Tenold, M.; Ravi, P.; Kumar, M.; Bowman, A.; Hammers, H.; Choueiri, T.K.; Lara, P.N., Jr. Current Approaches to the Treatment of Advanced or Metastatic Renal Cell Carcinoma. Am. Soc. Clin. Oncol. Educ. Book 2020, 40, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Ji, X.; Liu, J.; Fan, D.; Zhou, Q.; Chen, C.; Wang, W.; Wang, G.; Wang, H.; Yuan, W.; et al. Effects of exosomes on pre-metastatic niche formation in tumors. Mol. Cancer 2019, 18, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Cao, X.T. Characteristics and significance of the pre-metastatic niche. Cancer Cell. 2016, 30, 668–681. [Google Scholar] [CrossRef] [Green Version]
- Vautrot, V.; Bentayeb, H.; Causse, S.; Garrido, C.; Gobbo, J. Tumor-Derived Exosomes: Hidden Players in PD-1/PD-L1 Resistance. Cancers 2021, 13, 4537. [Google Scholar] [CrossRef]
- Teixeira, A.L.; Dias, F.; Gomes, M.; Fernandes, M.; Medeiros, R. Circulating biomarkers in renal cell carcinoma: The link between microRNAs and extracellular vesicles, where are we now? J. Kidney Cancer VHL 2014, 1, 84–98. [Google Scholar] [CrossRef]
- Li, X.; Corbett, A.L.; Taatizadeh, E.; Tasnim, N.; Little, J.P.; Garnis, C.; Daugaard, M.; Guns, E.; Hoorfar, M.; Li, I.T.S. Challenges and opportunities in exosome research-Perspectives from biology, engineering, and cancer therapy. APL Bioeng. 2019, 3, 011503. [Google Scholar] [CrossRef] [Green Version]
- Zomer, A.; Maynard, C.; Verweij, F.J.; Kamermans, A.; Schäfer, R.; Beerling, E.; Schiffelers, R.M.; de Wit, E.; Berenguer, J.; Ellenbroek, S.; et al. In Vivo imaging reveals extracellular ve2_3sicle-mediated phenocopying of metastatic behavior. Cell 2015, 161, 1046–1057. [Google Scholar] [CrossRef] [Green Version]
- Qu, Y.; Tan, H.Y.; Chan, Y.T.; Jiang, H.; Wang, N.; Wang, D. The functional role of long noncoding RNA in resistance to anticancer treatment. Ther. Adv. Med. Oncol. 2020, 12, 1758835920927850. [Google Scholar] [CrossRef]
- Behera, J.; Tyagi, N. Exosomes: Mediators of bone diseases, protection, and therapeutics potential. J. Oncosci. 2018, 5, 181–195. [Google Scholar] [CrossRef] [Green Version]
- Tominaga, N. Anti-Cancer Role and Therapeutic Potential of Extracellular Vesicles. Cancers 2021, 13, 6303. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Blockade of mutant RAS oncogenic signaling with a special emphasis on KRAS. Pharmacol. Res. 2021, 172, 105806. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zeng, H.; Zhang, H.; Han, Y. The role of exosomal PD-L1 in tumor immunotherapy. Transl. Oncol. 2021, 14, 101047. [Google Scholar] [CrossRef] [PubMed]
- Garje, R.; An, J.; Greco, A.; Vaddepally, R.K.; Zakharia, Y. The future of immunotherapy-based combination therapy in metastatic renal cell carcinoma. Cancers 2020, 12, 143. [Google Scholar] [CrossRef] [Green Version]
- Ahn, S.H.; Nguyen, S.L.; Petroff, M.G. Exploring the Origin and Antigenic Specificity of Maternal Regulatory T Cells in Pregnancy. Front. Immunol. 2020, 25, 1302. [Google Scholar] [CrossRef]
- Czernek, L.; Duchler, M. Functions of Cancer-derived extracellular vesicles in immunosuppression. Arch. Immunol. Ther. Exp. 2017, 65, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Li, C.W.; Chan, L.C.; Wei, Y.; Hsu, J.M.; Xia, W.; Cha, J.H.; Hou, J.; Hsu, J.L.; Sun, L.; et al. Exosomal PD-L1 harbors active defense function to suppress T cell killing of breast cancer cells and promote tumor growth. Cell Res. 2018, 28, 862–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef]
- Gong, J.; Chehrazi-Raffle, A.; Reddi, S.; Salgia, R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: A comprehensive review of registration trials and future considerations. J. Immunother. Cancer 2018, 6, 8. [Google Scholar] [CrossRef]
- Lu, L.; Hatipoglu, E.; Robert de Massy, M.; Litchfield, K.; Beattie, G.; Rowan, A.; Schnidrig, D.; Thompson, R.; Byrne, F.; Horswell, S.; et al. Determinants of anti-PD-1 response and resistance in clear cell renal cell carcinoma. Cancer Cell. 2021, 39, 1497.e11–1518.e11. [Google Scholar]
- Greenberg, J.W.; Kim, H.; Moustafa, A.A.; Datta, A.; Barata, P.C.; Boulares, A.H.; Abdel-Mageed, A.B.; Krane, L.S. Repurposing ketoconazole as an exosome directed adjunct to sunitinib in treating renal cell carcinoma. Sci. Rep. 2021, 11, 10200. [Google Scholar] [CrossRef] [PubMed]
- Maas, S.L.; Broekman, M.L.; de Vrij, J. Tunable Resistive Pulse Sensing for the Characterization of Extracellular Vesicles. Methods Mol. Biol. 2017, 1545, 21–33. [Google Scholar] [PubMed]
- Lee, S.M.; Ko, E.B.; Go, R.E.; Lee, H.K.; Choi, K.C. Effect of the phenylpyrrole fungicide fludioxonil on cell proliferation and cardiac differentiation in mouse embryonic stem cells. Reprod. Toxicol. 2021, 104, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Abd Elmageed, Z.Y.; Ju, J.; Naura, A.S.; Abdel-Mageed, A.B.; Varughese, S.; Paul, D.; Alahari, S.; Catling, A.; Kim, J.G.; et al. PDZK1 is a novel factor in breast cancer that is indirectly regulated by estrogen through IGF-1R and promotes estrogen-mediated growth. Mol. Med. 2013, 19, 253–262. [Google Scholar] [CrossRef]
- Datta, A.; Kim, H.; Lal, M.; McGee, L.; Johnson, A.; Moustafa, A.A.; Jones, J.C.; Mondal, D.; Ferrer, M.; Abdel-Mageed, A.B. Manumycin A suppresses exosome biogenesis and secretion via targeted inhibition of Ras/Raf/ERK1/2 signaling and hnRNP H1 in castration-resistant prostate cancer cells. Cancer Lett. 2017, 408, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Datta, A.; Kim, H.; McGee, L.; Johnson, A.E.; Talwar, S.; Marugan, J.; Southall, N.; Hu, X.; Lal, M.; Mondal, D.; et al. High-throughput screening identified selective inhibitors of exosome biogenesis and secretion: A drug repurposing strategy for advanced cancer. Sci. Rep. 2018, 8, 8161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrissey, S.M.; Yan, J. Exosomal PD-L1: Roles in Tumor Progression and Immunotherapy. Trends Cancer 2020, 6, 550–558. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, W.; Xu, Z.P.; Gu, W. PD-L1 Distribution and Perspective for Cancer Immunotherapy-Blockade, Knockdown, or Inhibition. Front. Immunol. 2019, 10, 2022. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef]
- Kim, D.H.; Kim, H.; Choi, Y.J.; Kim, S.Y.; Lee, J.E.; Sung, K.J.; Sung, Y.H.; Pack, C.G.; Jung, M.K.; Han, B.; et al. Exosomal PD-L1 promotes tumor growth through immune escape in non-small cell lung cancer. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Huang, Z.; Teng, F.; Xing, L.; Yu, J. Predictive biomarkers in PD-1/PD-L1 checkpoint blockade immunotherapy. Cancer Treat. Rev. 2015, 41, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Zhou, X.; Fang, M.; Li, H.; Su, P.; Tu, Y.; Zhang, L.; Zhou, F. Extracellular Vesicles in Cancer Immune Microenvironment and Cancer Immunotherapy. Adv. Sci. 2019, 6, 1901779. [Google Scholar] [CrossRef] [PubMed]
- Saeidi, A.; Zandi, K.; Cheok, Y.Y.; Saeidi, H.; Wong, W.F.; Lee, C.Y.Q.; Cheong, H.C.; Yong, Y.K.; Larsson, M.; Shankar, E.M. T-Cell Exhaustion in Chronic Infections: Reversing the State of Exhaustion and Reinvigorating Optimal Protective Immune Responses. Front. Immunol. 2018, 9, 2569. [Google Scholar] [CrossRef] [PubMed]
- Del Re, M.; van Schaik, R.H.N.; Fogli, S.; Mathijssen, R.H.J.; Cucchiara, F.; Capuano, A.; Scavone, C.; Jenster, G.W.; Danesi, R. Blood-based PD-L1 analysis in tumor-derived extracellular vesicles: Applications for optimal use of anti-PD-1/PD-L1 axis inhibitors. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188463. [Google Scholar] [CrossRef]
- Li, Z.; Fang, R.; Fang, J.; He, S.; Liu, T. Functional implications of Rab27 GTPases in Cancer. Cell Commun. Signal. 2018, 16, 44. [Google Scholar] [CrossRef] [Green Version]
- Tsuruda, M.; Yoshino, H.; Okamura, S.; Kuroshima, K.; Osako, Y.; Sakaguchi, T.; Sugita, S.; Tatarano, S.; Nakagawa, M.; Enokida, H. Oncogenic effects of RAB27B through exosome independent function in renal cell carcinoma including sunitinib-resistant. PLoS ONE 2020, 15, e0232545. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Liang, Y.; Chen, Y.; Wang, L.; Li, D.; Liang, Z.; Wang, X.; Tian, D.; Yang, X.; Niu, H. Glutamine Deprivation Induces PD-L1 Expression via Activation of EGFR/ERK/c-Jun Signaling in Renal Cancer. Mol. Cancer Res. 2020, 18, 324–339. [Google Scholar] [CrossRef] [Green Version]
- Ho, A.L.; Brana, I.; Haddad, R.; Bauman, J.; Bible, K.; Oosting, S.; Wong, D.J.; Ahn, M.J.; Boni, V.; Even, C.; et al. Tipifarnib in Head and Neck Squamous Cell Carcinoma with HRAS Mutations. J. Clin. Oncol. 2021, 39, 1856–1864. [Google Scholar] [CrossRef]
- de la Cruz, M.P.; Shabaka, A.; Mielgo-Rubio, X.; Guerrero-Marquez, C.; Gimenez-Moyano, S.; Fernandez-Juarez, G. Concomitant Acute Tubular Necrosis and Acute Interstitial Nephritis Induced by Tipifarnib in a Patient with Squamous Cell Carcinoma of the Lung. Am. J. Med. Sci. 2021, 362, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Sa, J.K.; Gualberto, A.; Scholz, C.; Sung, H.H.; Jeong, B.C.; Choi, H.Y.; Kwon, G.Y.; Park, S.H. A Phase II Trial of Tipifarnib for Patients with Previously Treated, Metastatic Urothelial Carcinoma Harboring HRAS Mutations. Clin. Cancer Res. 2020, 19, 5113–5119. [Google Scholar] [CrossRef] [PubMed]
- Hanna, G.J.; Guenette, J.P.; Chau, N.G.; Sayehli, C.M.; Wilhelm, C.; Metcalf, R.; Wong, D.J.; Brose, M.; Razaq, M.; Pérez-Ruiz, E.; et al. Tipifarnib in recurrent, metastatic HRAS-mutant salivary gland cancer. Cancer 2020, 17, 3972–3981. [Google Scholar] [CrossRef] [PubMed]
- Gilardi, M.; Wang, Z.; Proietto, M.; Chillà, A.; Calleja-Valera, J.L.; Goto, Y.; Vanoni, M.; Janes, M.R.; Mikulski, Z.; Gualberto, A.; et al. Tipifarnib as a Precision Therapy for HRAS-Mutant Head and Neck Squamous Cell Carcinomas. Mol. Cancer Ther. 2020, 9, 1784–1796. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Alonso, R.; Mondéjar, R.; Martínez, N.; García-Diaz, N.; Pérez, C.; Merino, D.; Rodríguez, M.; Esteve-Codina, A.; Fuste, B.; Gut, M.; et al. Identification of tipifarnib sensitivity biomarkers in T-cell acute lymphoblastic leukemia and T-cell lymphoma. Sci. Rep. 2020, 10, 6721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muus, P.; Langemeijer, S.; van Bijnen, S.; Blijlevens, N.; de Witte, T. A phase I clinical trial to study the safety of treatment with tipifarnib combined with bortezomib in patients with advanced stages of myelodysplastic syndrome and oligoblastic acute myeloid leukemia. Leuk. Res. 2021, 105, 106573. [Google Scholar] [CrossRef]
- Yam, C.; Murthy, R.K.; Valero, V.; Szklaruk, J.; Shroff, G.S.; Stalzer, C.J.; Buzdar, A.U.; Murray, J.L.; Yang, W.; Hortobagyi, G.N.; et al. A phase II study of tipifarnib and gemcitabine in metastatic breast cancer. Investig. New Drugs 2018, 36, 299–306. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; Gannon, A.; Figlin, R.A. Sunitinib: Ten Years of Successful Clinical Use and Study in Advanced Renal Cell Carcinoma. Oncologist 2017, 22, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Morais, C. Sunitinib resistance in renal cell carcinoma. J. Kidney Cancer VHL 2014, 1, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Qu, L.; Ding, J.; Chen, C.; Wu, Z.J.; Liu, B.; Gao, Y.; Chen, W.; Liu, F.; Sun, W.; Li, X.F.; et al. Exosome-Transmitted lncAR SR Promotes Sunitinib Resistance in Renal Cancer by Acting as a Competing Endogenous RNA. Cancer Cell. 2016, 29, 653–668. [Google Scholar] [CrossRef]
- Zheng, J.; Sun, X.; Chen, J.; Jiang, F.; Li, W.; Xie, S. Mechanism of immune escape in renal cell carcinoma. Zhonghua Zhong Liu Za Zhi 2002, 24, 24–26. [Google Scholar]
- Das, T.; Sa, G.; Paszkiewicz-Kozik, E.; Hilston, C.; Molto, L.; Rayman, P.; Kudo, D.; Biswas, K.; Bukowski, R.M.; Finke, J.H.; et al. Renal cell carcinoma tumors induce T cell apoptosis through receptor-dependent and receptor-independent pathways. J. Immunol. 2008, 180, 4687–4696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uzzo, R.G.; Rayman, P.; Kolenko, V.; Clark, P.E.; Bloom, T.; Ward, A.M.; Molto, L.; Tannenbaum, C.; Worford, L.J.; Bukowski, R.; et al. Mechanisms of apoptosis in T cells from patients with renal cell carcinoma. Clin. Cancer Res. 1999, 5, 1219–1229. [Google Scholar] [PubMed]
- Yang, L.; Wu, X.; Wang, D.; Luo, C.; Chen, L. Renal carcinoma cell-derived exosomes induce human immortalized line of Jurkat T lymphocyte apoptosis in vitro. Urol. Int. 2013, 91, 363–369. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Greenberg, J.W.; Kim, H.; Ahn, M.; Moustafa, A.A.; Zhou, H.; Barata, P.C.; Boulares, A.H.; Abdel-Mageed, A.B.; Krane, L.S. Combination of Tipifarnib and Sunitinib Overcomes Renal Cell Carcinoma Resistance to Tyrosine Kinase Inhibitors via Tumor-Derived Exosome and T Cell Modulation. Cancers 2022, 14, 903. https://doi.org/10.3390/cancers14040903
Greenberg JW, Kim H, Ahn M, Moustafa AA, Zhou H, Barata PC, Boulares AH, Abdel-Mageed AB, Krane LS. Combination of Tipifarnib and Sunitinib Overcomes Renal Cell Carcinoma Resistance to Tyrosine Kinase Inhibitors via Tumor-Derived Exosome and T Cell Modulation. Cancers. 2022; 14(4):903. https://doi.org/10.3390/cancers14040903
Chicago/Turabian StyleGreenberg, Jacob W., Hogyoung Kim, Miae Ahn, Ahmed A. Moustafa, He Zhou, Pedro C. Barata, A. Hamid Boulares, Asim B. Abdel-Mageed, and Louis S. Krane. 2022. "Combination of Tipifarnib and Sunitinib Overcomes Renal Cell Carcinoma Resistance to Tyrosine Kinase Inhibitors via Tumor-Derived Exosome and T Cell Modulation" Cancers 14, no. 4: 903. https://doi.org/10.3390/cancers14040903