
Bone Marrow Stroma-Induced Transcriptome and Regulome Signatures of Multiple Myeloma


 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Cell Culture
2.3. Cell Isolation
2.4. RNA-Seq
2.5. Omni-ATAC
2.6. Data Analysis
2.6.1. RNA-Seq Data Analysis
2.6.2. Omni-ATAC Data Analysis
2.6.3. Overall Survival Analysis
3. Results
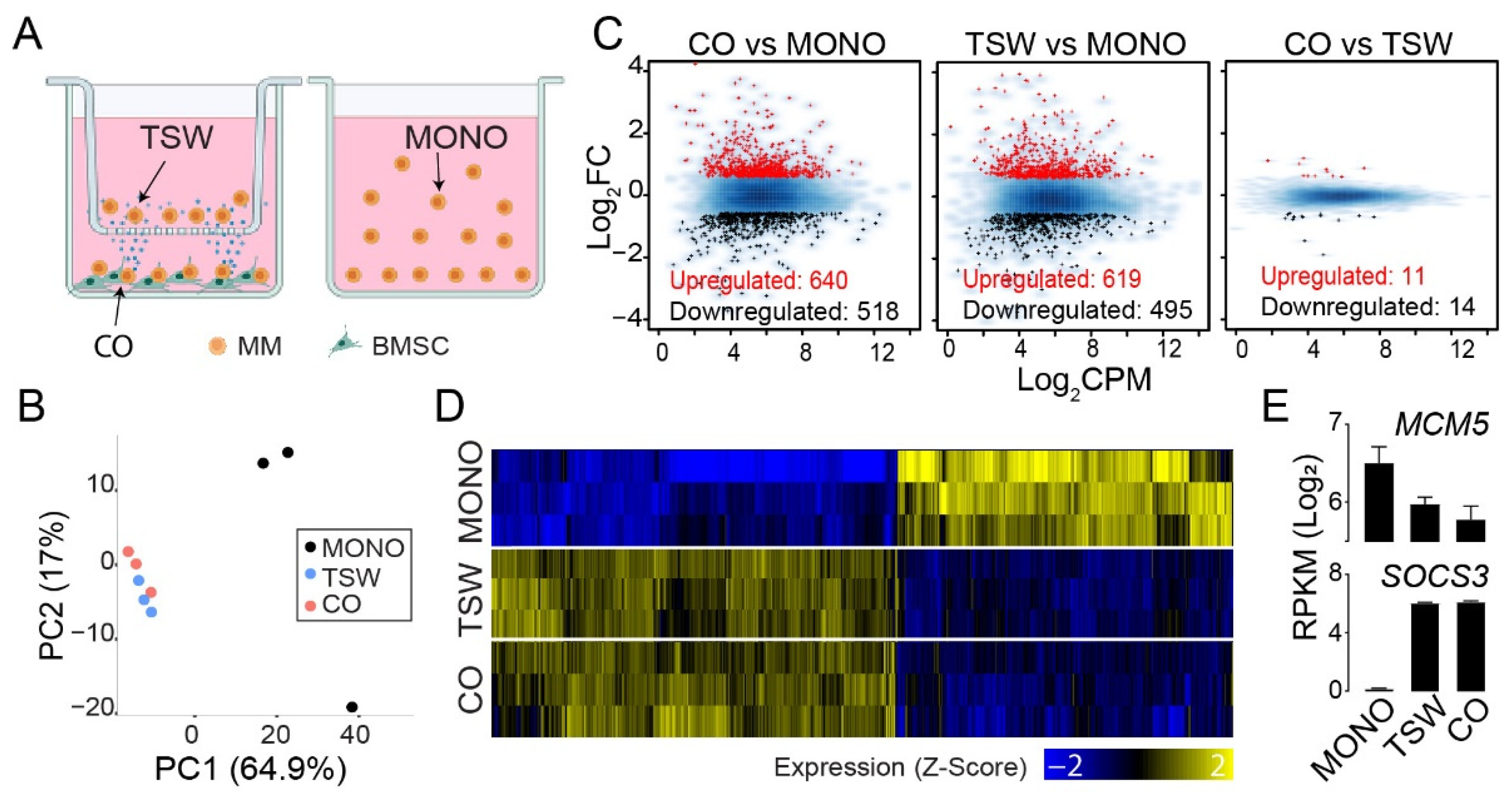
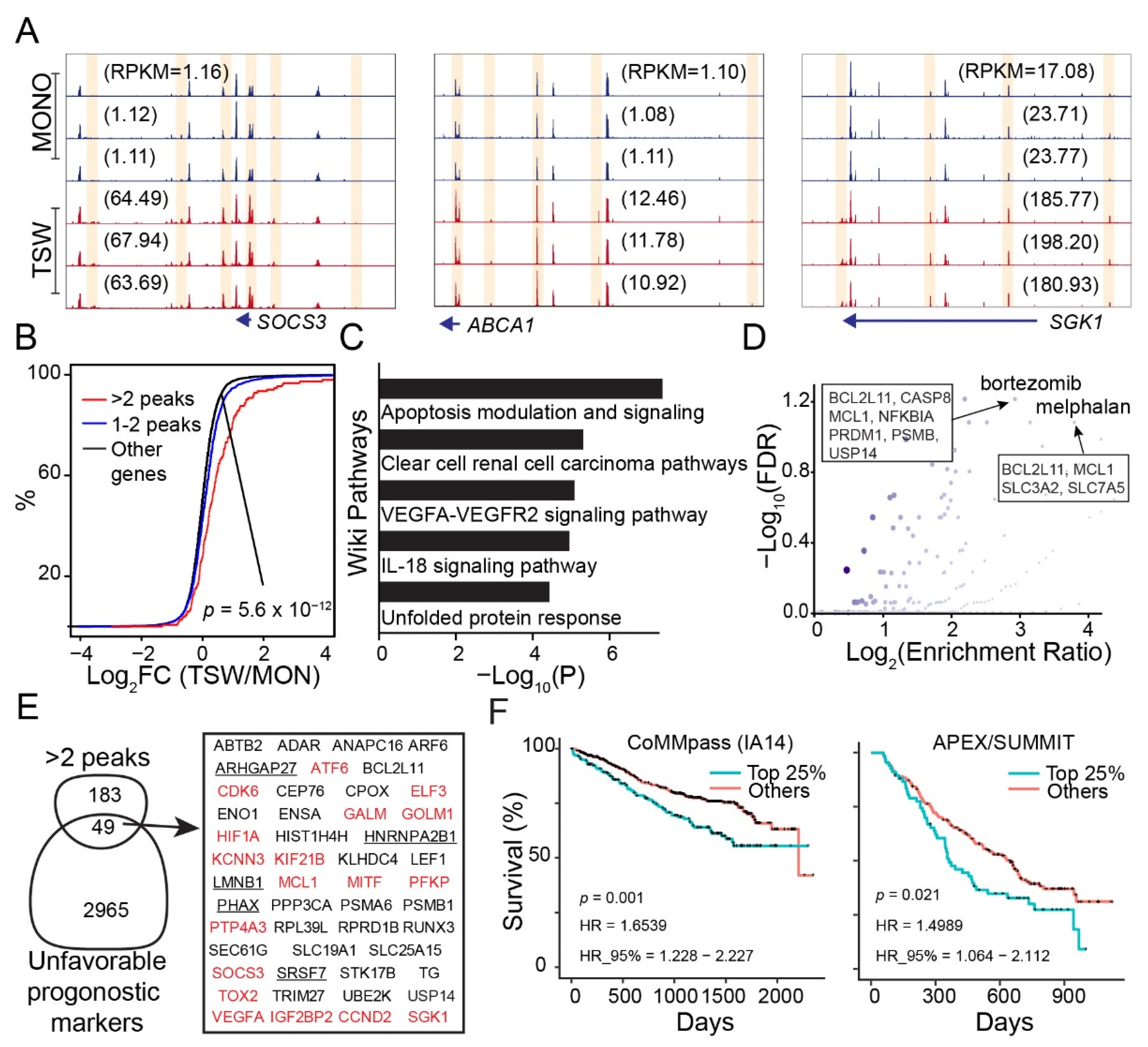
3.1. An Overall View of BMSC-Induced Expression Changes in MM Cells
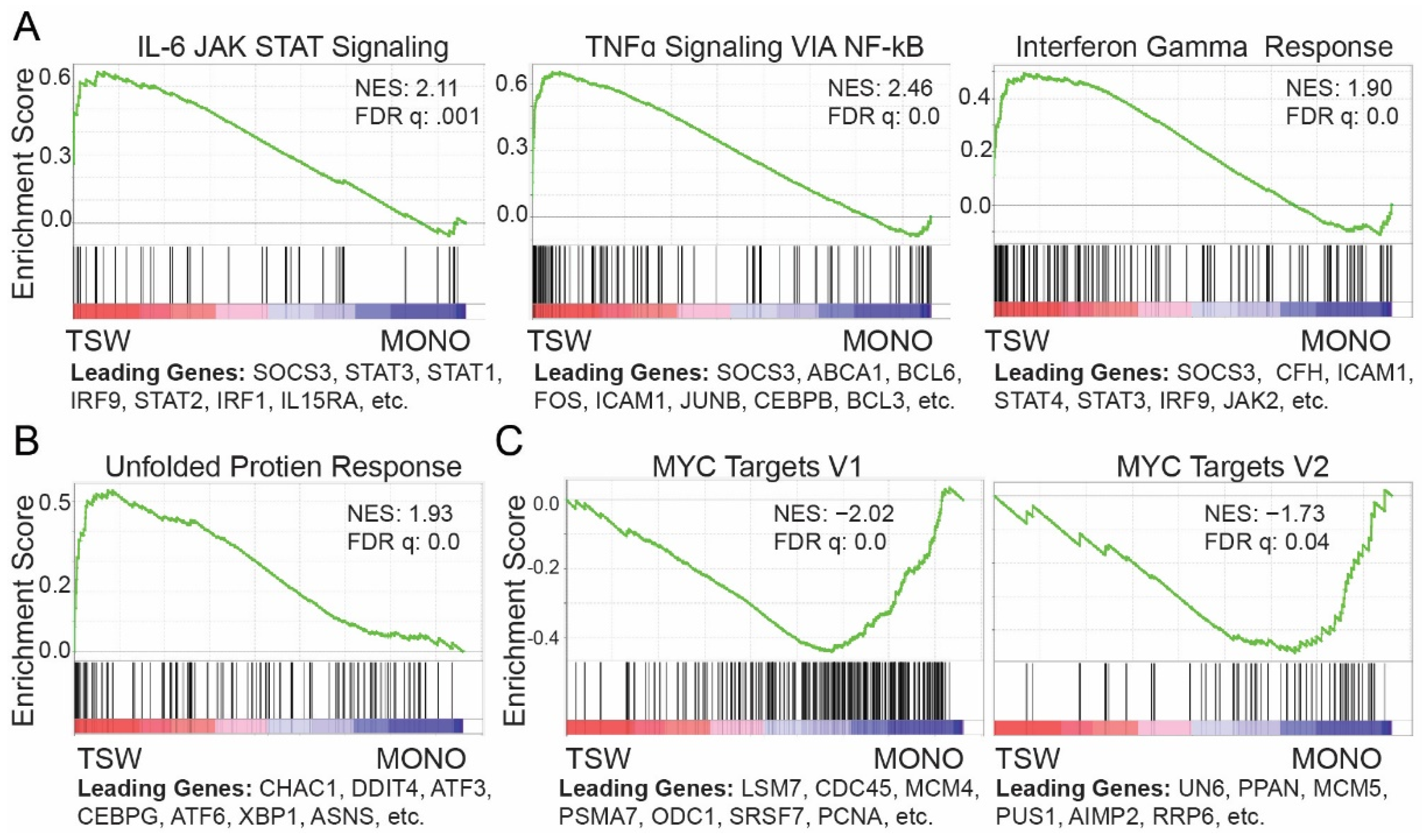
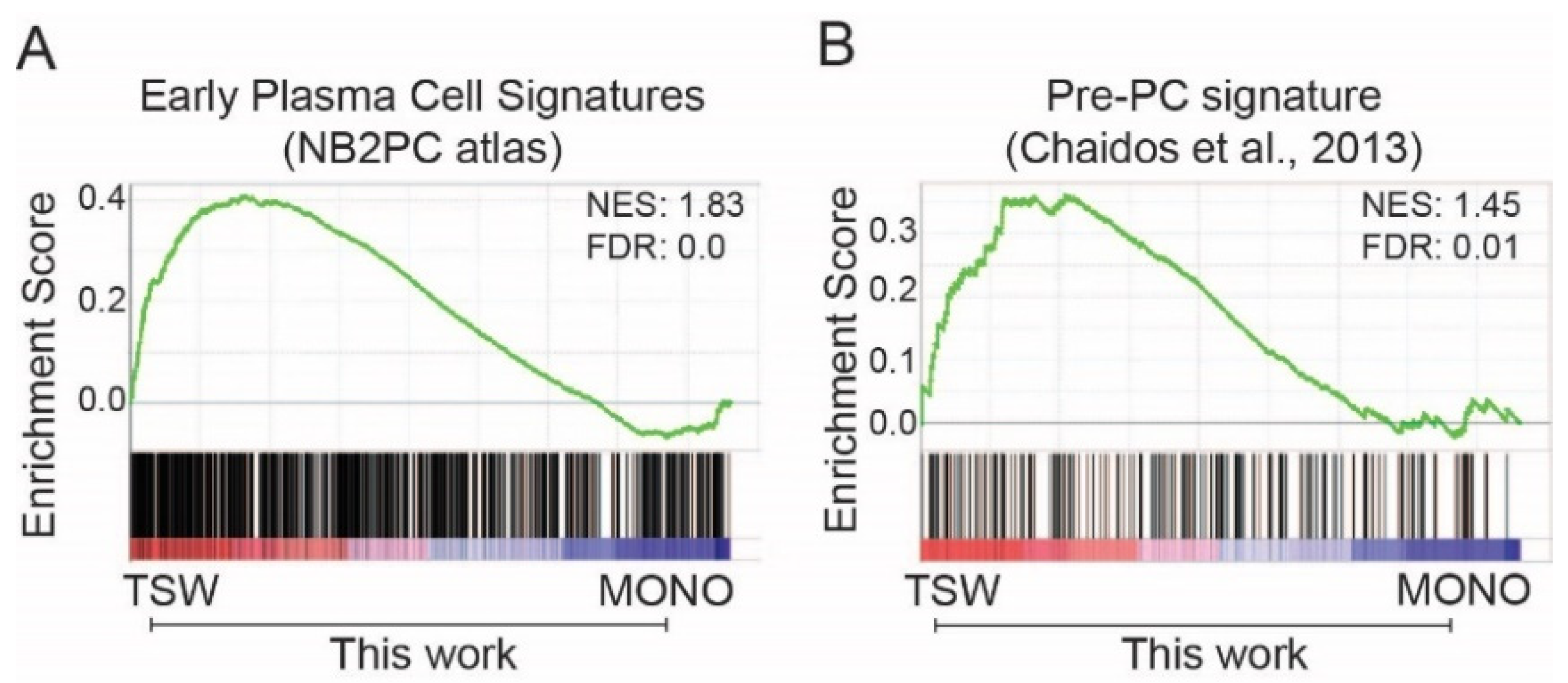
3.2. BMSC-Induced Transcriptome Signatures Associated with Soluble Factors
3.3. BMSC-Induced Transcriptome Signatures Associated with Physical Adhesion
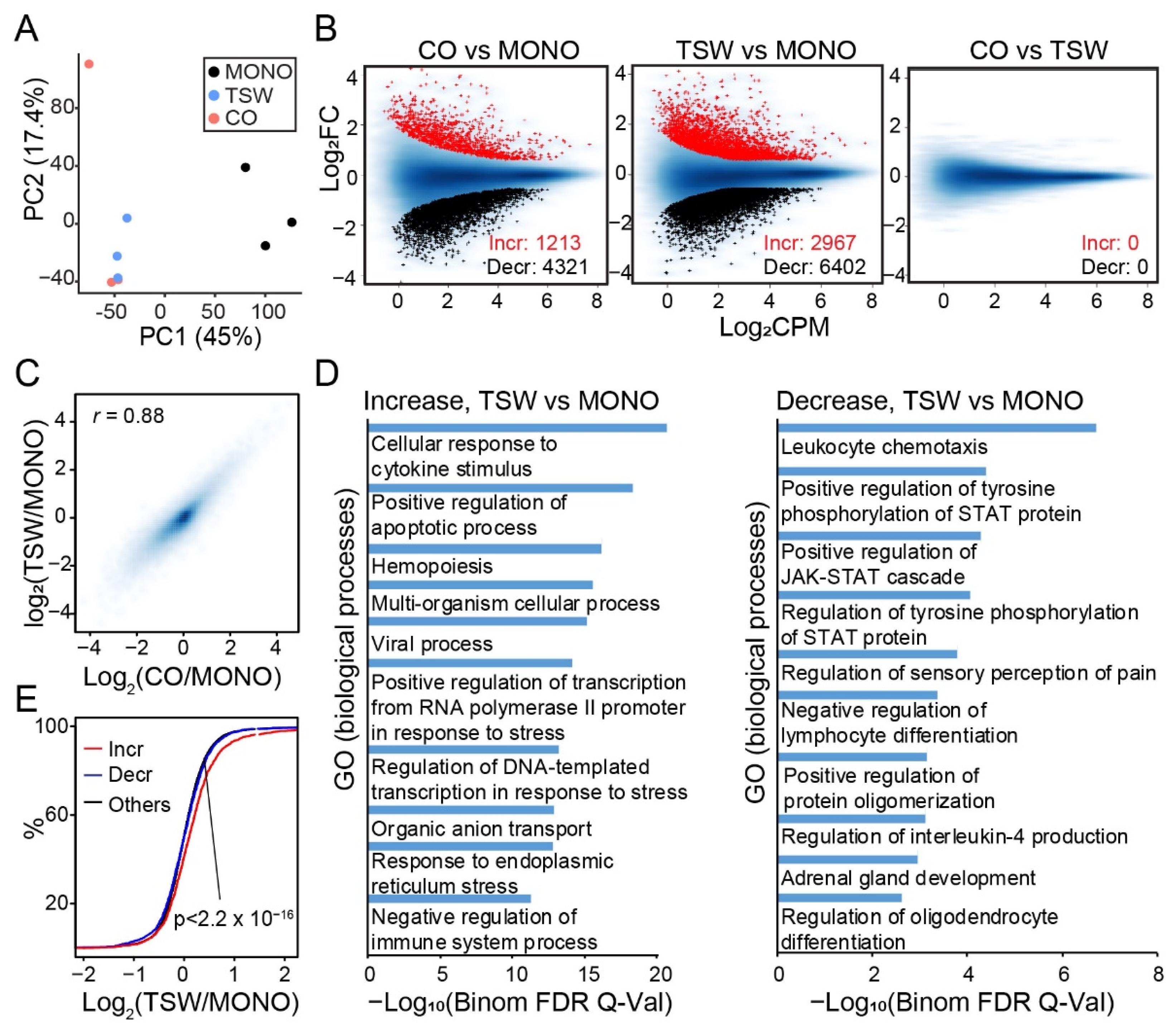
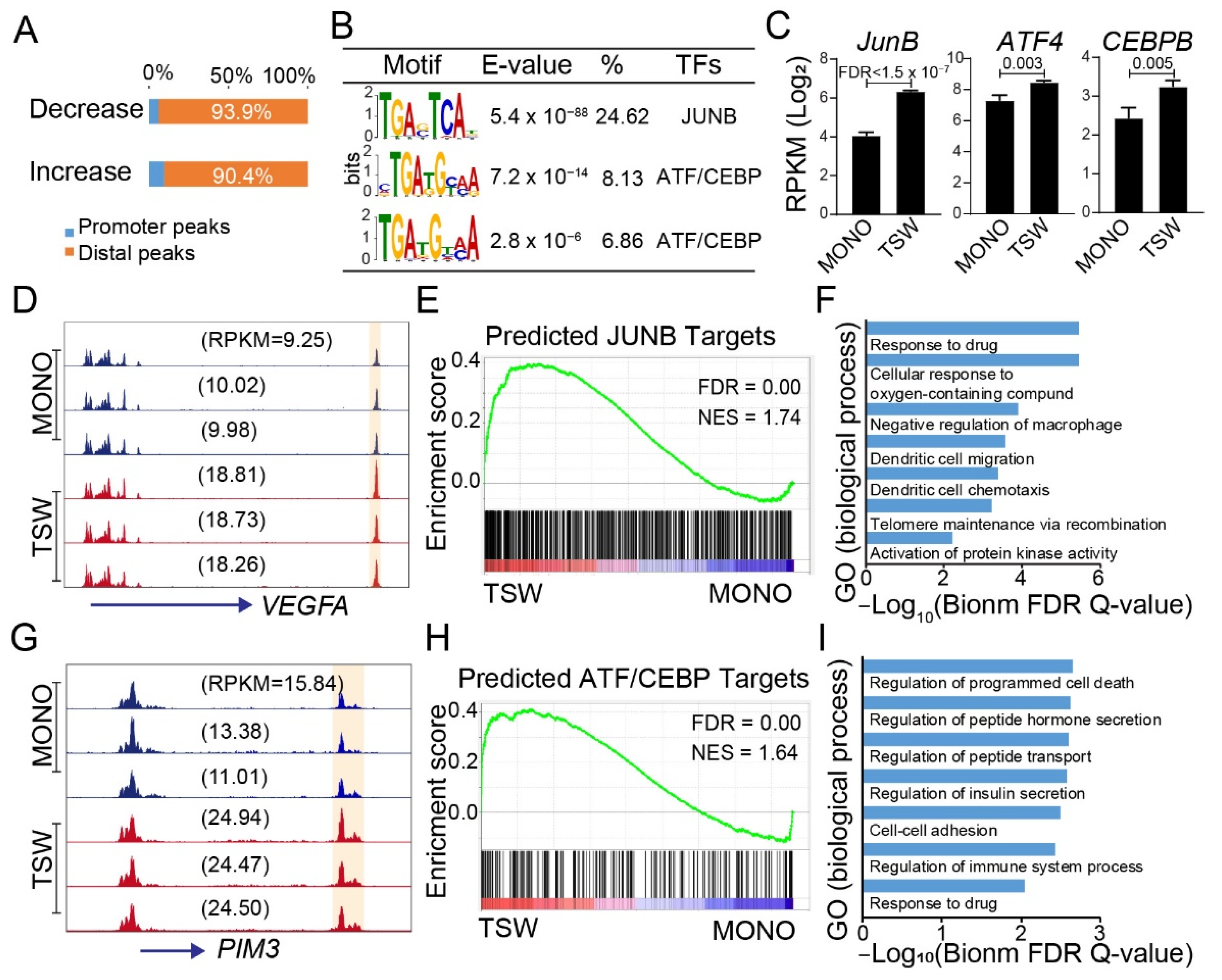
3.4. An Overall View of BMSC-Induced Transformation of Regulome in MM Cells
3.5. Genes Increasing Accessibility at Multiple Regulatory Sites Are Clinically Relevant to MM
3.6. Candidate Regulators of BMSC-Induced Transformation of Regulome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aung, T.N.; Bickell, N.A.; Jagannath, S.; Kamath, G.; Meltzer, J.; Kunzel, B.; Egorova, N.N. Do Patients with Multiple Myeloma Enrolled in Clinical Trials Live Longer? Am. J. Clin. Oncol. 2021, 44, 603–612. [Google Scholar] [CrossRef]
- Meads, M.B.; Hazlehurst, L.A.; Dalton, W.S. The bone marrow microenvironment as a tumor sanctuary and contributor to drug resistance. Clin. Cancer Res. 2008, 14, 2519–2526. [Google Scholar] [CrossRef]
- Chen, W.C.; Hu, G.; Hazlehurst, L.A. Contribution of the bone marrow stromal cells in mediating drug resistance in hematopoietic tumors. Curr. Opin. Pharmacol. 2020, 54, 36–43. [Google Scholar] [CrossRef]
- Hideshima, T.; Mitsiades, C.; Tonon, G.; Richardson, P.G.; Anderson, K.C. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat. Rev. Cancer 2007, 7, 585–598. [Google Scholar] [CrossRef]
- Kikuchi, J.; Koyama, D.; Wada, T.; Izumi, T.; Hofgaard, P.O.; Bogen, B.; Furukawa, Y. Phosphorylation-mediated EZH2 inactivation promotes drug resistance in multiple myeloma. J. Clin. Investig. 2015, 125, 4375–4390. [Google Scholar] [CrossRef]
- Meads, M.B.; Gatenby, R.A.; Dalton, W.S. Environment-mediated drug resistance: A major contributor to minimal residual disease. Nat. Rev. Cancer 2009, 9, 665–674. [Google Scholar] [CrossRef]
- Farrell, M.L.; Reagan, M.R. Soluble and Cell-Cell-Mediated Drivers of Proteasome Inhibitor Resistance in Multiple Myeloma. Front. Endocrinol. 2018, 9, 218. [Google Scholar] [CrossRef]
- Hideshima, T.; Anderson, K.C. Signaling Pathway Mediating Myeloma Cell Growth and Survival. Cancers 2021, 13, 216. [Google Scholar] [CrossRef]
- Ohguchi, H.; Hideshima, T.; Anderson, K.C. The biological significance of histone modifiers in multiple myeloma: Clinical applications. Blood Cancer J. 2018, 8, 83. [Google Scholar] [CrossRef]
- Ren, Z.; Ahn, J.H.; Liu, H.; Tsai, Y.H.; Bhanu, N.V.; Koss, B.; Allison, D.F.; Ma, A.; Storey, A.J.; Wang, P.; et al. PHF19 promotes multiple myeloma tumorigenicity through PRC2 activation and broad H3K27me3 domain formation. Blood 2019, 134, 1176–1189. [Google Scholar] [CrossRef]
- Yu, T.; Du, C.; Ma, X.; Sui, W.; Yu, Z.; Liu, L.; Zhao, L.; Li, Z.; Xu, J.; Wei, X.; et al. Polycomb-like Protein 3 Induces Proliferation and Drug Resistance in Multiple Myeloma and Is Regulated by miRNA-15a. Mol. Cancer Res. 2020, 18, 1063–1073. [Google Scholar] [CrossRef]
- Ohguchi, H.; Hideshima, T.; Bhasin, M.K.; Gorgun, G.T.; Santo, L.; Cea, M.; Samur, M.K.; Mimura, N.; Suzuki, R.; Tai, Y.T.; et al. The KDM3A-KLF2-IRF4 axis maintains myeloma cell survival. Nat. Commun. 2016, 7, 10258. [Google Scholar] [CrossRef]
- Minami, J.; Suzuki, R.; Mazitschek, R.; Gorgun, G.; Ghosh, B.; Cirstea, D.; Hu, Y.; Mimura, N.; Ohguchi, H.; Cottini, F.; et al. Histone deacetylase 3 as a novel therapeutic target in multiple myeloma. Leukemia 2014, 28, 680–689. [Google Scholar] [CrossRef]
- Ohguchi, H.; Park, P.M.C.; Wang, T.; Gryder, B.E.; Ogiya, D.; Kurata, K.; Zhang, X.; Li, D.; Pei, C.; Masuda, T.; et al. Lysine Demethylase 5A is Required for MYC Driven Transcription in Multiple Myeloma. Blood Cancer Discov. 2021, 2, 370–387. [Google Scholar] [CrossRef]
- Lhoumaud, P.; Badri, S.; Rodriguez-Hernaez, J.; Sakellaropoulos, T.; Sethia, G.; Kloetgen, A.; Cornwell, M.; Bhattacharyya, S.; Ay, F.; Bonneau, R.; et al. NSD2 overexpression drives clustered chromatin and transcriptional changes in a subset of insulated domains. Nat. Commun. 2019, 10, 4843. [Google Scholar] [CrossRef]
- Ohguchi, H.; Harada, T.; Sagawa, M.; Kikuchi, S.; Tai, Y.T.; Richardson, P.G.; Hideshima, T.; Anderson, K.C. KDM6B modulates MAPK pathway mediating multiple myeloma cell growth and survival. Leukemia 2017, 31, 2661–2669. [Google Scholar] [CrossRef]
- Corces, M.R.; Trevino, A.E.; Hamilton, E.G.; Greenside, P.G.; Sinnott-Armstrong, N.A.; Vesuna, S.; Satpathy, A.T.; Rubin, A.J.; Montine, K.S.; Wu, B.; et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 2017, 14, 959–962. [Google Scholar] [CrossRef]
- Rellick, S.L.; Hu, G.; Piktel, D.; Martin, K.H.; Geldenhuys, W.J.; Nair, R.R.; Gibson, L.F. Co-culture model of B-cell acute lymphoblastic leukemia recapitulates a transcription signature of chemotherapy-refractory minimal residual disease. Sci. Rep. 2021, 11, 15840. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Puente-Santamaria, L.; Wasserman, W.W.; Del Peso, L. TFEA.ChIP: A tool kit for transcription factor binding site enrichment analysis capitalizing on ChIP-seq datasets. Bioinformatics 2019, 35, 5339–5340. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Chaidos, A.; Barnes, C.P.; Cowan, G.; May, P.C.; Melo, V.; Hatjiharissi, E.; Papaioannou, M.; Harrington, H.; Doolittle, H.; Terpos, E.; et al. Clinical drug resistance linked to interconvertible phenotypic and functional states of tumor-propagating cells in multiple myeloma. Blood 2013, 121, 318–328. [Google Scholar] [CrossRef]
- Fagerli, U.M.; Ullrich, K.; Stuhmer, T.; Holien, T.; Kochert, K.; Holt, R.U.; Bruland, O.; Chatterjee, M.; Nogai, H.; Lenz, G.; et al. Serum/glucocorticoid-regulated kinase 1 (SGK1) is a prominent target gene of the transcriptional response to cytokines in multiple myeloma and supports the growth of myeloma cells. Oncogene 2011, 30, 3198–3206. [Google Scholar] [CrossRef]
- Brocke-Heidrich, K.; Kretzschmar, A.K.; Pfeifer, G.; Henze, C.; Loffler, D.; Koczan, D.; Thiesen, H.J.; Burger, R.; Gramatzki, M.; Horn, F. Interleukin-6-dependent gene expression profiles in multiple myeloma INA-6 cells reveal a Bcl-2 family-independent survival pathway closely associated with Stat3 activation. Blood 2004, 103, 242–251. [Google Scholar] [CrossRef]
- Croonquist, P.A.; Linden, M.A.; Zhao, F.; Van Ness, B.G. Gene profiling of a myeloma cell line reveals similarities and unique signatures among IL-6 response, N-ras-activating mutations, and coculture with bone marrow stromal cells. Blood 2003, 102, 2581–2592. [Google Scholar] [CrossRef]
- McMillin, D.W.; Delmore, J.; Weisberg, E.; Negri, J.M.; Geer, D.C.; Klippel, S.; Mitsiades, N.; Schlossman, R.L.; Munshi, N.C.; Kung, A.L.; et al. Tumor cell-specific bioluminescence platform to identify stroma-induced changes to anticancer drug activity. Nat. Med. 2010, 16, 483–489. [Google Scholar] [CrossRef]
- Lam, C.; Ferguson, I.D.; Mariano, M.C.; Lin, Y.T.; Murnane, M.; Liu, H.; Smith, G.A.; Wong, S.W.; Taunton, J.; Liu, J.O.; et al. Repurposing tofacitinib as an anti-myeloma therapeutic to reverse growth-promoting effects of the bone marrow microenvironment. Haematologica 2018, 103, 1218–1228. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Meyer, C.A.; Wang, Q.; Liu, J.S.; Shirley Liu, X.; Zhang, Y. GFOLD: A generalized fold change for ranking differentially expressed genes from RNA-seq data. Bioinformatics 2012, 28, 2782–2788. [Google Scholar] [CrossRef]
- McLean, C.Y.; Bristor, D.; Hiller, M.; Clarke, S.L.; Schaar, B.T.; Lowe, C.B.; Wenger, A.M.; Bejerano, G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010, 28, 495–501. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Liao, Y.X.; Wang, J.; Jaehnig, E.J.; Shi, Z.A.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef]
- Jourquin, J.; Duncan, D.; Shi, Z.; Zhang, B. GLAD4U: Deriving and prioritizing gene lists from PubMed literature. BMC Genom. 2012, 13 (Suppl. S8), S20. [Google Scholar] [CrossRef]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.E.; Bailey, T.L.; Noble, W.S. FIMO: Scanning for occurrences of a given motif. Bioinformatics 2011, 27, 1017–1018. [Google Scholar] [CrossRef] [PubMed]
- Kulakovskiy, I.V.; Vorontsov, I.E.; Yevshin, I.S.; Sharipov, R.N.; Fedorova, A.D.; Rumynskiy, E.I.; Medvedeva, Y.A.; Magana-Mora, A.; Bajic, V.B.; Papatsenko, D.A.; et al. HOCOMOCO: Towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res. 2018, 46, D252–D259. [Google Scholar] [CrossRef] [PubMed]
- Keats, J.J.; Craig, D.W.; Liang, W.; Venkata, Y.; Kurdoglu, A.; Aldrich, J.; Auclair, D.; Allen, K.; Harrison, B.; Jewell, S.; et al. Interim Analysis of The Mmrf Commpass Trial, a Longitudinal Study in Multiple Myeloma Relating Clinical Outcomes to Genomic and Immunophenotypic Profiles. Blood 2013, 122. [Google Scholar] [CrossRef]
- Shi, L.; Campbell, G.; Jones, W.D.; Campagne, F.; Wen, Z.; Walker, S.J.; Su, Z.; Chu, T.M.; Goodsaid, F.M.; Pusztai, L.; et al. The MicroArray Quality Control (MAQC)-II study of common practices for the development and validation of microarray-based predictive models. Nat. Biotechnol. 2010, 28, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, G.; Mitsiades, C.; Bryant, B.; Zhan, F.; Chng, W.J.; Roels, S.; Koenig, E.; Fergus, A.; Huang, Y.; Richardson, P.; et al. Gene expression profiling and correlation with outcome in clinical trials of the proteasome inhibitor bortezomib. Blood 2007, 109, 3177–3188. [Google Scholar] [CrossRef]
- Driscoll, J.J.; Pelluru, D.; Lefkimmiatis, K.; Fulciniti, M.; Prabhala, R.H.; Greipp, P.R.; Barlogie, B.; Tai, Y.T.; Anderson, K.C.; Shaughnessy, J.D.; et al. The sumoylation pathway is dysregulated in multiple myeloma and is associated with adverse patient outcome. Blood 2010, 115, 2827–2834. [Google Scholar] [CrossRef]
- Heuck, C.J.; Qu, P.; van Rhee, F.; Waheed, S.; Usmani, S.Z.; Epstein, J.; Zhang, Q.; Edmondson, R.; Hoering, A.; Crowley, J.; et al. Five gene probes carry most of the discriminatory power of the 70-gene risk model in multiple myeloma. Leukemia 2014, 28, 2410–2413. [Google Scholar] [CrossRef][Green Version]
- Schulze, A.; Oshi, M.; Endo, I.; Takabe, K. MYC Targets Scores Are Associated with Cancer Aggressiveness and Poor Survival in ER-Positive Primary and Metastatic Breast Cancer. Int. J. Mol. Sci. 2020, 21, 8127. [Google Scholar] [CrossRef]
- Dezorella, N.; Pevsner-Fischer, M.; Deutsch, V.; Kay, S.; Baron, S.; Stern, R.; Tavor, S.; Nagler, A.; Naparstek, E.; Zipori, D.; et al. Mesenchymal stromal cells revert multiple myeloma cells to less differentiated phenotype by the combined activities of adhesive interactions and interleukin-6. Exp. Cell Res. 2009, 315, 1904–1913. [Google Scholar] [CrossRef]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J., Jr.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.H.; Ferguson, I.D.; Thornton, A.M.; Bastola, P.; Lam, C.; Lin, Y.H.T.; Choudhry, P.; Mariano, M.C.; Marcoulis, M.D.; Teo, C.F.; et al. Proteasome inhibitor-induced modulation reveals the spliceosome as a specific therapeutic vulnerability in multiple myeloma. Nat. Commun. 2020, 11, 1931. [Google Scholar] [CrossRef] [PubMed]
- Le Moigne, R.; Aftab, B.T.; Djakovic, S.; Dhimolea, E.; Valle, E.; Murnane, M.; King, E.M.; Soriano, F.; Menon, M.K.; Wu, Z.Y.; et al. The p97 Inhibitor CB-5083 Is a Unique Disrupter of Protein Homeostasis in Models of Multiple Myeloma. Mol. Cancer Ther. 2017, 16, 2375–2386. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.K.; Li, M.; Tea, M.N.; Pitman, M.R.; Toubia, J.; Wang, P.P.; Anderson, D.; Creek, D.J.; Orlowski, R.Z.; Gliddon, B.L.; et al. Resensitising proteasome inhibitor-resistant myeloma with sphingosine kinase 2 inhibition. Neoplasia 2022, 24, 1–11. [Google Scholar] [CrossRef]
- Shuai, K.; Liu, B. Regulation of JAK-STAT signalling in the immune system. Nat. Rev. Immunol. 2003, 3, 900–911. [Google Scholar] [CrossRef]
- Pasello, M.; Giudice, A.M.; Scotlandi, K. The ABC subfamily A transporters: Multifaceted players with incipient potentialities in cancer. Semin. Cancer Biol. 2020, 60, 57–71. [Google Scholar] [CrossRef]
- Al-Odat, O.; von Suskil, M.; Chitren, R.; Elbezanti, W.; Srivastava, S.; Budak-Alpddogan, T.; Jonnalagadda, S.; Aggarwal, B.; Pandey, M. Mcl-1 Inhibition: Managing Malignancy in Multiple Myeloma. Front. Pharmacol. 2021, 12, 699629. [Google Scholar] [CrossRef]
- Ng, Y.L.D.; Bohl, S.; Ramberger, E.; Popp, O.; Bauhuf, I.; Dolnik, A.; Steinebach, C.; Gutschow, M.; Bullinger, L.; Mertins, P.; et al. Quantitative Proteomic Analysis of Relapsed Multiple Myeloma Identifies CDK6 Upregulation As a Potential Targetable Resistance Mechanism for Lenalidomide. Blood 2019, 134. [Google Scholar] [CrossRef]
- Maiso, P.; Huynh, D.; Moschetta, M.; Sacco, A.; Aljawai, Y.; Mishima, Y.; Asara, J.M.; Roccaro, A.M.; Kimmelman, A.C.; Ghobrial, I.M. Metabolic Signature Identifies Novel Targets for Drug Resistance in Multiple Myeloma. Cancer Res. 2015, 75, 2071–2082. [Google Scholar] [CrossRef]
- Pei, X.Y.; Dai, Y.; Felthousen, J.; Chen, S.; Takabatake, Y.; Zhou, L.; Youssefian, L.E.; Sanderson, M.W.; Bodie, W.W.; Kramer, L.B.; et al. Circumvention of Mcl-1-dependent drug resistance by simultaneous Chk1 and MEK1/2 inhibition in human multiple myeloma cells. PLoS ONE 2014, 9, e89064. [Google Scholar] [CrossRef]
- Zhang, B.P.; Ma, L.; Wei, J.; Hu, J.Y.; Zhao, Z.C.; Wang, Y.P.; Chen, Y.; Zhao, F. miR-137 Suppresses the Phosphorylation of AKT and Improves the Dexamethasone Sensitivity in Multiple Myeloma Cells Via Targeting MITF. Curr. Cancer Drug Tar. 2016, 16, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Ohta, E.; Takeda, S.; Sunamura, S.; Ishibashi, M.; Tamura, H.; Wang, Y.H.; Deguchi, A.; Tanaka, J.; Maru, Y.; et al. Histone deacetylase inhibitor panobinostat induces calcineurin degradation in multiple myeloma. JCI Insight 2016, 1, e85061. [Google Scholar] [CrossRef] [PubMed]
- Chong, P.S.Y.; Zhou, J.B.; Lim, J.S.L.; Hee, Y.T.; Chooi, J.Y.; Chung, T.H.; Tan, Z.T.; Zeng, Q.; Waller, D.D.; Sebag, M.; et al. IL6 Promotes a STAT3-PRL3 Feedforward Loop via SHP2 Repression in Multiple Myeloma. Cancer Res. 2019, 79, 4679–4688. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Gutierrez-Uzquiza, A.; Zheng, X.Y.; Chang, R.X.; Vogl, D.T.; Garfall, A.L.; Bernabei, L.; Saraf, A.; Florens, L.; Washburn, M.P.; et al. RUNX proteins desensitize multiple myeloma to lenalidomide via protecting IKZFs from degradation. Leukemia 2019, 33, 2006–2021. [Google Scholar] [CrossRef] [PubMed]
- Tsubaki, M.; Takeda, T.; Matsuda, T.; Seki, S.; Tomonari, Y.; Koizumi, S.; Nagatakiya, M.; Katsuyama, M.; Yamamoto, Y.; Tsurushima, K.; et al. Activation of Serum/Glucocorticoid Regulated Kinase 1/Nuclear Factor-kappaB Pathway Are Correlated with Low Sensitivity to Bortezomib and Ixazomib in Resistant Multiple Myeloma Cells. Biomedicines 2021, 9, 33. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.H.; Liu, J.; Shen, C.Y.; Ding, L.L.; Zhong, F.; Ouyang, Y.; Wang, Y.C.; He, S. The role of ubiquitin-specific protease 14 (USP14) in cell adhesion-mediated drug resistance (CAM-DR) of multiple myeloma cells. Eur. J. Haematol. 2017, 98, 4–12. [Google Scholar] [CrossRef]
- Fan, F.J.; Malvestiti, S.; Vallet, S.; Lind, J.; Garcia-Manteiga, J.M.; Morelli, E.; Jiang, Q.Y.; Seckinger, A.; Hose, D.; Goldschmidt, H.; et al. JunB is a key regulator of multiple myeloma bone marrow angiogenesis. Leukemia 2021, 35, 3509–3525. [Google Scholar] [CrossRef]
- Fan, F.; Bashari, M.H.; Morelli, E.; Tonon, G.; Malvestiti, S.; Vallet, S.; Jarahian, M.; Seckinger, A.; Hose, D.; Bakiri, L.; et al. The AP-1 transcription factor JunB is essential for multiple myeloma cell proliferation and drug resistance in the bone marrow microenvironment. Leukemia 2017, 31, 1570–1581. [Google Scholar] [CrossRef]
- Wortel, I.M.N.; van der Meer, L.T.; Kilberg, M.S.; van Leeuwen, F.N. Surviving Stress: Modulation of ATF4-Mediated Stress Responses in Normal and Malignant Cells. Trends Endocrinol. Metab. 2017, 28, 794–806. [Google Scholar] [CrossRef]
- Mukaida, N.; Wang, Y.Y.; Li, Y.Y. Roles of Pim-3, a novel survival kinase, in tumorigenesis. Cancer Sci. 2011, 102, 1437–1442. [Google Scholar] [CrossRef]
- Maiso, P.; Mogollon, P.; Ocio, E.M.; Garayoa, M. Bone Marrow Mesenchymal Stromal Cells in Multiple Myeloma: Their Role as Active Contributors to Myeloma Progression. Cancers 2021, 13, 2542. [Google Scholar] [CrossRef] [PubMed]
- De la Puente, P.; Quan, N.; Hoo, R.S.; Muz, B.; Gilson, R.C.; Luderer, M.; King, J.; Achilefu, S.; Salama, N.N.; Vij, R.; et al. Newly established myeloma-derived stromal cell line MSP-1 supports multiple myeloma proliferation, migration, and adhesion and induces drug resistance more than normal-derived stroma. Haematologica 2016, 101, e307–e311. [Google Scholar] [CrossRef] [PubMed]
- Belloni, D.; Heltai, S.; Ponzoni, M.; Villa, A.; Vergani, B.; Pecciarini, L.; Marcatti, M.; Girlanda, S.; Tonon, G.; Ciceri, F.; et al. Modeling multiple myeloma-bone marrow interactions and response to drugs in a 3D surrogate microenvironment. Haematologica 2018, 103, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Benayas, J.; Trasanidis, N.; Katsarou, A.; Ponnusamy, K.; Chaidos, A.; May, P.C.; Xiao, X.; Bua, M.; Atta, M.; Roberts, I.A.G.; et al. Chromatin-based, in cis and in trans regulatory rewiring underpins distinct oncogenic transcriptomes in multiple myeloma. Nat. Commun. 2021, 12, 5450. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.A.; Barwick, B.G.; Matulis, S.M.; Shirasaki, R.; Jaye, D.L.; Keats, J.J.; Oberlton, B.; Joseph, N.S.; Hofmeister, C.C.; Heffner, L.T.; et al. Venetoclax sensitivity in multiple myeloma is associated with B-cell gene expression. Blood 2021, 137, 3604–3615. [Google Scholar] [CrossRef]
- Barwick, B.G.; Gupta, V.A.; Matulis, S.M.; Patton, J.C.; Powell, D.R.; Gu, Y.; Jaye, D.L.; Conneely, K.N.; Lin, Y.C.; Hofmeister, C.C.; et al. Chromatin Accessibility Identifies Regulatory Elements Predictive of Gene Expression and Disease Outcome in Multiple Myeloma. Clin. Cancer Res. 2021, 27, 3178–3189. [Google Scholar] [CrossRef]
- Frede, J.; Anand, P.; Sotudeh, N.; Pinto, R.A.; Nair, M.S.; Stuart, H.; Yee, A.J.; Vijaykumar, T.; Waldschmidt, J.M.; Potdar, S.; et al. Dynamic transcriptional reprogramming leads to immunotherapeutic vulnerabilities in myeloma. Nat. Cell Biol. 2021, 23, 1199–1211. [Google Scholar] [CrossRef]
- Jin, Y.; Chen, K.; De Paepe, A.; Hellqvist, E.; Krstic, A.D.; Metang, L.; Gustafsson, C.; Davis, R.E.; Levy, Y.M.; Surapaneni, R.; et al. Active enhancer and chromatin accessibility landscapes chart the regulatory network of primary multiple myeloma. Blood 2018, 131, 2138–2150. [Google Scholar] [CrossRef]
- He, H.H.; Meyer, C.A.; Hu, S.S.; Chen, M.W.; Zang, C.; Liu, Y.; Rao, P.K.; Fei, T.; Xu, H.; Long, H.; et al. Refined DNase-seq protocol and data analysis reveals intrinsic bias in transcription factor footprint identification. Nat. Methods 2014, 11, 73–78. [Google Scholar] [CrossRef]
- Li, Z.J.; Schulz, M.H.; Look, T.; Begemann, M.; Zenke, M.; Costa, I.G. Identification of transcription factor binding sites using ATAC-seq. Genome Biol. 2019, 20, 45. [Google Scholar] [CrossRef] [PubMed]
- Gusmao, E.G.; Allhoff, M.; Zenke, M.; Costa, I.G. Analysis of computational footprinting methods for DNase sequencing experiments. Nat. Methods 2016, 13, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Beisaw, A.; Kuenne, C.; Guenther, S.; Dallmann, J.; Wu, C.C.; Bentsen, M.; Looso, M.; Stainier, D.Y.R. AP-1 Contributes to Chromatin Accessibility to Promote Sarcomere Disassembly and Cardiomyocyte Protrusion during Zebrafish Heart Regeneration. Circ. Res. 2020, 126, 1760–1778. [Google Scholar] [CrossRef]
- Biddie, S.C.; John, S.; Sabo, P.J.; Thurman, R.E.; Johnson, T.A.; Schiltz, R.L.; Miranda, T.B.; Sung, M.H.; Trump, S.; Lightman, S.L.; et al. Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol. Cell 2011, 43, 145–155. [Google Scholar] [CrossRef]
- Yukawa, M.; Jagannathan, S.; Vallabh, S.; Kartashov, A.V.; Chen, X.T.; Weirauch, M.T.; Barski, A. AP-1 activity induced by co-stimulation is required for chromatin opening during T cell activation. J. Exp. Med. 2020, 217, e20182009. [Google Scholar] [CrossRef]
- Vierbuchen, T.; Ling, E.; Cowley, C.J.; Couch, C.H.; Wang, X.; Harmin, D.A.; Roberts, C.W.M.; Greenberg, M.E. AP-1 Transcription Factors and the BAF Complex Mediate Signal-Dependent Enhancer Selection. Mol. Cell 2017, 68, 1067–1082. [Google Scholar] [CrossRef]
- Ito, T.; Yamauchi, M.; Nishina, M.; Yamamichi, N.; Mizutani, T.; Ui, M.; Murakami, M.; Iba, H. Identification of SWI center dot SNF complex subunit BAF60a as a determinant of the transactivation potential of Fos/Jun dimers. J. Biol. Chem. 2001, 276, 2852–2857. [Google Scholar] [CrossRef]
- Cohen, D.M.; Won, K.J.; Nguyen, N.; Lazar, M.A.; Chen, C.S.; Steger, D.J. ATF4 licenses C/EBP beta activity in human mesenchymal stem cells primed for adipogenesis. eLife 2015, 4, e06821. [Google Scholar] [CrossRef]
- Lin, Q.X.X.; Thieffry, D.; Jha, S.; Benoukraf, T. TFregulomeR reveals transcription factors’ context-specific features and functions. Nucleic Acids Res. 2020, 48, e10. [Google Scholar] [CrossRef]
- Mann, I.K.; Chatterjee, R.; Zhao, J.; He, X.; Weirauch, M.T.; Hughes, T.R.; Vinson, C. CG methylated microarrays identify a novel methylated sequence bound by the CEBPB|ATF4 heterodimer that is active in vivo. Genome Res. 2013, 23, 988–997. [Google Scholar] [CrossRef]
- Torrence, M.E.; MacArthur, M.R.; Hosios, A.M.; Valvezan, A.J.; Asara, J.M.; Mitchell, J.R.; Manning, B.D. The mTORC1-mediated activation of ATF4 promotes protein and glutathione synthesis downstream of growth signals. eLife 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Narita, T.; Ri, M.; Masaki, A.; Mori, F.; Ito, A.; Kusumoto, S.; Ishida, T.; Komatsu, H.; Iida, S. Lower expression of activating transcription factors 3 and 4 correlates with shorter progression-free survival in multiple myeloma patients receiving bortezomib plus dexamethasone therapy. Blood Cancer J. 2015, 5, e373. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Huang, S.; Peng, J.; Gao, X.; Li, M.; Yu, S.; Liu, Z.; Qie, P.; Wang, Y.; Yu, S.; et al. Targeting lipid metabolism in multiple myeloma cells: Rational development of a synergistic strategy with proteasome inhibitors. Br. J. Pharmacol. 2021, 178, 4741–4757. [Google Scholar] [CrossRef] [PubMed]
- Shan, J.; Fu, L.; Balasubramanian, M.N.; Anthony, T.; Kilberg, M.S. ATF4-dependent regulation of the JMJD3 gene during amino acid deprivation can be rescued in Atf4-deficient cells by inhibition of deacetylation. J. Biol. Chem. 2012, 287, 36393–36403. [Google Scholar] [CrossRef]
- Zhao, E.; Ding, J.; Xia, Y.; Liu, M.; Ye, B.; Choi, J.H.; Yan, C.; Dong, Z.; Huang, S.; Zha, Y.; et al. KDM4C and ATF4 Cooperate in Transcriptional Control of Amino Acid Metabolism. Cell Rep. 2016, 14, 506–519. [Google Scholar] [CrossRef]
- Pal, R.; Janz, M.; Galson, D.L.; Gries, M.; Li, S.; Johrens, K.; Anagnostopoulos, I.; Dorken, B.; Mapara, M.Y.; Borghesi, L.; et al. C/EBPbeta regulates transcription factors critical for proliferation and survival of multiple myeloma cells. Blood 2009, 114, 3890–3898. [Google Scholar] [CrossRef][Green Version]
- Li, S.R.; Pal, R.; Monaghan, S.; Schafer, P.; Ouyang, H.; Mapara, M.Y.; Lentzsch, S. C/EBPb Is a Critical Mediator of Resistance to IMiD (R) Immunomodulatory Compounds and Affected by IMiD Compounds Via Control of Protein Translation. Blood 2010, 116, 64–65. [Google Scholar] [CrossRef]
- Nerlov, C. C/EBPs: Recipients of extracellular signals through proteome modulation. Curr. Opin. Cell Biol. 2008, 20, 180–185. [Google Scholar] [CrossRef]
- Grontved, L.; John, S.; Baek, S.; Liu, Y.; Buckley, J.R.; Vinson, C.; Aguilera, G.; Hager, G.L. C/EBP maintains chromatin accessibility in liver and facilitates glucocorticoid receptor recruitment to steroid response elements. EMBO J. 2013, 32, 1568–1583. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dziadowicz, S.A.; Wang, L.; Akhter, H.; Aesoph, D.; Sharma, T.; Adjeroh, D.A.; Hazlehurst, L.A.; Hu, G. Bone Marrow Stroma-Induced Transcriptome and Regulome Signatures of Multiple Myeloma. Cancers 2022, 14, 927. https://doi.org/10.3390/cancers14040927
Dziadowicz SA, Wang L, Akhter H, Aesoph D, Sharma T, Adjeroh DA, Hazlehurst LA, Hu G. Bone Marrow Stroma-Induced Transcriptome and Regulome Signatures of Multiple Myeloma. Cancers. 2022; 14(4):927. https://doi.org/10.3390/cancers14040927
Chicago/Turabian StyleDziadowicz, Sebastian A., Lei Wang, Halima Akhter, Drake Aesoph, Tulika Sharma, Donald A. Adjeroh, Lori A. Hazlehurst, and Gangqing Hu. 2022. "Bone Marrow Stroma-Induced Transcriptome and Regulome Signatures of Multiple Myeloma" Cancers 14, no. 4: 927. https://doi.org/10.3390/cancers14040927
APA StyleDziadowicz, S. A., Wang, L., Akhter, H., Aesoph, D., Sharma, T., Adjeroh, D. A., Hazlehurst, L. A., & Hu, G. (2022). Bone Marrow Stroma-Induced Transcriptome and Regulome Signatures of Multiple Myeloma. Cancers, 14(4), 927. https://doi.org/10.3390/cancers14040927