
Broad Transcriptomic Impact of Sorafenib and Its Relation to the Antitumoral Properties in Liver Cancer Cells

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines, Culture Conditions, and Sorafenib Treatment
2.2. Cell Proliferation
2.3. RNA Extraction
2.4. mRNA Library Preparation
2.5. RNA Sequencing and Data Analyses
2.6. Gene Set Enrichment Analysis and Over-Representation Analysis
2.7. Real-Time Quantitative PCR
2.8. Western Blot Analysis
2.9. Statistical Analyses
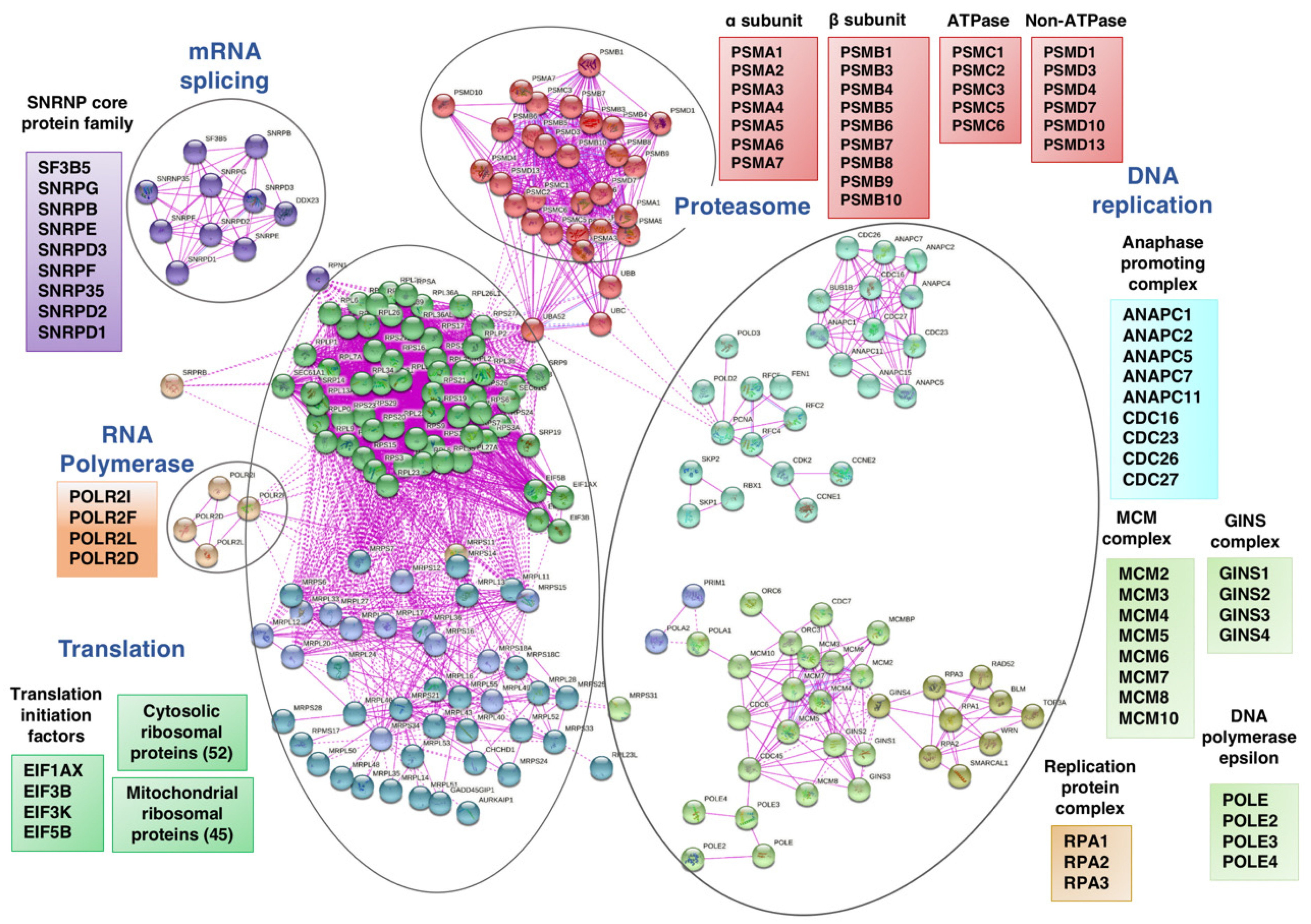
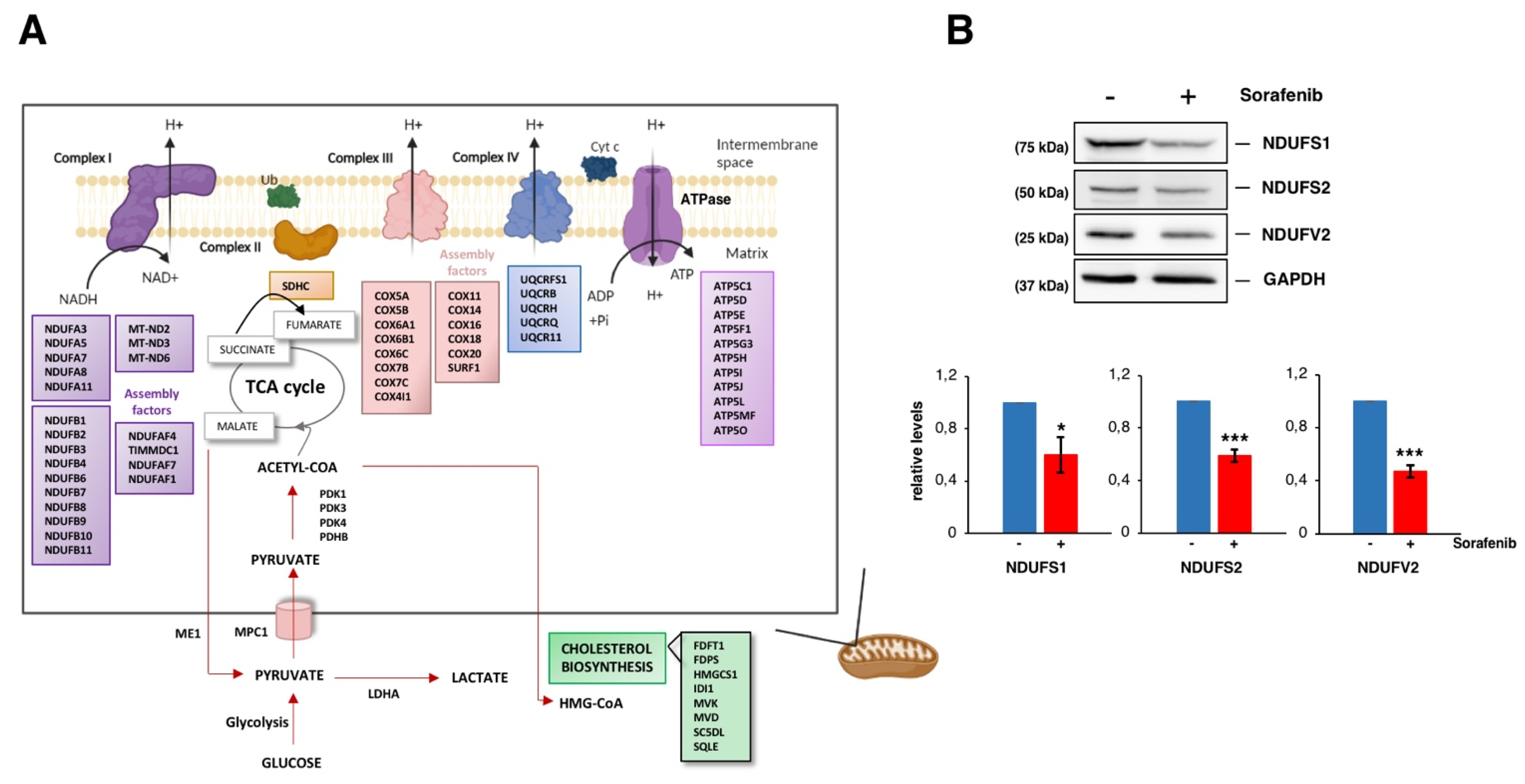
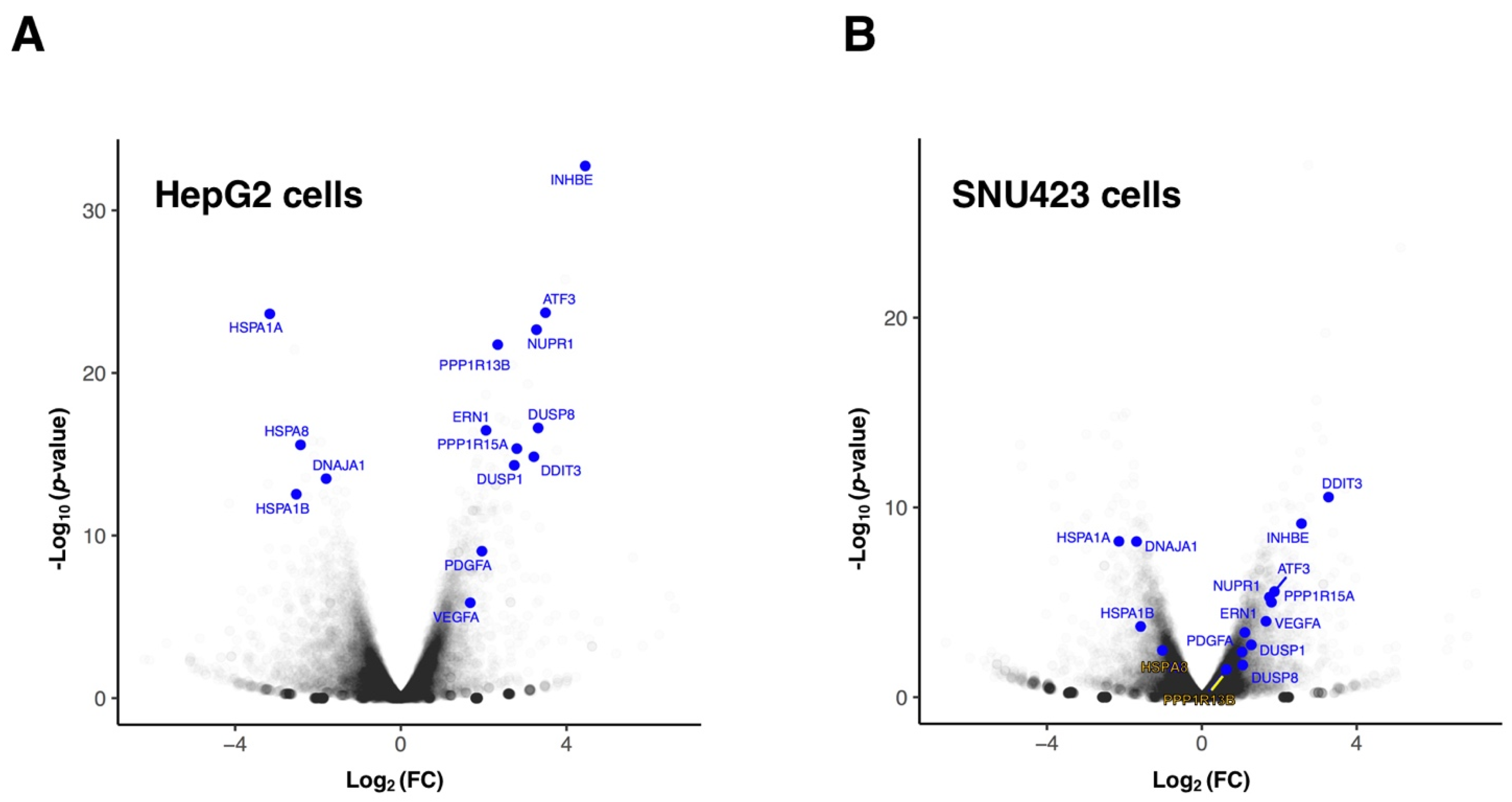
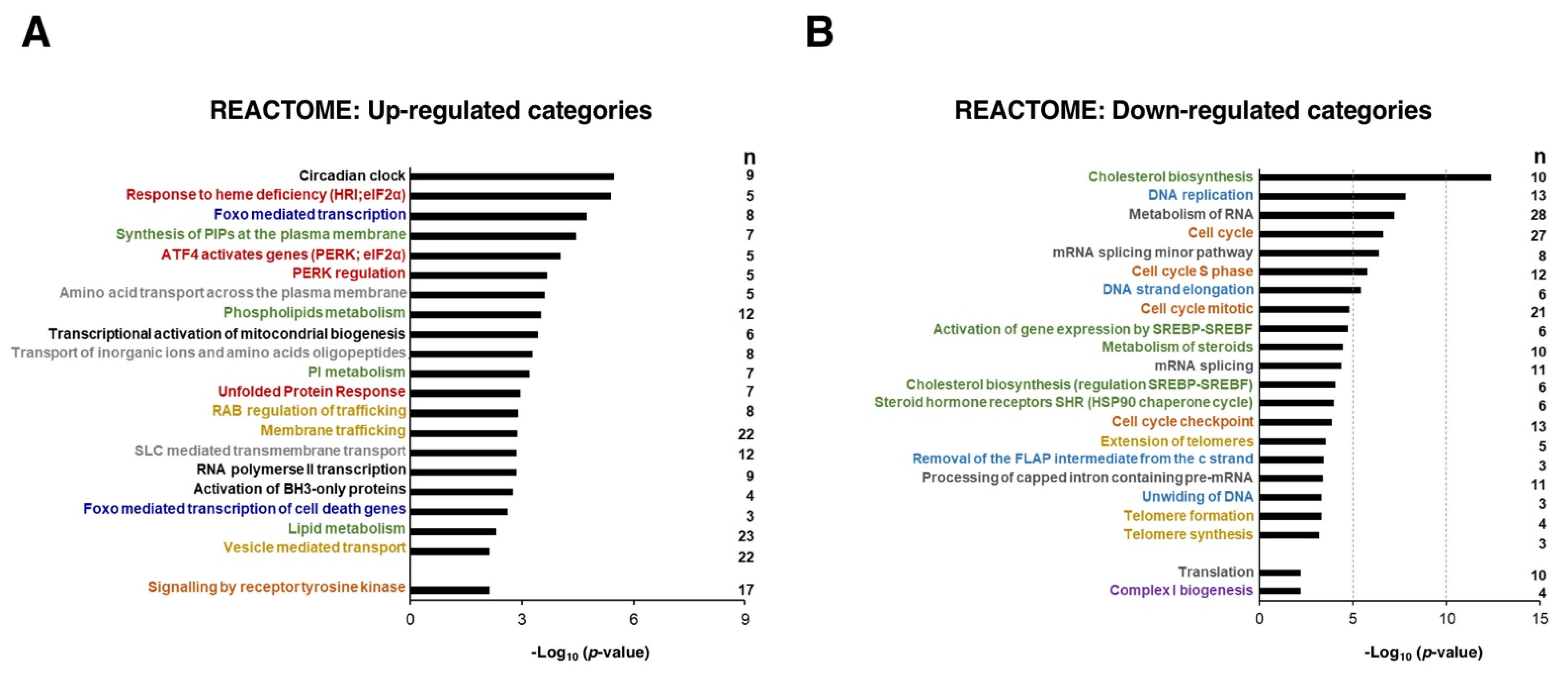
3. Results and Discussion
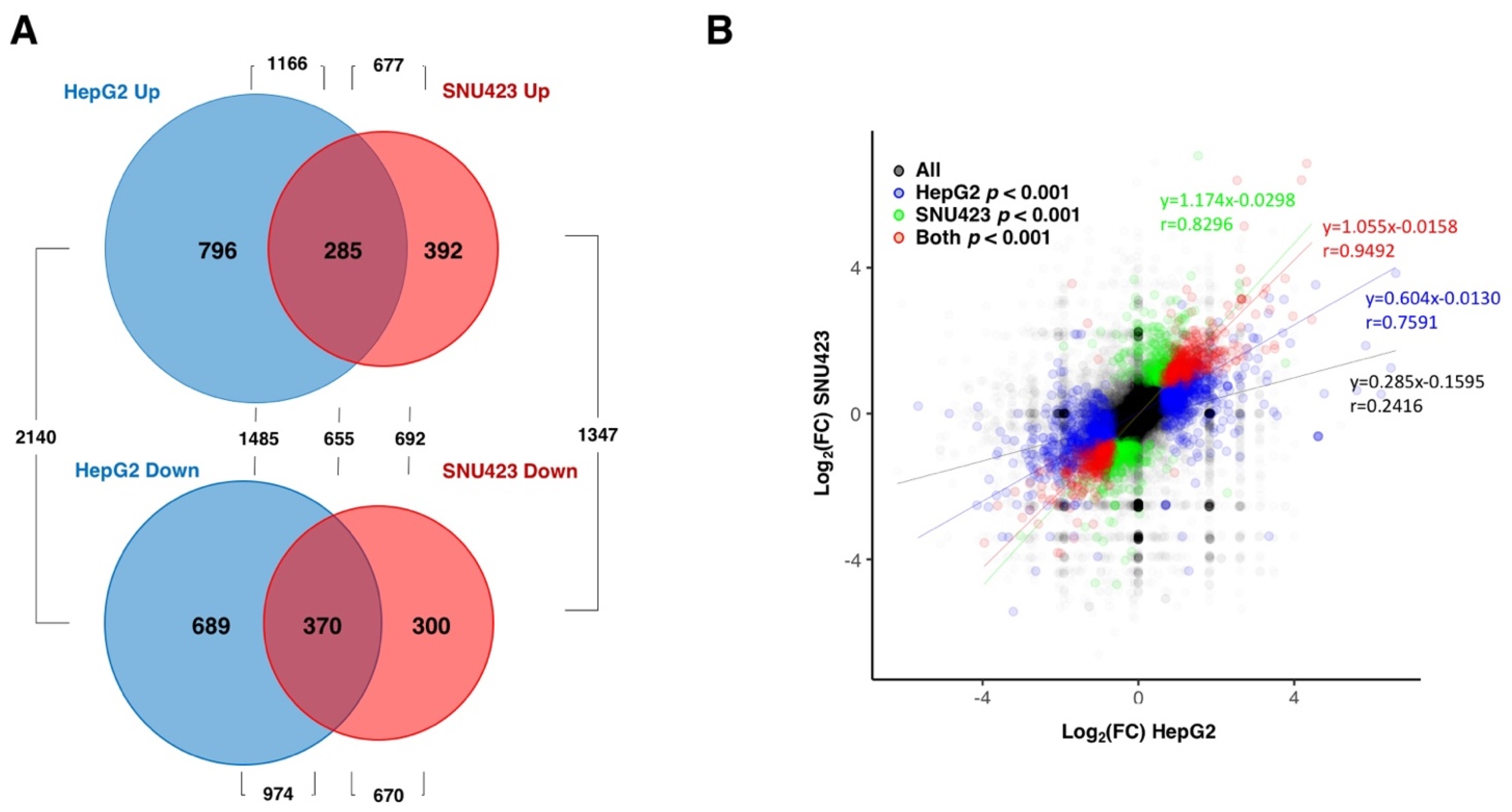
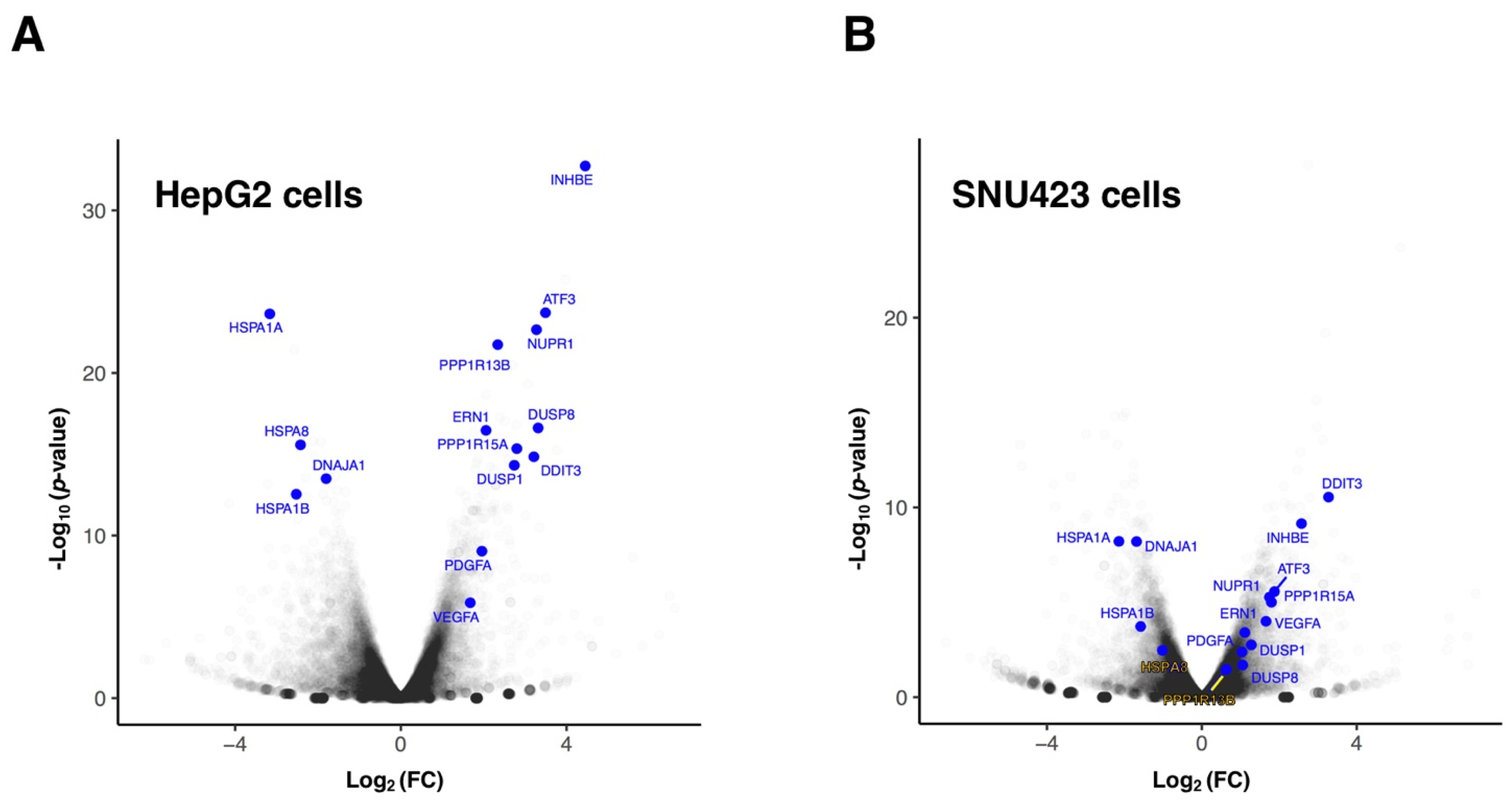
3.1. Transcriptional Changes Caused by Sorafenib in Hepg2 Hepatoblastoma and SNU423 Hepatocellular Carcinoma Cell Lines
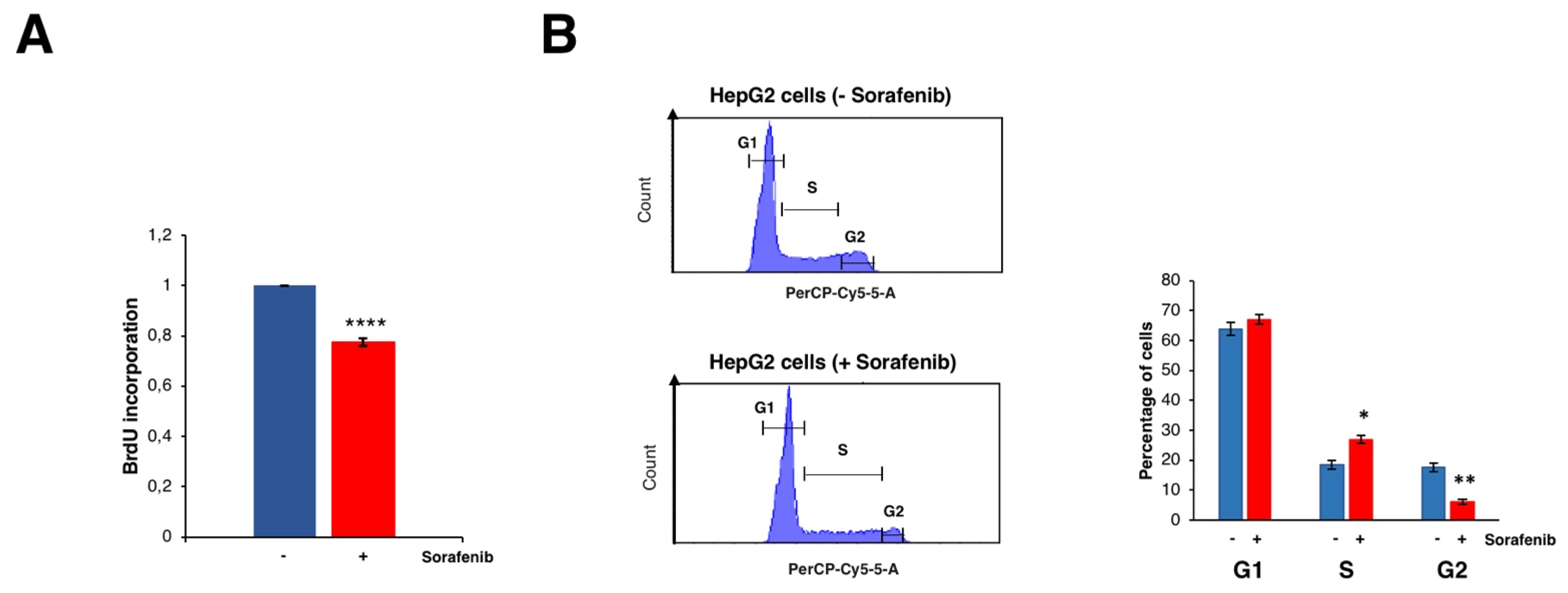
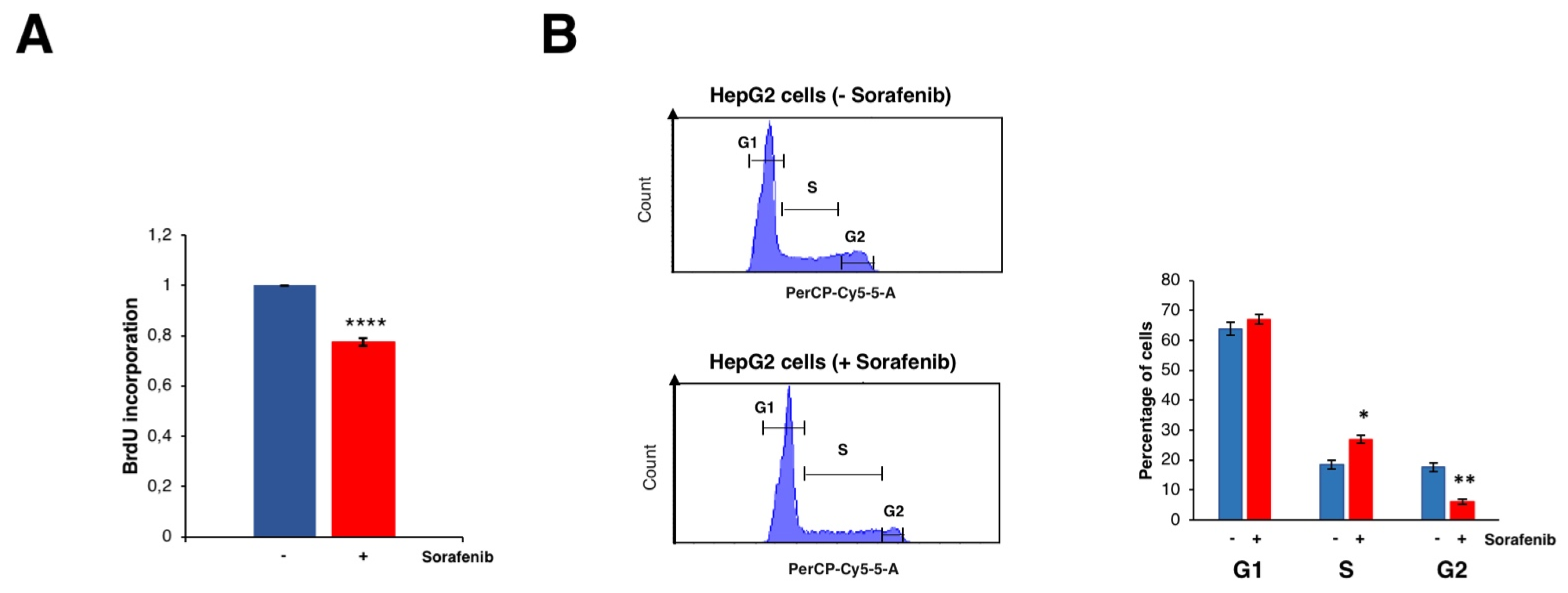
3.2. Sorafenib Inhibits Cell Growth
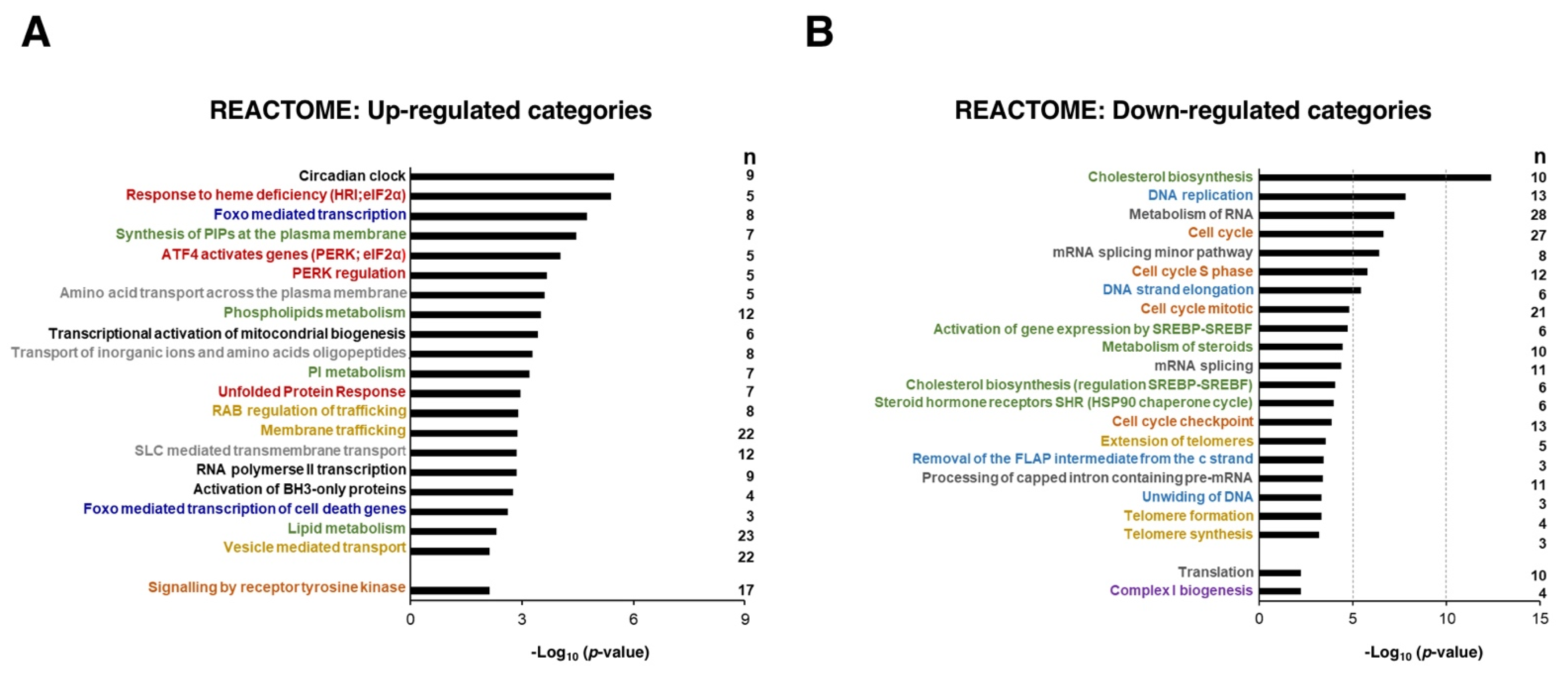
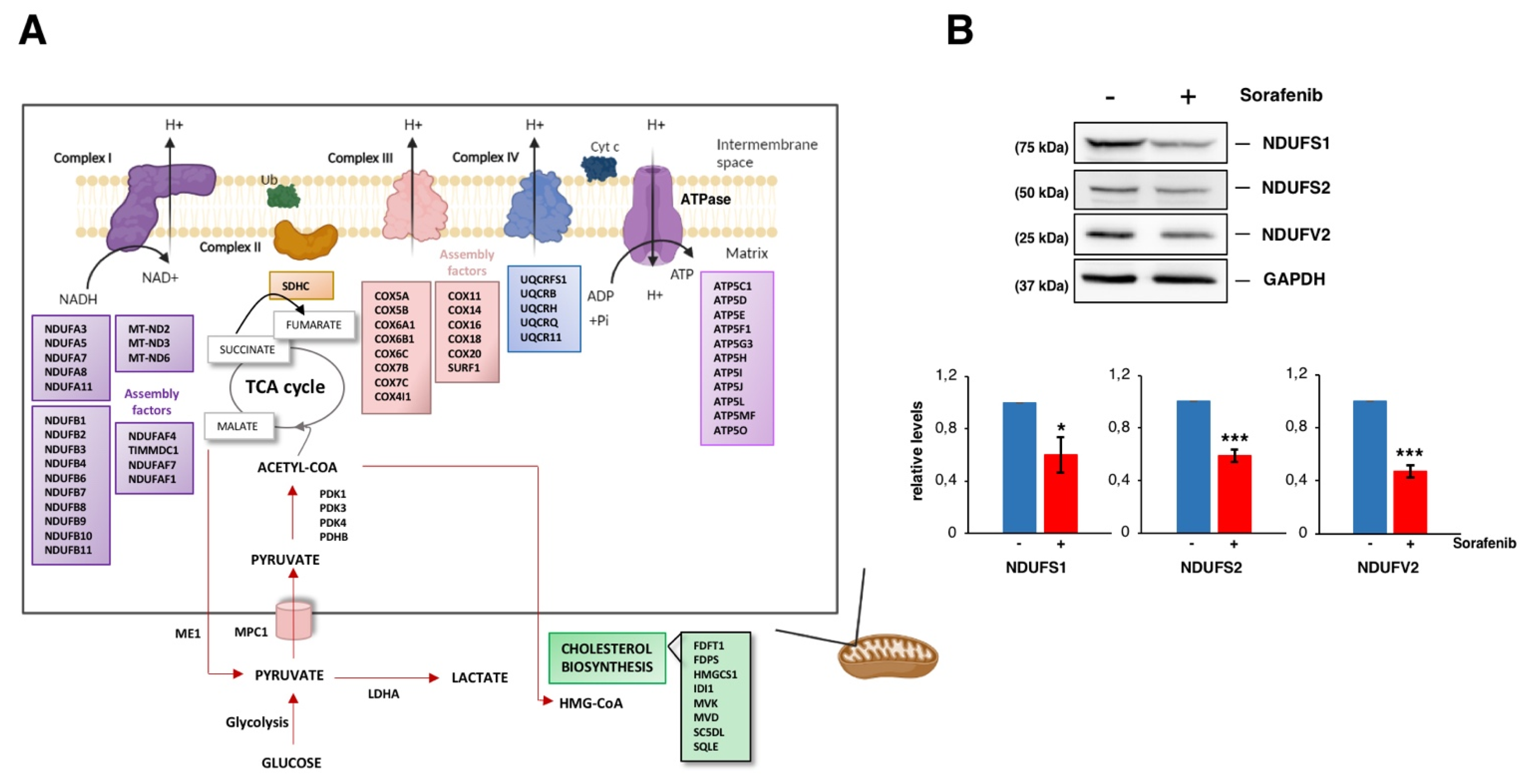
3.3. Sorafenib Downregulates Mitochondrial Functions, Especially the Oxidative Phosphorylation Pathway
3.4. Lipid Metabolism in Sorafenib-Treated Hepg2 Cells
3.5. Upregulation of the Circadian Clock in Sorafenib-Treated Hepg2 Cells
3.6. Sorafenib at the Global Transcriptomic Level
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Forner, A.; Llovet, J.M.; Bruix, J. Hepatocellular carcinoma. Lancet 2012, 379, 1245–1255. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. EASL clinical practice guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar]
- Rossi, L.; Zoratto, F.; Papa, A.; Iodice, F.; Minozzi, M.; Frati, L.; Tomao, S. Current approach in the treatment of hepatocellular carcinoma. World J. Gastrointest. Oncol. 2010, 2, 348–359. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Bruix, J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology 2008, 48, 1312–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Rahman, O.; Fouad, M. Sorafenib-based combination as a first line treatment for advanced hepatocellular carcinoma: A systematic review of the literature. Crit. Rev. Oncol. Hematol. 2014, 91, 1–8. [Google Scholar] [CrossRef]
- Reyes, R.; Wani, N.A.; Ghoshal, K.; Jacob, S.T.; Motiwala, T. Sorafenib and 2-deoxyglucose synergistically inhibit proliferation of both Sorafenib-sensitive and -resistant HCC cells by Inhibiting ATP production. Gene Expr. 2017, 17, 129–140. [Google Scholar] [CrossRef] [Green Version]
- Cervello, M.; Bachvarov, D.; Lampiasi, N.; Cusimano, A.; Azzolina, A.; McCubrey, J.A.; Montalto, G. Molecular mechanisms of sorafenib action in liver cancer cells. Cell Cycle 2012, 11, 2843–2855. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Cao, Y.; Chen, C.; Zhang, X.; McNabola, A.; Wilkie, D.; Wilhelm, S.; Lynch, M.; Carter, C. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006, 66, 11851–11858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asati, V.; Mahapatra, D.K.; Bharti, S.K. PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: Structural and pharmacological perspectives. Eur. J. Med. Chem. 2016, 109, 314–341. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Hernández, M.A.; González, R.; de la Rosa, A.J.; Gallego, P.; Ordoñez, R.; Navarro-Villarán, E.; Contreras, L.; Rodríguez-Arribas, M.; González-Gallego, J.; Álamo-Martínez, J.M.; et al. Molecular characterization of autophagic and apoptotic signaling induced by sorafenib in liver cancer cells. J. Cell. Physiol. 2018, 234, 692–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernando, J.; Sancho, P.; Fernández-Rodríguez, C.M.; Lledó, J.L.; Caja, L.; Campbell, J.S.; Fausto, N.; Fabregat, I. Sorafenib sensitizes hepatocellular carcinoma cells to physiological apoptotic stimuli. J. Cell. Physiol. 2012, 227, 1319–1325. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, A.; Ho, M. Role of sorafenib in the treatment of advanced hepatocellular carcinoma: An update. Hepatol. Res. 2013, 43, 147–154. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Hernández, M.A.; Chapresto-Garzón, R.; Cadenas, M.; Navarro-VillarÁn, E.; Negrete, M.; Gómez-Bravo, M.A.; Victor, V.M.; Padillo, F.J.; Muntané, J. Differential effectiveness of tyrosine kinase inhibitors in 2D/3D culture according to cell differentiation, p53 status and mitochondrial respiration in liver cancer cells. Cell Death Dis. 2020, 11, 339. [Google Scholar] [CrossRef]
- López-Terrada, D.; Gunaratne, P.H.; Adesina, A.M.; Pulliam, J.; Hoang, D.M.; Nguyen, Y.; Mistretta, T.A.; Margolin, J.; Finegold, M.J. Histologic subtypes of hepatoblastoma are characterized by differential canonical Wnt and Notch pathway activation in DLK+ precursors. Hum. Pathol. 2009, 40, 783–794. [Google Scholar] [CrossRef]
- Park, J.G.; Lee, J.H.; Kang, M.S.; Park, K.J.; Jeon, Y.M.; Lee, H.J.; Kwon, H.S.; Park, H.S.; Yeo, K.S.; Lee, K.U.; et al. Characterization of cell lines established from human hepatocellular carcinoma. Int. J. Cancer 1995, 62, 276–282. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef] [PubMed]
- Morine, M.J.; McMonagle, J.; Toomey, S.; Reynolds, C.M.; Moloney, A.P.; Gormley, I.C.; Gaora, P.O.; Roche, H.M. Bi-directional gene set enrichment and canonical correlation analysis identify key diet-sensitive pathways and biomarkers of metabolic syndrome. BMC Bioinform. 2010, 11, 499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micallef, L.; Rodgers, P. eulerAPE: Drawing area-proportional 3-Venn diagrams using ellipses. PLoS ONE 2014, 9, e101717. [Google Scholar]
- Molina-Ruiz, F.J.; González, R.; Rodríguez-Hernández, M.A.; Navarro-Villarán, E.; Padillo, F.J.; Muntané, J. Antitumoral activity of Sorafenib in hepatocellular carcinoma: Effects on cell survival and death pathways, cell metabolism reprogramming, and nitrosative and oxidative stress. Crit. Rev. Oncog. 2016, 21, 413–432. [Google Scholar] [CrossRef]
- Brüning, A.; Matsingou, C.; Brem, G.J.; Rahmeh, M.; Mylonas, I. Inhibin beta E is upregulated by drug-induced endoplasmic reticulum stress as a transcriptional target gene of ATF4. Toxicol. Appl. Pharmacol. 2012, 264, 300–304. [Google Scholar] [CrossRef]
- Phillips, D.J.; Woodruff, T.K. Inhibin: Actions and signalling. Growth Factors 2004, 22, 13–18. [Google Scholar] [CrossRef]
- Bernard, D.J.; Chapman, S.C.; Woodruff, T.K. Mechanisms of inhibin signal transduction. Recent Prog. Horm. Res. 2001, 56, 417–450. [Google Scholar] [CrossRef]
- Chen, H.F.; Chuang, H.C.; Tan, T.H. Regulation of Dual-Specificity Phosphatase (DUSP) ubiquitination and protein stability. Int. J. Mol. Sci. 2019, 20, 2668. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.; Chen, J.; Hai, T. The regulation of ATF3 gene expression by mitogen-activated protein kinases. Biochem. J. 2007, 401, 559–567. [Google Scholar] [CrossRef] [Green Version]
- Cai, D.; Huang, E.; Luo, B.; Yang, Y.; Zhang, F.; Liu, C.; Lin, Z.; Xie, W.B.; Wang, H. Nupr1/Chop signal axis is involved in mitochondrion-related endothelial cell apoptosis induced by methamphetamine. Cell Death Dis. 2016, 7, e2161. [Google Scholar] [CrossRef] [Green Version]
- Wolfgang, C.D.; Chen, B.P.; Martindale, J.L.; Holbrook, N.J.; Hai, T. gadd153/Chop10, a potential target gene of the transcriptional repressor ATF3. Mol. Cell. Biol. 1997, 17, 6700–6707. [Google Scholar] [CrossRef] [Green Version]
- Verfaillie, T.; Salazar, M.; Velasco, G.; Agostinis, P. Linking ER stress to sutophagy: Potential implications for cancer therapy. Int. J. Cell Biol. 2010, 2010, 930509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Brandizzi, F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 2013, 23, 547–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.E.; Hipp, M.S.; Bracher, A.; Hayer-Hartl, M.; Hartl, F.U. Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 2013, 82, 323–355. [Google Scholar] [CrossRef] [PubMed]
- Bollard, J.; Miguela, V.; Ruiz de Galarreta, M.; Venkatesh, A.; Bian, C.B.; Roberto, M.P.; Tovar, V.; Sia, D.; Molina-Sánchez, P.; Nguyen, C.B.; et al. Palbociclib (PD-0332991), a selective CDK4/6 inhibitor, restricts tumour growth in preclinical models of hepatocellular carcinoma. Gut 2017, 66, 1286–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smits, V.A.J.; Cabrera, E.; Freire, R.; Gillespie, D.A. Claspin-checkpoint adaptor and DNA replication factor. FEBS J. 2019, 286, 441–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maréchal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Krokan, H.E.; Bjørås, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef]
- Richly, H.; Schultheis, B.; Adamietz, I.A.; Kupsch, P.; Grubert, M.; Hilger, R.A.; Ludwig, M.; Brendel, E.; Christensen, O.; Strumberg, D. Combination of sorafenib and doxorubicin in patients with advanced hepatocellular carcinoma: Results from a phase I extension trial. Eur. J. Cancer 2009, 45, 579–587. [Google Scholar] [CrossRef]
- Yang, X.D.; Kong, F.E.; Qi, L.; Lin, J.X.; Yan, Q.; Loong, J.H.C.; Xi, S.Y.; Zhao, Y.; Zhang, Y.; Yuan, Y.F.; et al. PARP inhibitor Olaparib overcomes Sorafenib resistance through reshaping the pluripotent transcriptome in hepatocellular carcinoma. Mol. Cancer 2021, 20, 20. [Google Scholar] [CrossRef]
- Yu, W.; Gu, K.; Yu, Z.; Yuan, D.; He, M.; Ma, N.; Lai, S.; Zhao, J.; Ren, Z.; Zhang, X.; et al. Sorafenib potentiates irradiation effect in hepatocellular carcinoma in vitro and in vivo. Cancer Lett. 2013, 329, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, H.; Lieftink, C.; du Chatinier, A.; Gao, D.; Jin, G.; Jin, H.; Beijersbergen, R.L.; Qin, W.; Bernards, R. CDK12 inhibition mediates DNA damage and is synergistic with sorafenib treatment in hepatocellular carcinoma. Gut 2020, 69, 727–736. [Google Scholar] [CrossRef] [PubMed]
- In der Stroth, L.; Tharehalli, U.; Günes, C.; Lechel, A. Telomeres and Telomerase in the Development of Liver Cancer. Cancers 2020, 12, 2048. [Google Scholar] [CrossRef]
- Zhou, T.; Lv, X.; Guo, X.; Ruan, B.; Liu, D.; Ding, R.; Gao, Y.; Ding, J.; Dou, K.; Chen, Y. RACK1 modulates apoptosis induced by sorafenib in HCC cells by interfering with the IRE1/XBP1 axis. Oncol. Rep. 2015, 33, 3006–3014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.H.; Ding, Z.B.; Zhou, J.; Hui, B.; Shi, G.M.; Ke, A.W.; Wang, X.Y.; Dai, Z.; Peng, Y.F.; Gu, C.Y.; et al. Targeting autophagy enhances sorafenib lethality for hepatocellular carcinoma via ER stress-related apoptosis. Autophagy 2011, 7, 1159–1172. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Xie, H.; Zhao, J.; Wang, W.; Hou, H.; Zhang, B.; Zhou, D.; Geng, X. eIF4E-eIF4G complex inhibition synergistically enhances the effect of sorafenib in hepatocellular carcinoma. Anticancer. Drugs 2021, 32, 822–828. [Google Scholar] [CrossRef]
- Honma, Y.; Shimizu, S.; Takehara, T.; Harada, M. Sorafenib enhances proteasome inhibitor-induced cell death via inactivation of Akt and stress-activated protein kinases. J. Gastroenterol. 2014, 49, 517–526. [Google Scholar] [CrossRef]
- Jiang, C.; Xu, R.; Li, X.X.; Zhou, Y.F.; Xu, X.Y.; Yang, Y.; Wang, H.Y.; Zheng, X.F.S. Sorafenib and Carfilzomib synergistically inhibit the proliferation, survival, and metastasis of hepatocellular carcinoma. Mol. Cancer Ther. 2018, 17, 2610–2621. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.F.; Yu, H.C.; Liu, T.H.; Lee, S.S.; Chen, P.J.; Cheng, A.L. Synergistic interactions between sorafenib and bortezomib in hepatocellular carcinoma involve PP2A-dependent Akt inactivation. J. Hepatol. 2010, 52, 88–95. [Google Scholar] [CrossRef]
- Rodríguez-Hernández, M.A.; de la Cruz-Ojeda, P.; López-Grueso, M.J.; Navarro-Villarán, E.; Requejo-Aguilar, R.; Castejón-Vega, B.; Negrete, M.; Gallego, P.; Vega-Ochoa, A.; Victor, V.M.; et al. Integrated molecular signaling involving mitochondrial dysfunction and alteration of cell metabolism induced by tyrosine kinase inhibitors in cancer. Redox Biol. 2020, 36, 101510. [Google Scholar] [CrossRef]
- Fiume, L.; Manerba, M.; Vettraino, M.; Di Stefano, G. Effect of sorafenib on the energy metabolism of hepatocellular carcinoma cells. Eur. J. Pharmacol. 2011, 670, 39–43. [Google Scholar] [CrossRef]
- Tesori, V.; Piscaglia, A.C.; Samengo, D.; Barba, M.; Bernardini, C.; Scatena, R.; Pontoglio, A.; Castellini, L.; Spelbrink, J.N.; Maulucci, G.; et al. The multikinase inhibitor Sorafenib enhances glycolysis and synergizes with glycolysis blockade for cancer cell killing. Sci. Rep. 2015, 5, 9149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Liu, Z.; Bunker, E.; Ramírez, A.; Lee, S.; Peng, Y.; Tan, A.C.; Eckhardt, S.G.; Chapnick, D.A.; Liu, X. Sorafenib targets the mitochondrial electron transport chain complexes and ATP synthase to activate the PINK1-Parkin pathway and modulate cellular drug response. J. Biol. Chem. 2017, 292, 15105–15120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Hernández, M.A.; de la Cruz-Ojeda, P.; Gallego, P.; Navarro-Villarán, E.; Stankova, P.; del Campo, J.A.; Kucera, O.; Elkalaf, M.; Maseko, T.E.; Cervinkova, Z.; et al. Dose-dependent regulation of mitochondrial function and cell death pathway by sorafenib in liver cancer cells. Biochem. Pharmacol. 2020, 176, 113902. [Google Scholar] [CrossRef]
- Paech, F.; Mingard, C.; Grunig, D.; Abegg, V.F.; Bouitbir, J.; Krähenbühl, S. Mechanisms of mitochondrial toxicity of the kinase inhibitors ponatinib, regorafenib and sorafenib in human hepatic HepG2 cells. Toxicology 2018, 395, 34–44. [Google Scholar] [CrossRef]
- Li, Y.; Xia, J.; Shao, F.; Zhou, Y.; Yu, J.; Wu, H.; Du, J.; Ren, X. Sorafenib induces mitochondrial dysfunction and exhibits synergistic effect with cysteine depletion by promoting HCC cells ferroptosis. Biochem. Biophys. Res. Commun. 2021, 534, 877–884. [Google Scholar] [CrossRef]
- Fan, G.; Wei, X.; Xu, X. Is the era of sorafenib over? A review of the literature. Ther. Adv. Med. Oncol. 2020, 12, 1758835920927602. [Google Scholar] [CrossRef]
- Liu, G.; Kuang, S.; Cao, R.; Wang, J.; Peng, Q.; Sun, C. Sorafenib kills liver cancer cells by disrupting SCD1-mediated synthesis of monounsaturated fatty acids via the ATP-AMPK-mTOR-SREBP1 signaling pathway. FASEB J. 2019, 33, 10089–10103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, F.; Feng, F.; Wang, L.; Wang, X.; Li, Z.; Cao, Y. SREBP-1 inhibitor Betulin enhances the antitumor effect of Sorafenib on hepatocellular carcinoma via restricting cellular glycolytic activity. Cell Death Dis. 2019, 10, 672. [Google Scholar] [CrossRef] [Green Version]
- Eun, H.S.; Cho, S.Y.; Lee, B.S.; Seong, I.O.; Kim, K.H. Profiling cytochrome P450 family 4 gene expression in human hepatocellular carcinoma. Mol. Med. Rep. 2018, 18, 4865–4876. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.C.; Chowdhury, K.; Huang, M.; Kim, H.G. Signal transduction and molecular regulation in fatty liver disease. Antioxid. Redox Signal. 2021, 35, 689–717. [Google Scholar] [CrossRef] [PubMed]
- Che, L.; Chi, W.; Qiao, Y.; Zhang, J.; Song, X.; Liu, Y.; Li, L.; Jia, J.; Pilo, M.G.; Wang, J.; et al. Cholesterol biosynthesis supports the growth of hepatocarcinoma lesions depleted of fatty acid synthase in mice and humans. Gut 2020, 69, 177–186. [Google Scholar] [CrossRef] [Green Version]
- Di Paolo, G.; De Camilli, P. Phosphoinositides in cell regulation and membrane dynamics. Nature 2006, 443, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Reppert, S.M.; Weaver, D.R. Coordination of circadian timing in mammals. Nature 2002, 418, 935–941. [Google Scholar] [CrossRef]
- Bass, J.; Takahashi, J.S. Circadian integration of metabolism and energetics. Science 2010, 330, 1349–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinke, H.; Asher, G. Circadian clock control of liver metabolic functions. Gastroenterology 2016, 150, 574–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobi, D.; Liu, S.; Burkewitz, K.; Kory, N.; Knudsen, N.H.; Alexander, R.K.; Unluturk, U.; Li, X.; Kong, X.; Hyde, A.L.; et al. Hepatic Bmal1 regulates rhythmic mitochondrial dynamics and promotes metabolic fitness. Cell Metab. 2015, 22, 709–720. [Google Scholar] [CrossRef] [Green Version]
- Sahar, S.; Sassone-Corsi, P. Metabolism and cancer: The circadian clock connection. Nat. Rev. Cancer 2009, 9, 886–896. [Google Scholar] [CrossRef]
- Fuchs, B.C.; Fujii, T.; Dorfman, J.D.; Goodwin, J.M.; Zhu, A.X.; Lanuti, M.; Tanabe, K.K. Epithelial-to-mesenchymal transition and integrin-linked kinase mediate sensitivity to epidermal growth factor receptor inhibition in human hepatoma cells. Cancer Res. 2008, 68, 2391–2399. [Google Scholar] [CrossRef] [Green Version]
- Hirschfield, H.; Bian, C.B.; Higashi, T.; Nakagawa, S.; Zeleke, T.Z.; Nair, V.D.; Fuchs, B.C.; Hoshida, Y. In Vitro modeling of hepatocellular carcinoma molecular subtypes for anti-cancer drug assessment. Exp. Mol. Med. 2018, 50, e419. [Google Scholar] [CrossRef]
- Clayton, N.S.; Ridley, A.J. Targeting Rho GTPase signaling networks in cancer. Front. Cell Dev. Biol. 2020, 8, 222. [Google Scholar] [CrossRef]
- Bruix, J.; Gores, G.J.; Mazzaferro, V. Hepatocellular carcinoma: Clinical frontiers and perspectives. Gut 2014, 63, 844–855. [Google Scholar] [CrossRef] [PubMed]
- Paech, F.; Bouitbir, J.; Krähenbaühl, S. Hepatocellular toxicity associated with tyrosine kinase inhibitors: Mitochondrial damage and inhibition of glycolysis. Front. Pharmacol. 2017, 8, 367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asghar, U.; Meyer, T. Are there opportunities for chemotherapy in the treatment of hepatocellular cancer? J. Hepatol. 2012, 56, 686–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regan-Fendt, K.; Li, D.; Reyes, R.; Yu, L.; Wani, N.A.; Hu, P.; Jacob, S.T.; Ghoshal, K.; Payne, P.R.O.; Motiwala, T. Transcriptomics-based drug repurposing approach identifies novel drugs against Sorafenib-resistant hepatocellular carcinoma. Cancers 2020, 12, 2730. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Mayerle, J.; Ziesch, A.; Reiter, F.P.; Gerbes, A.L.; De Toni, E.N. The PI3K inhibitor copanlisib synergizes with sorafenib to induce cell death in hepatocellular carcinoma. Cell. Death Discov. 2019, 5, 86. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Chen, J.; Li, J.; Zheng, Y.; Zhong, X.; Huang, S.; Chen, B.; Peng, B.; Zou, X.; Chen, X. Pristimerin synergistically sensitizes conditionally reprogrammed patient derived-primary hepatocellular carcinoma cells to sorafenib through endoplasmic reticulum stress and ROS generation by modulating Akt/FoxO1/p27(kip1) signaling pathway. Phytomedicine 2021, 86, 153563. [Google Scholar] [CrossRef]
- Şirin, N.; Elmas, L.; Secme, M.; Dodurga, Y. Investigation of possible effects of apigenin, sorafenib and combined applications on apoptosis and cell cycle in hepatocellular cancer cells. Gene 2020, 737, 144428. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Contreras, L.; Rodríguez-Gil, A.; Muntané, J.; de la Cruz, J. Broad Transcriptomic Impact of Sorafenib and Its Relation to the Antitumoral Properties in Liver Cancer Cells. Cancers 2022, 14, 1204. https://doi.org/10.3390/cancers14051204
Contreras L, Rodríguez-Gil A, Muntané J, de la Cruz J. Broad Transcriptomic Impact of Sorafenib and Its Relation to the Antitumoral Properties in Liver Cancer Cells. Cancers. 2022; 14(5):1204. https://doi.org/10.3390/cancers14051204
Chicago/Turabian StyleContreras, Laura, Alfonso Rodríguez-Gil, Jordi Muntané, and Jesús de la Cruz. 2022. "Broad Transcriptomic Impact of Sorafenib and Its Relation to the Antitumoral Properties in Liver Cancer Cells" Cancers 14, no. 5: 1204. https://doi.org/10.3390/cancers14051204
APA StyleContreras, L., Rodríguez-Gil, A., Muntané, J., & de la Cruz, J. (2022). Broad Transcriptomic Impact of Sorafenib and Its Relation to the Antitumoral Properties in Liver Cancer Cells. Cancers, 14(5), 1204. https://doi.org/10.3390/cancers14051204