
Superoxide Dismutase-3 Downregulates Laminin α5 Expression in Tumor Endothelial Cells via the Inhibition of Nuclear Factor Kappa B Signaling

 , ,
, , 
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines, Transfection and Stimulation
2.2. Tumor Samples
2.3. RT-qPCR
2.4. Microarray Analysis
2.5. IPA and GSEA Analyses
2.6. Immunofluorescence Staining
2.7. Immunoblot and Chase Experiments
2.8. SOD Activity Assays
2.9. NF-κB Promoter Assay
2.10. ELISA and Flow Cytometry
2.11. Bioinformatic Analyses
2.12. Statistical Analyses
3. Results
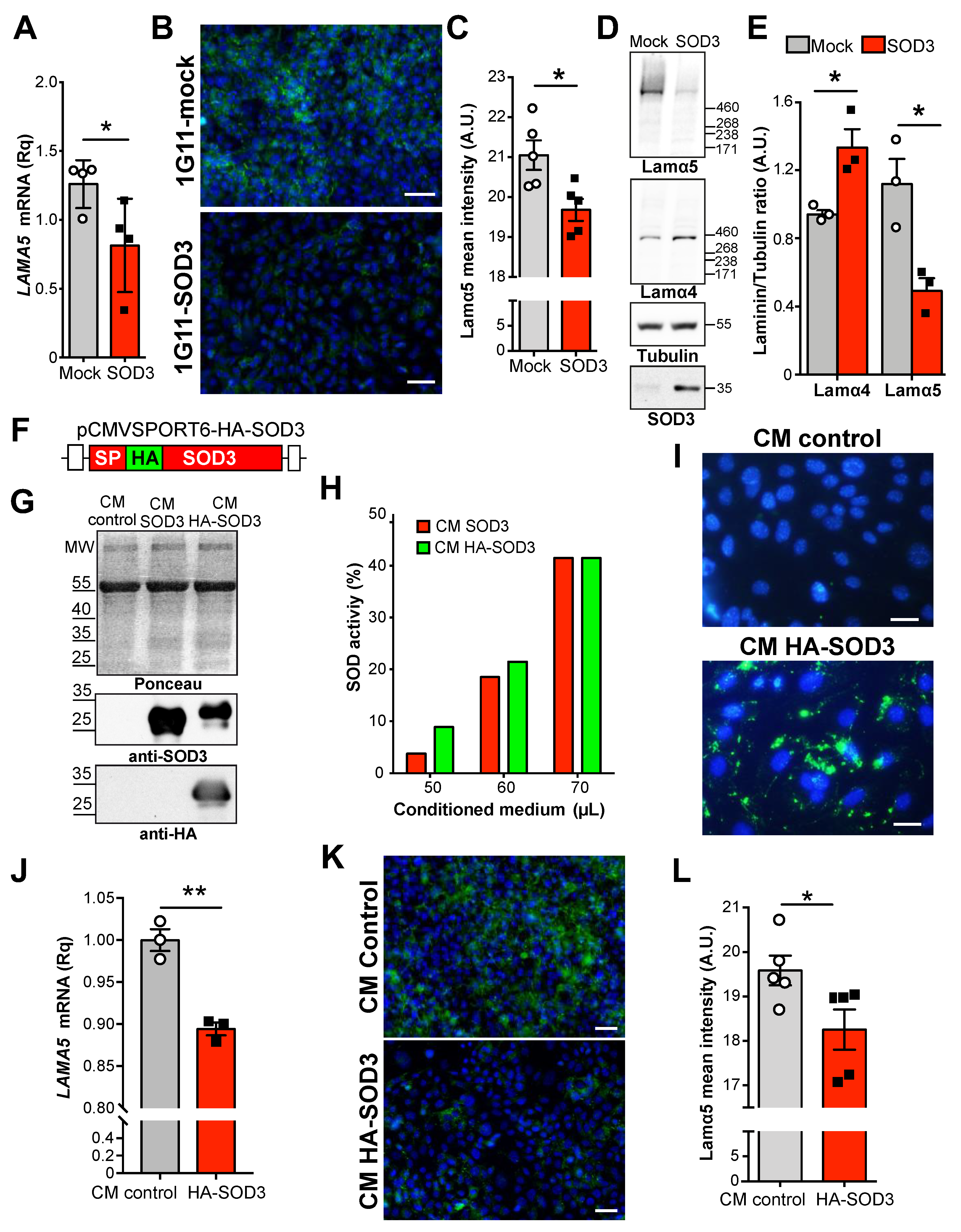
3.1. SOD3 Downregulates Laminin α5 Expression in the Tumor Vasculature
3.2. Extracellular SOD3 Represses LAMA5 Transcription in Endothelial Cells
3.3. LAMA5 Transcription Is Not Affected by SOD1
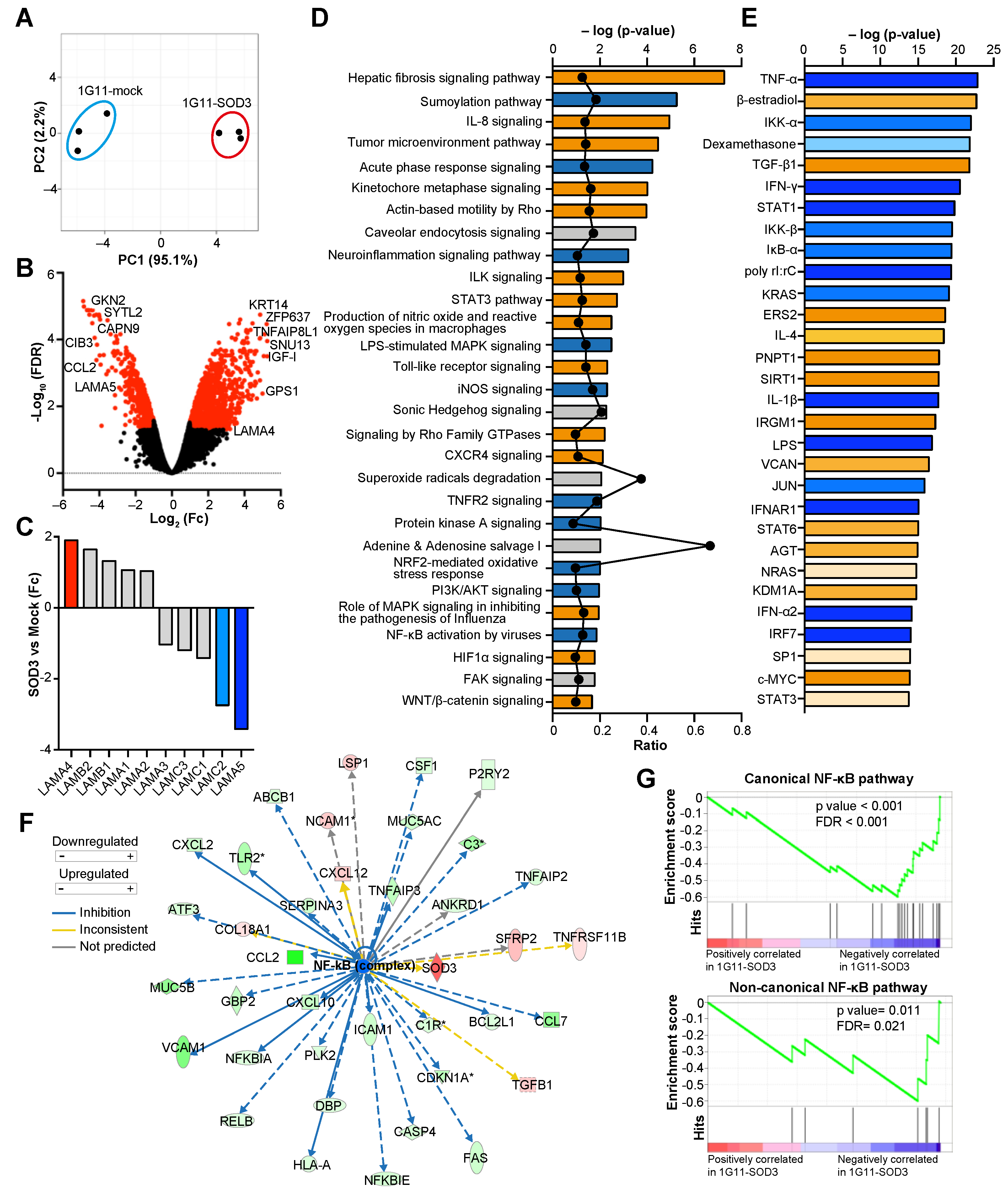
3.4. SOD3 Modifies the Transcriptional Program of Endothelial Cells
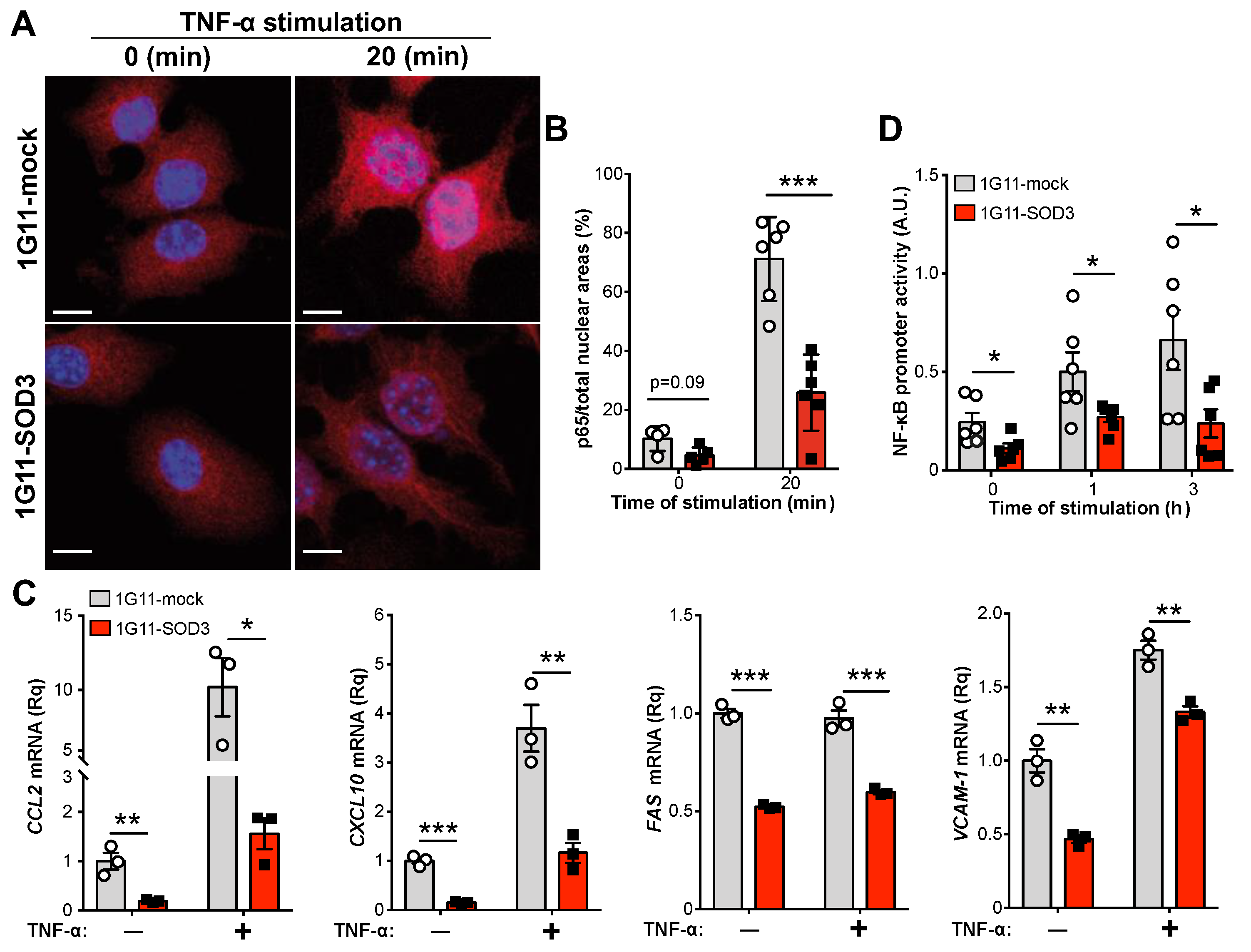
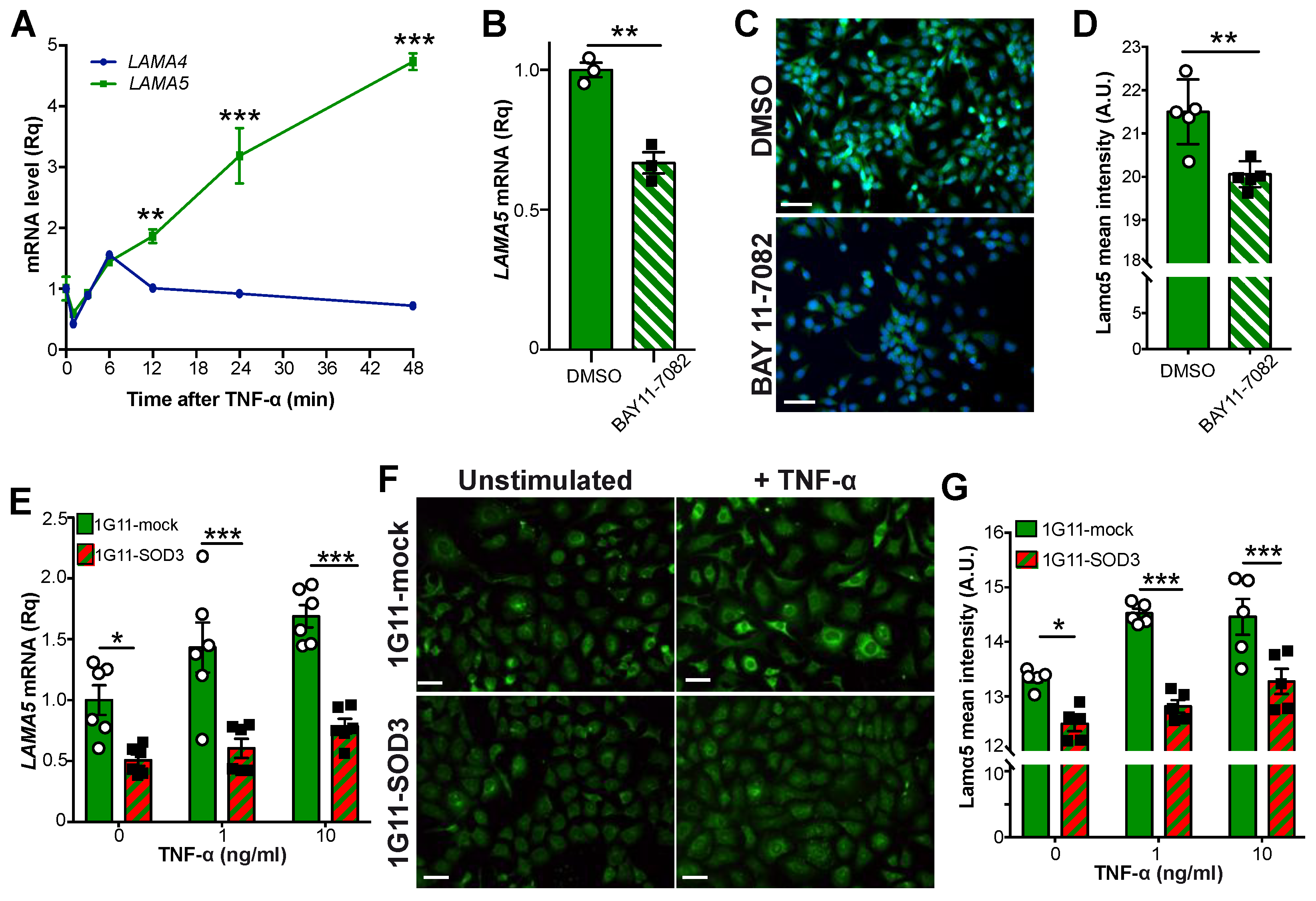
3.5. SOD3 Attenuates the NF-κB Pathway in Endothelial Cells
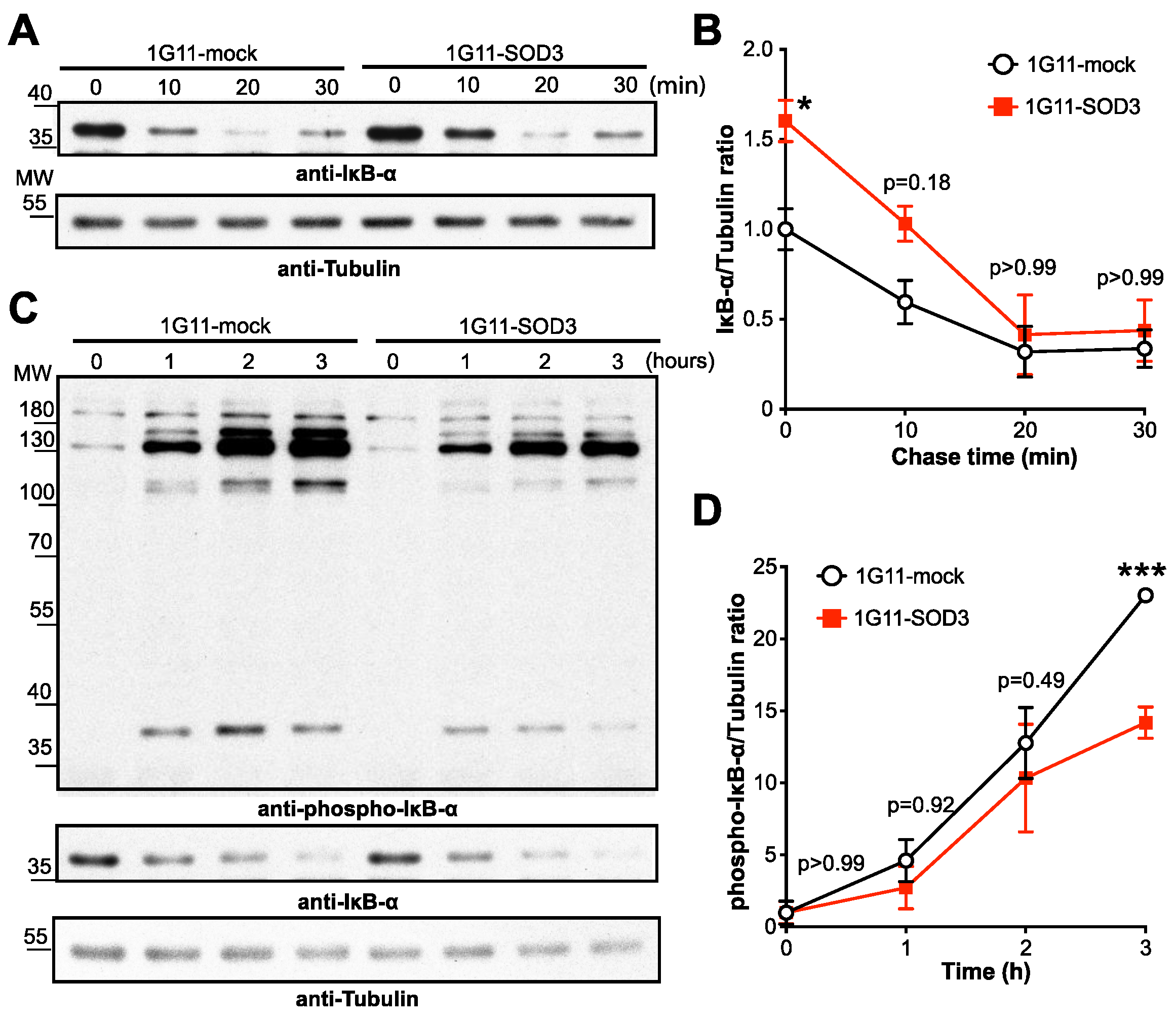
3.6. SOD3 Inhibits the NF-κB Pathway by Stabilizing IκBα
3.7. SOD3 Regulates LAMA5 Expression via the NF-κB Pathway
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Song, Y.; Fu, Y.; Xie, Q.; Zhu, B.; Wang, J.; Zhang, B. Anti-angiogenic Agents in Combination with Immune Checkpoint Inhibitors: A Promising Strategy for Cancer Treatment. Front. Immunol. 2020, 11, 1956. [Google Scholar] [CrossRef] [PubMed]
- Ganss, R. Tumour vessel remodelling: New opportunities in cancer treatment. Vasc. Biol. 2020, 2, R35–R43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintela-Fandino, M.; Holgado, E.; Manso, L.; Morales, S.; Bermejo, B.; Colomer, R.; Apala, J.V.; Blanco, R.; Muñoz, M.; Caleiras, E.; et al. Immuno-priming durvalumab with bevacizumab in HER2-negative advanced breast cancer: A pilot clinical trial. Breast Cancer Res. 2020, 22, 124. [Google Scholar] [CrossRef]
- Yang, T.; Xiao, H.; Liu, X.; Wang, Z.; Zhang, Q.; Wei, N.; Guo, X. Vascular Normalization: A New Window Opened for Cancer Therapies. Front. Oncol. 2021, 11, 719836. [Google Scholar] [CrossRef]
- Matuszewska, K.; Pereira, M.; Petrik, D.; Lawler, J.; Petrik, J. Normalizing Tumor Vasculature to Reduce Hypoxia, Enhance Perfusion, and Optimize Therapy Uptake. Cancers 2021, 13, 4444. [Google Scholar] [CrossRef] [PubMed]
- Nagy, J.A.; Chang, S.-H.; Dvorak, A.M.; Dvorak, H.F. Why are tumour blood vessels abnormal and why is it important to know? Br. J. Cancer 2009, 100, 865–869. [Google Scholar] [CrossRef]
- Goel, S.; Wong, A.H.-K.; Jain, R.K. Vascular Normalization as a Therapeutic Strategy for Malignant and Nonmalignant Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006486. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427. [Google Scholar] [CrossRef]
- Zhou, W.; Yang, L.; Nie, L.; Lin, H. Unraveling the molecular mechanisms between inflammation and tumor angiogenesis. Am. J. Cancer Res. 2021, 11, 301–317. [Google Scholar]
- Dorrington, M.G.; Fraser, I.D.C.; Dorrington, M.G.; Fraser, I.D.C. NF-κB Signaling in Macrophages: Dynamics, Crosstalk, and Signal Integration. Front. Immunol. 2019, 10, 705. [Google Scholar] [CrossRef]
- Mussbacher, M.; Salzmann, M.; Brostjan, C.; Hoesel, B.; Schoergenhofer, C.; Datler, H.; Hohensinner, P.; Basílio, J.; Petzelbauer, P.; Assinger, A.; et al. Cell Type-Specific Roles of NF-κB Linking Inflammation and Thrombosis. Front. Immunol. 2019, 10, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moorthy, A.K.; Savinova, O.V.; Ho, J.Q.; Wang, V.Y.-F.; Vu, D.; Ghosh, G. The 20S proteasome processes NF-κB1 p105 into p50 in a translation-independent manner. EMBO J. 2006, 25, 1945–1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [Green Version]
- Napetschnig, J.; Wu, H. Molecular Basis of NF-κB Signaling. Annu. Rev. Biophys. 2013, 42, 443–468. [Google Scholar] [CrossRef] [Green Version]
- Kisseleva, T.; Song, L.; Vorontchikhina, M.; Feirt, N.; Kitajewski, J.; Schindler, C. NF-κB regulation of endothelial cell function during LPS-induced toxemia and cancer. J. Clin. Investig. 2006, 116, 2955–2963. [Google Scholar] [CrossRef]
- Denk, A.; Goebeler, M.; Schmid, S.; Berberich, I.; Ritz, O.; Lindemann, D.; Ludwig, S.; Wirth, T. Activation of NF-κB via the IκB Kinase Complex Is Both Essential and Sufficient for Proinflammatory Gene Expression in Primary Endothelial Cells. J. Biol. Chem. 2001, 276, 28451–28458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhab, L.J.B.; Saber-Ayad, M.M.; Al-Hakm, R.; Nair, V.A.; Paliogiannis, P.; Pintus, G.; Abdel-Rahman, W.M. Chronic Inflammation and Cancer: The Role of Endothelial Dysfunction and Vascular Inflammation. Curr. Pharm. Des. 2021, 27, 2156–2169. [Google Scholar] [CrossRef]
- Hallmann, R.; Hannocks, M.-J.; Song, J.; Zhang, X.; Di Russo, J.; Luik, A.-L.; Burmeister, M.; Gerwien, H.; Sorokin, L. The role of basement membrane laminins in vascular function. Int. J. Biochem. Cell Biol. 2020, 127, 105823. [Google Scholar] [CrossRef]
- Di Russo, J.; Luik, A.; Yousif, L.; Budny, S.; Oberleithner, H.; Hofschröer, V.; Klingauf, J.; van Bavel, E.; Bakker, E.; Hellstrand, P.; et al. Endothelial basement membrane laminin 511 is essential for shear stress response. EMBO J. 2016, 36, 183–201. [Google Scholar] [CrossRef]
- Baluk, P.; Morikawa, S.; Haskell, A.; Mancuso, M.; McDonald, D.M. Abnormalities of Basement Membrane on Blood Vessels and Endothelial Sprouts in Tumors. Am. J. Pathol. 2003, 163, 1801–1815. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Rey, D.; Carmona-Rodríguez, L.; Fernández-Aceñero, M.J.; Mira, E.; Mañes, S. Extracellular Superoxide Dismutase, the Endothelial Basement Membrane, and the WNT Pathway: New Players in Vascular Normalization and Tumor Infiltration by T-Cells. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Pozzi, A.; Yurchenco, P.; Iozzo, R.V. The nature and biology of basement membranes. Matrix Biol. 2017, 57–58, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousif, L.F.; Di Russo, J.; Sorokin, L. Laminin isoforms in endothelial and perivascular basement membranes. Cell Adhes. Migr. 2013, 7, 101–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, M.; Sekiguchi, K. Molecular Basis of Laminin—Integrin Interactions. Curr. Top. Membr. 2015, 76, 197–229. [Google Scholar] [CrossRef] [PubMed]
- Arimori, T.; Miyazaki, N.; Mihara, E.; Takizawa, M.; Taniguchi, Y.; Cabañas, C.; Sekiguchi, K.; Takagi, J. Structural mechanism of laminin recognition by integrin. Nat. Commun. 2021, 12, 4012. [Google Scholar] [CrossRef]
- Sixt, M.; Engelhardt, B.; Pausch, F.; Hallmann, R.; Wendler, O.; Sorokin, L.M. Endothelial Cell Laminin Isoforms, Laminins 8 and 10, Play Decisive Roles in T Cell Recruitment across the Blood-Brain Barrier in Experimental Autoimmune Encephalomyelitis. J. Cell Biol. 2001, 153, 933–946. [Google Scholar] [CrossRef]
- Warren, K.J.; Iwami, D.; Harris, D.; Bromberg, J.S.; Burrell, B.E. Laminins affect T cell trafficking and allograft fate. J. Clin. Investig. 2014, 124, 2204–2218. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Ivars, F.; Anderson, P.; Hallmann, R.; Vestweber, D.; Nilsson, P.; Robenek, H.; Tryggvason, K.; Song, J.; Korpos, E.; et al. Endothelial basement membrane laminin α5 selectively inhibits T lymphocyte extravasation into the brain. Nat. Med. 2009, 15, 519–527. [Google Scholar] [CrossRef]
- Kenne, E.; Soehnlein, O.; Genové, G.; Rotzius, P.; Eriksson, E.E.; Lindbom, L. Immune cell recruitment to inflammatory loci is impaired in mice deficient in basement membrane protein laminin α4. J. Leukoc. Biol. 2010, 88, 523–528. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y.; Song, J.; Gerwien, H.; Chuquisana, O.; Chashchina, A.; Denz, C.; Sorokin, L. The endothelial basement membrane acts as a checkpoint for entry of pathogenic T cells into the brain. J. Exp. Med. 2020, 217, e20191339. [Google Scholar] [CrossRef]
- Song, J.; Zhang, X.; Buscher, K.; Wang, Y.; Wang, H.; Di Russo, J.; Li, L.; Lütke-Enking, S.; Zarbock, A.; Stadtmann, A.; et al. Endothelial basement membrane laminin 511 contributes to endothelial junctional tightness and thereby inhibits leukocyte transmigration. Cell Rep. 2017, 18, 1256–1269. [Google Scholar] [CrossRef] [Green Version]
- Carmona-Rodríguez, L.; Martínez-Rey, D.; Fernández-Aceñero, M.J.; González-Martín, A.; Cabezas, M.P.; Rodriguez-Rodriguez, N.; Pérez-Villamil, B.; Sáez, M.E.; Díaz-Rubio, E.; Mira, E.; et al. SOD3 induces a HIF-2α-dependent program in endothelial cells that provides a selective signal for tumor infiltration by T cells. J. Immunother. Cancer 2020, 8, e000432. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Rodríguez, L.; Martínez-Rey, D.; Mira, E.; Mañes, S. SOD3 boosts T cell infiltration by normalizing the tumor endothelium and inducing laminin-α4. OncoImmunology 2020, 9, 1794163. [Google Scholar] [CrossRef] [PubMed]
- Petersen, S.V.; Olsen, D.A.; Kenney, J.M.; Oury, T.D.; Valnickova, Z.; Thøgersen, I.B.; Crapo, J.D.; Enghild, J.J. The high concentration of Arg213→Gly extracellular superoxide dismutase (EC-SOD) in plasma is caused by a reduction of both heparin and collagen affinities. Biochem. J. 2005, 385, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Piper, R.; Richardson, S.; Watanabe, Y.; Patel, P.; Heistad, D.D. Endocytosis of Extracellular Superoxide Dismutase into Endothelial Cells: Role of the heparin-binding domain. Arter. Thromb. Vasc. Biol. 2006, 26, 1985–1990. [Google Scholar] [CrossRef] [Green Version]
- O’Leary, B.; Carroll, R.; Steers, G.; Hrabe, J.; Domann, F.; Cullen, J. Impact of EcSOD Perturbations in Cancer Progression. Antioxidants 2021, 10, 1219. [Google Scholar] [CrossRef]
- Mira, E.; Carmona-Rodríguez, L.; Pérez-Villamil, B.; Casas, J.; Fernández-Aceñero, M.J.; Martinez-Rey, D.; Martín-González, P.; Heras-Murillo, I.; Cabezas, M.P.; Tardáguila, M.; et al. SOD3 improves the tumor response to chemotherapy by stabilizing endothelial HIF-2α. Nat. Commun. 2018, 9, 575. [Google Scholar] [CrossRef] [Green Version]
- González-Martín, A.; Gómez, L.; Lustgarten, J.; Mira, E.; Mañes, S.; Rechsteiner, M.P.; von Teichman, A.; Nowicka, A.; Sulser, T.; Schraml, P.; et al. Maximal T Cell-Mediated Antitumor Responses Rely upon CCR5 Expression in Both CD4+ and CD8+ T Cells. Cancer Res. 2011, 71, 5455–5466. [Google Scholar] [CrossRef] [Green Version]
- Stevens, J.C.; Chia, R.; Hendriks, W.T.; Bros-Facer, V.; Van Minnen, J.; Martin, J.E.; Jackson, G.S.; Greensmith, L.; Schiavo, G.; Fisher, E.M.C. Modification of Superoxide Dismutase 1 (SOD1) Properties by a GFP Tag—Implications for Research into Amyotrophic Lateral Sclerosis (ALS). PLoS ONE 2010, 5, e9541. [Google Scholar] [CrossRef]
- Bolstad, B.; Irizarry, R.; Astrand, M.; Speed, T. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Smyth, G. Limma: Linear models for microarray data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Gentleman, R., Carey, V., Dudoit, S., Irizarry, R., Huber, W., Eds.; Springer: New York, NY, USA, 2005; pp. 397–420. [Google Scholar]
- Benjamini, Y.; Drai, D.; Elmer, G.; Kafkafi, N.; Golani, I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 2001, 125, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Hannocks, M.-J.; Pizzo, M.; Huppert, J.; Deshpande, T.; Abbott, N.J.; Thorne, R.G.; Sorokin, L. Molecular characterization of perivascular drainage pathways in the murine brain. J. Cereb. Blood Flow Metab. 2017, 38, 669–686. [Google Scholar] [CrossRef]
- Lánczky, A.; Győrffy, B. Web-Based Survival Analysis Tool Tailored for Medical Research (KMplot): Development and Implementation. J. Med. Internet Res. 2021, 23, e27633. [Google Scholar] [CrossRef]
- Gordon-Weeks, A.; Lim, S.Y.; Yuzhalin, A.; Lucotti, S.; Vermeer, J.A.; Jones, K.; Chen, J.; Muschel, R.J. Tumour-Derived Laminin α5 (LAMA5) Promotes Colorectal Liver Metastasis Growth, Branching Angiogenesis and Notch Pathway Inhibition. Cancers 2019, 11, 630. [Google Scholar] [CrossRef] [Green Version]
- Pouliot, N.; Kusuma, N. Laminin-511. Cell Adhes. Migr. 2013, 7, 142–149. [Google Scholar] [CrossRef] [Green Version]
- Laurila, J.P.; Laatikainen, L.E.; Castellone, M.D.; Laukkanen, M.O. SOD3 Reduces Inflammatory Cell Migration by Regulating Adhesion Molecule and Cytokine Expression. PLoS ONE 2009, 4, e5786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Froio, R.M.; Sciuto, T.E.; Dvorak, A.M.; Alon, R.; Luscinskas, F.W. ICAM-1 regulates neutrophil adhesion and transcellular migration of TNF-α-activated vascular endothelium under flow. Blood 2005, 106, 584–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abadier, M.; Jahromi, N.H.; Alves, L.C.; Boscacci, R.; Vestweber, D.; Barnum, S.; Deutsch, U.; Engelhardt, B.; Lyck, R. Cell surface levels of endothelial ICAM-1 influence the transcellular or paracellular T-cell diapedesis across the blood-brain barrier. Eur. J. Immunol. 2015, 45, 1043–1058. [Google Scholar] [CrossRef] [PubMed]
- Schoppmeyer, R.; van Steen, A.C.; Kempers, L.; Timmerman, A.L.; Nolte, M.A.; Hombrink, P.; van Buul, J.D. The endothelial diapedesis synapse regulates transcellular migration of human T lymphocytes in a CX3CL1- and SNAP23-dependent manner. Cell Rep. 2022, 38, 110243. [Google Scholar] [CrossRef]
- Tak, L.-J.; Kim, H.-Y.; Ham, W.-K.; Agrahari, G.; Seo, Y.; Yang, J.; An, E.-J.; Bang, C.; Lee, M.; Kim, H.-S.; et al. Superoxide Dismutase 3-Transduced Mesenchymal Stem Cells Preserve Epithelial Tight Junction Barrier in Murine Colitis and Attenuate Inflammatory Damage in Epithelial Organoids. Int. J. Mol. Sci. 2021, 22, 6431. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, B.H.; Lee, H.; Jeon, B.; Lee, Y.S.; Kwon, M.-J.; Kim, T.-Y. Regulation of skin inflammation and angiogenesis by EC-SOD via HIF-1α and NF-κB pathways. Free Radic. Biol. Med. 2011, 51, 1985–1995. [Google Scholar] [CrossRef] [PubMed]
- Agrahari, G.; Sah, S.K.; Bang, C.H.; Kim, Y.H.; Kim, T.-Y. Superoxide Dismutase 3 Controls the Activation and Differentiation of CD4+T Cells. Front. Immunol. 2021, 12, 628117. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.-J.; Kim, B.; Lee, Y.S.; Kim, T.-Y. Role of superoxide dismutase 3 in skin inflammation. J. Dermatol. Sci. 2012, 67, 81–87. [Google Scholar] [CrossRef]
- Birbach, A.; Gold, P.; Binder, B.R.; Hofer, E.; de Martin, R.; Schmid, J. Signaling Molecules of the NF-κB Pathway Shuttle Constitutively between Cytoplasm and Nucleus. J. Biol. Chem. 2002, 277, 10842–10851. [Google Scholar] [CrossRef] [Green Version]
- Carlotti, F.; Dower, S.K.; Qwarnstrom, E. Dynamic Shuttling of Nuclear Factor κB between the Nucleus and Cytoplasm as a Consequence of Inhibitor Dissociation. J. Biol. Chem. 2000, 275, 41028–41034. [Google Scholar] [CrossRef] [Green Version]
- Katsuyama, K.; Shichiri, M.; Marumo, F.; Hirata, Y. NO Inhibits Cytokine-Induced iNOS Expression and NF-κB Activation by Interfering with Phosphorylation and Degradation of IκB-α. Arter. Thromb. Vasc. Biol. 1998, 18, 1796–1802. [Google Scholar] [CrossRef] [Green Version]
- Reynaert, N.L.; Ckless, K.; Korn, S.H.; Vos, N.; Guala, A.S.; Wouters, E.F.; van der Vliet, A.; Janssen-Heininger, Y.M. Nitric oxide represses inhibitory κB kinase through S-nitrosylation. Proc. Natl. Acad. Sci. USA 2004, 101, 8945–8950. [Google Scholar] [CrossRef] [Green Version]
- Bernard-Marissal, N.; Moumen, A.; Sunyach, C.; Pellegrino, C.; Dudley, K.; Henderson, C.E.; Raoul, C.; Pettmann, B. Reduced Calreticulin Levels Link Endoplasmic Reticulum Stress and Fas-Triggered Cell Death in Motoneurons Vulnerable to ALS. J. Neurosci. 2012, 32, 4901–4912. [Google Scholar] [CrossRef]
- Hickok, J.R.; Vasudevan, D.; Antholine, W.E.; Thomas, D.D. Nitric Oxide Modifies Global Histone Methylation by Inhibiting Jumonji C Domain-containing Demethylases. J. Biol. Chem. 2013, 288, 16004–16015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogdan, C. Nitric oxide and the regulation of gene expression. Trends Cell Biol. 2001, 11, 66–75. [Google Scholar] [CrossRef]
- García-Giménez, J.-L.; Garcés, C.; Romá-Mateo, C.; Pallardó, F.V. Oxidative stress-mediated alterations in histone post-translational modifications. Free Radic. Biol. Med. 2021, 170, 6–18. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carmona-Rodríguez, L.; Martínez-Rey, D.; Martín-González, P.; Franch, M.; Sorokin, L.; Mira, E.; Mañes, S. Superoxide Dismutase-3 Downregulates Laminin α5 Expression in Tumor Endothelial Cells via the Inhibition of Nuclear Factor Kappa B Signaling. Cancers 2022, 14, 1226. https://doi.org/10.3390/cancers14051226
Carmona-Rodríguez L, Martínez-Rey D, Martín-González P, Franch M, Sorokin L, Mira E, Mañes S. Superoxide Dismutase-3 Downregulates Laminin α5 Expression in Tumor Endothelial Cells via the Inhibition of Nuclear Factor Kappa B Signaling. Cancers. 2022; 14(5):1226. https://doi.org/10.3390/cancers14051226
Chicago/Turabian StyleCarmona-Rodríguez, Lorena, Diego Martínez-Rey, Paula Martín-González, Mónica Franch, Lydia Sorokin, Emilia Mira, and Santos Mañes. 2022. "Superoxide Dismutase-3 Downregulates Laminin α5 Expression in Tumor Endothelial Cells via the Inhibition of Nuclear Factor Kappa B Signaling" Cancers 14, no. 5: 1226. https://doi.org/10.3390/cancers14051226
APA StyleCarmona-Rodríguez, L., Martínez-Rey, D., Martín-González, P., Franch, M., Sorokin, L., Mira, E., & Mañes, S. (2022). Superoxide Dismutase-3 Downregulates Laminin α5 Expression in Tumor Endothelial Cells via the Inhibition of Nuclear Factor Kappa B Signaling. Cancers, 14(5), 1226. https://doi.org/10.3390/cancers14051226