
Targeting Ribosome Biogenesis to Combat Tamoxifen Resistance in ER+ve Breast Cancer


Abstract
:Simple Summary
Abstract
1. Introduction
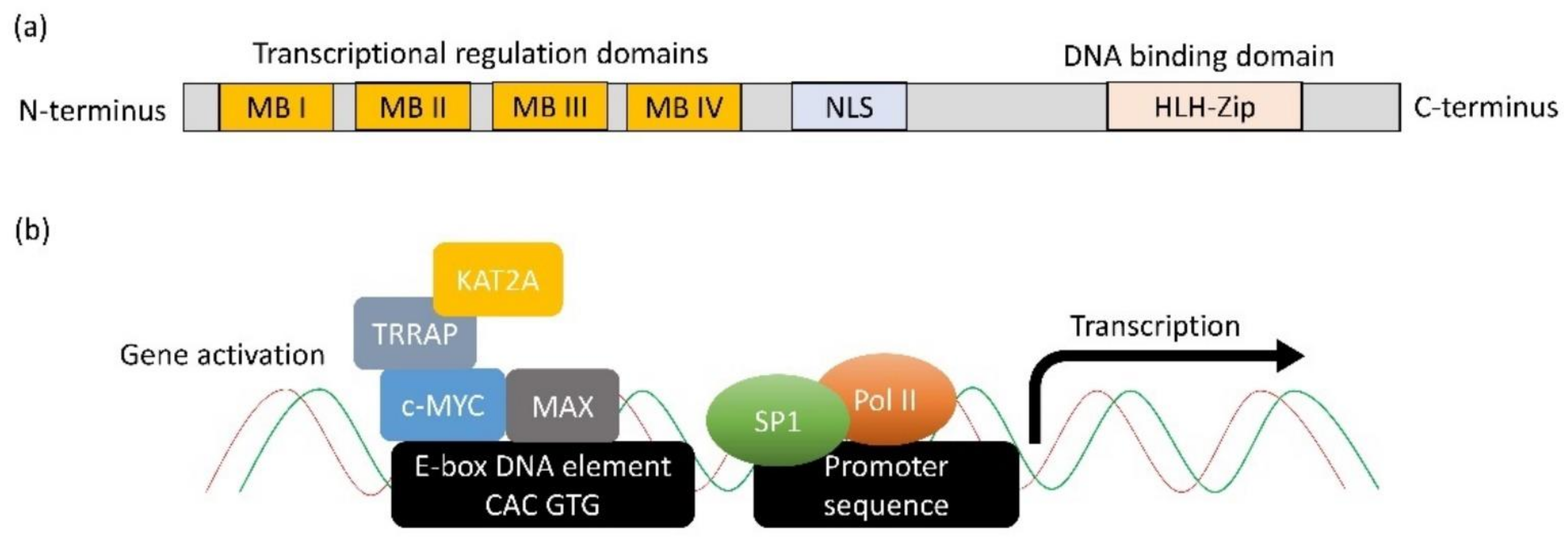
2. c-MYC in ER+ve Breast Cancer
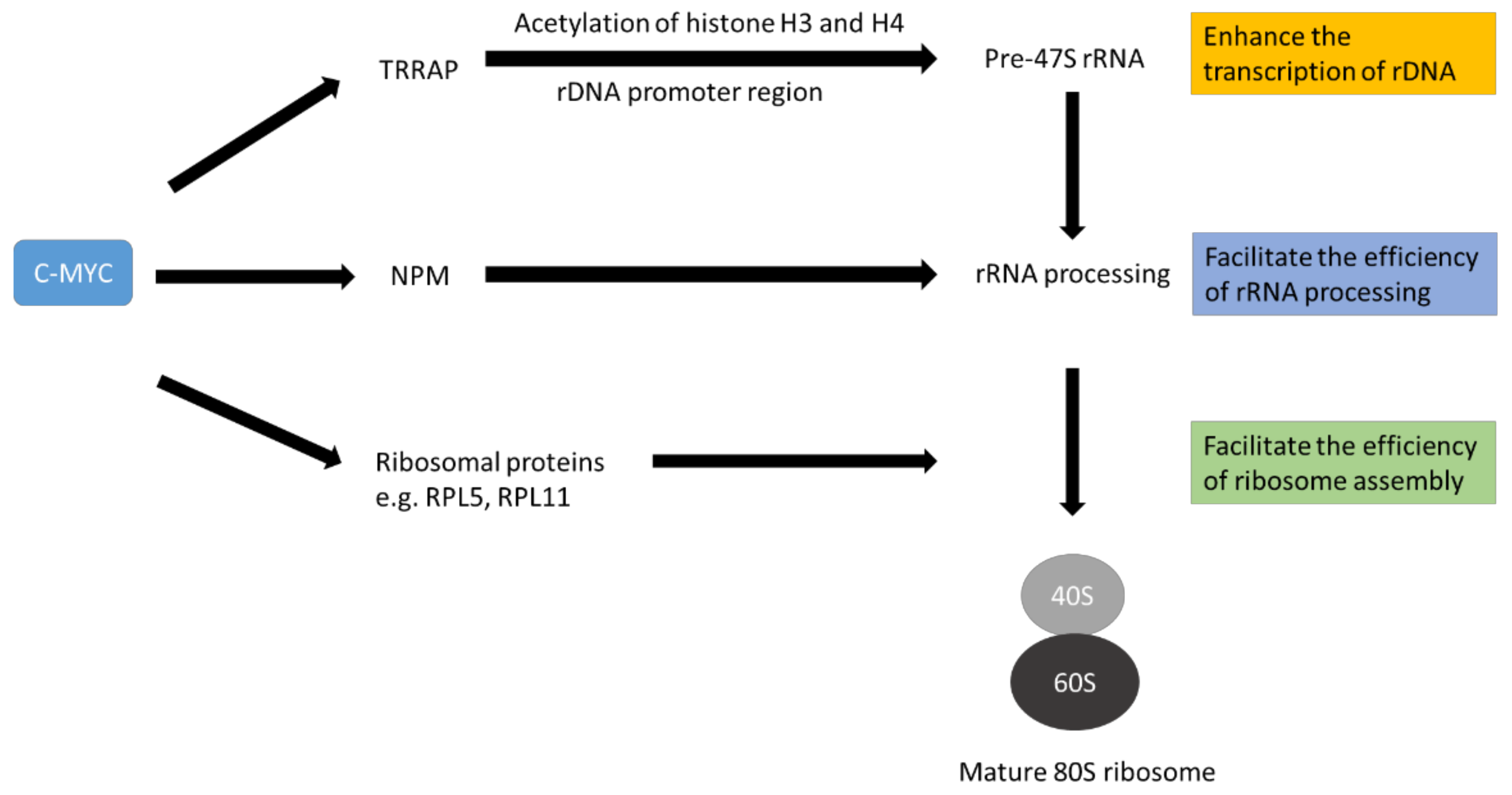
3. The Role of c-MYC on Ribosome Biogenesis
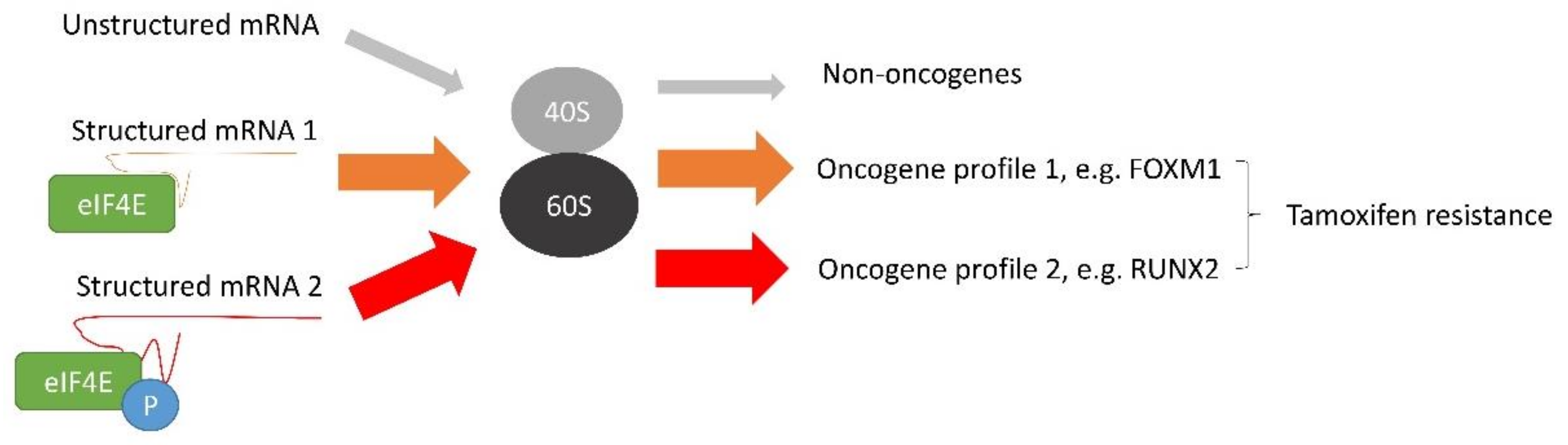
4. The Effect of c-MYC on Tamoxifen Resistance
5. Feasibility of Targeting Ribosome Biogenesis Enhanced by c-MYC Overexpression to Reverse Tamoxifen Resistance in Breast Cancers
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siersbaek, R.; Kumar, S.; Carroll, J.S. Signaling pathways and steroid receptors modulating estrogen receptor alpha function in breast cancer. Genes Dev. 2018, 32, 1141–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasar, P.; Ayaz, G.; User, S.D.; Gupur, G.; Muyan, M. Molecular mechanism of estrogen-estrogen receptor signaling. Reprod. Med. Biol. 2017, 16, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Liao, Q.; Su, M.; Huang, K.; Jin, J.; Cao, D. AKT and ERK dual inhibitors: The way forward? Cancer Lett. 2019, 459, 30–40. [Google Scholar] [CrossRef]
- Shang, Y.F.; Hu, X.; DiRenzo, J.; Lazar, M.A.; Brown, M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell 2000, 103, 843–852. [Google Scholar] [CrossRef] [Green Version]
- Davies, C.; Godwin, J.; Gray, R.; Clarke, M.; Darby, S.; McGale, P.; Wang, Y.C.; Peto, R.; Pan, H.C.; Cutter, D.; et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: Patient-level meta-analysis of randomised trials. Lancet 2011, 378, 771–784. [Google Scholar] [CrossRef] [Green Version]
- Ring, A.; Dowsett, M. Mechanisms of tamoxifen resistance. Endocr.-Relat. Cancer 2004, 11, 643–658. [Google Scholar] [CrossRef]
- Shou, J.; Massarweh, S.; Osborne, C.K.; Wakeling, A.E.; Ali, S.; Weiss, H.; Schiff, R. Mechanisms of tamoxifen resistance: Increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J. Natl. Cancer Inst. 2004, 96, 926–935. [Google Scholar] [CrossRef] [Green Version]
- Riggins, R.B.; Schrecengost, R.S.; Guerrero, M.S.; Bouton, A.H. Pathways to tamoxifen resistance. Cancer Lett. 2007, 256, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Sette, C.; Ladomery, M.; Ghigna, C. Alternative splicing: Role in cancer development and progression. Int. J. Cell Biol. 2013, 2013, 421606. [Google Scholar] [CrossRef] [Green Version]
- Omarjee, S.; Jacquemetton, J.; Poulard, C.; Rochel, N.; Dejaegere, A.; Chebaro, Y.; Treilleux, I.; Marangoni, E.; Corbo, L.; Le Romancer, M. The molecular mechanisms underlying the ER alpha-36-mediated signaling in breast cancer. Oncogene 2017, 36, 2503–2514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, C.; Man, E.P.S.; Tsoi, H.; Lee, T.K.W.; Lee, P.; Ma, S.T.; Wong, L.S.; Luk, M.Y.; Rakha, E.A.; Green, A.R.; et al. BQ323636.1, a Novel Splice Variant to NCOR2, as a Predictor for Tamoxifen-Resistant Breast Cancer. Clin. Cancer Res. 2018, 24, 3681–3691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhanasekaran, R.; Deutzmann, A.; Mahauad-Fernandez, W.D.; Hansen, A.S.; Gouw, A.M.; Felsher, D.W. The MYC oncogene—The grand orchestrator of cancer growth and immune evasion. Nat. Rev. Clin. Oncol. 2022, 19, 23–36. [Google Scholar] [CrossRef]
- Felsher, D.W. MYC Inactivation Elicits Oncogene Addiction through Both Tumor Cell-Intrinsic and Host-Dependent Mechanisms. Genes Cancer 2010, 1, 597–604. [Google Scholar] [CrossRef]
- Llombart, V.; Mansour, M.R. Therapeutic targeting of “undruggable” MYC. EBioMedicine 2021, 75, 103756. [Google Scholar] [CrossRef] [PubMed]
- Duesberg, P.H.; Vogt, P.K. Avian acute leukemia viruses MC29 and MH2 share specific RNA sequences: Evidence for a second class of transforming genes. Proc. Natl. Acad. Sci. USA 1979, 76, 1633–1637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef]
- Blackwood, E.M.; Eisenman, R.N. Max—A Helix-Loop-Helix Zipper Protein That Forms a Sequence-Specific DNA-Binding Complex with Myc. Science 1991, 251, 1211–1217. [Google Scholar] [CrossRef]
- Blackwell, T.K.; Kretzner, L.; Blackwood, E.M.; Eisenman, R.N.; Weintraub, H. Sequence-Specific DNA-Binding by the C-Myc Protein. Science 1990, 250, 1149–1151. [Google Scholar] [CrossRef]
- Zeller, K.I.; Zhao, X.D.; Lee, C.W.H.; Chiu, K.P.; Yao, F.; Yustein, J.T.; Ooi, H.S.; Orlov, Y.L.; Shahab, A.; Yong, H.C.; et al. Global mapping of c-Myc binding sites and target gene networks in human B cells. Proc. Natl. Acad. Sci. USA 2006, 103, 17834–17839. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, P.C.; Frank, S.R.; Wang, L.Q.; Schroeder, M.; Liu, S.X.; Greene, J.; Cocito, A.; Amati, B. Genomic targets of the human c-Myc protein. Gene Dev. 2003, 17, 1115–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalkat, M.; Resetca, D.; Lourenco, C.; Chan, P.K.; Wei, Y.; Shiah, Y.J.; Vitkin, N.; Tong, Y.F.; Sunnerhagen, M.; Done, S.J.; et al. MYC Protein Interactome Profiling Reveals Functionally Distinct Regions that Cooperate to Drive Tumorigenesis. Mol. Cell 2018, 72, 836–848.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.Q.; Cetinkaya, C.; Munoz-Alonso, M.J.; von der Lehr, N.; Bahram, F.; Beuger, V.; Eilers, M.; Leon, J.; Larsson, L.G. Myc represses differentiation-induced p21CIP1 expression via Miz-1-dependent interaction with the p21 core promoter. Oncogene 2003, 22, 351–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seoane, J.; Pouponnot, C.; Staller, P.; Schader, M.; Eilers, M.; Massague, J. TGF beta influences Myc, Miz-1 and Smad to control the CDK inhibitor p15(INK4b). Nat. Cell Biol. 2001, 3, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Gao, X.; Yuan, M.; Yang, B.; He, Q.; Cao, J. Targeting Myc Interacting Proteins as a Winding Path in Cancer Therapy. Front. Pharmacol. 2021, 12, 748852. [Google Scholar] [CrossRef] [PubMed]
- Andre, F.; Arnedos, M.; Baras, A.S.; Baselga, J.; Bedard, P.L.; Berger, M.F.; Bierkens, M.; Calvo, F.; Cerami, E.; Chakravarty, D.; et al. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef] [Green Version]
- Fallah, Y.; Brundage, J.; Allegakoen, P.; Shajahan-Haq, A.N. MYC-Driven Pathways in Breast Cancer Subtypes. Biomolecules 2017, 7, 53. [Google Scholar] [CrossRef]
- Qu, J.; Zhao, X.; Wang, J.; Liu, X.; Yan, Y.; Liu, L.; Cai, H.; Qu, H.; Lu, N.; Sun, Y.; et al. MYC overexpression with its prognostic and clinicopathological significance in breast cancer. Oncotarget 2017, 8, 93998–94008. [Google Scholar] [CrossRef]
- Wolfer, A.; Wittner, B.S.; Irimia, D.; Flavin, R.J.; Lupien, M.; Gunawardane, R.N.; Meyer, C.A.; Lightcap, E.S.; Tamayo, P.; Mesirov, J.P.; et al. MYC regulation of a “poor-prognosis” metastatic cancer cell state. Proc. Natl. Acad. Sci. USA 2010, 107, 3698–3703. [Google Scholar] [CrossRef] [Green Version]
- Nair, R.; Roden, D.L.; Teo, W.S.; McFarland, A.; Junankar, S.; Ye, S.; Nguyen, A.; Yang, J.; Nikolic, I.; Hui, M.; et al. c-Myc and Her2 cooperate to drive a stem-like phenotype with poor prognosis in breast cancer. Oncogene 2014, 33, 3992–4002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naab, T.J.; Gautam, A.; Ricks-Santi, L.; Esnakula, A.K.; Kanaan, Y.M.; DeWitty, R.L.; Asgedom, G.; Makambi, K.H.; Abawi, M.; Blancato, J.K. MYC amplification in subtypes of breast cancers in African American women. BMC Cancer 2018, 18, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razavi, P.; Chang, M.T.; Xu, G.T.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018, 34, 427–438.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.R.; Van Calcar, S.; Qu, C.X.; Cavenee, W.K.; Zhang, M.Q.; Ren, B. A global transcriptional regulatory role for c-Myc in Burkitt’s lymphoma cells. Proc. Natl. Acad. Sci. USA 2003, 100, 8164–8169. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.V. MYC, Metabolism, Cell Growth, and Tumorigenesis. CSH Perspect. Med. 2013, 3, a014217. [Google Scholar] [CrossRef]
- Jin, K.; Park, S.; Teo, W.W.; Korangath, P.; Cho, S.S.; Yoshida, T.; Gyorffy, B.; Goswami, C.P.; Nakshatri, H.; Cruz, L.A.; et al. HOXB7 Is an ER alpha Cofactor in the Activation of HER2 and Multiple ER Target Genes Leading to Endocrine Resistance. Cancer Discov. 2015, 5, 944–959. [Google Scholar] [CrossRef] [Green Version]
- Jin, K.; Kong, X.J.; Shah, T.; Penet, M.F.; Wildes, F.; Sgroi, D.C.; Ma, X.J.; Huang, Y.; Kallioniemi, A.; Landberg, G.; et al. The HOXB7 protein renders breast cancer cells resistant to tamoxifen through activation of the EGFR pathway. Proc. Natl. Acad. Sci. USA 2012, 109, 2736–2741. [Google Scholar] [CrossRef] [Green Version]
- Ali, R.; Wendt, M.K. The paradoxical functions of EGFR during breast cancer progression. Signal Transduct. Target. 2017, 2, 16042. [Google Scholar] [CrossRef]
- Hasson, S.P.; Brezis, M.R.; Shachar, E.; Shachar, S.S.; Wolf, I.; Sonnenblick, A. Adjuvant endocrine therapy in HER2-positive breast cancer patients: Systematic review and meta-analysis. ESMO Open 2021, 6, 100088. [Google Scholar] [CrossRef]
- Smith, A.P.; Verrecchia, A.; Faga, G.; Doni, M.; Perna, D.; Martinato, F.; Guccione, E.; Amati, B. A positive role for Myc in TGF beta-induced Snail transcription and epithelial-to-mesenchymal transition. Oncogene 2009, 28, 422–430. [Google Scholar] [CrossRef] [Green Version]
- Viswanathan, S.R.; Powers, J.T.; Einhorn, W.; Hoshida, Y.; Ng, T.L.; Toffanin, S.; O’Sullivan, M.; Lu, J.; Phillips, L.A.; Lockhart, V.L.; et al. Lin28 promotes transformation and is associated with advanced human malignancies. Nat. Genet. 2009, 41, 843–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, S.; Zhou, C.Q.; Lou, X.M.; Xiao, Z.F.; Zhu, H.X.; Wang, Q.F.; Wang, Y.H.; Lu, N.; He, S.; Zhan, Q.M.; et al. PTTG Overexpression Promotes Lymph Node Metastasis in Human Esophageal Squamous Cell Carcinoma. Cancer Res. 2009, 69, 3283–3290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, N.M.A.; Masui, O.; Newsted, D.; Scorilas, A.; Romaschin, A.D.; Bjarnason, G.A.; Siu, K.W.M.; Yousef, G.M. Galectin-1 has potential prognostic significance and is implicated in clear cell renal cell carcinoma progression through the HIF/mTOR signaling axis (vol 110, pg 1250, 2014). Br. J. Cancer 2017, 116, E3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, C.H.; Lee, S.W.; Li, C.F.; Wang, J.; Yang, W.L.; Wu, C.Y.; Wu, J.; Nakayama, K.I.; Kang, H.Y.; Huang, H.Y.; et al. Deciphering the transcriptional complex critical for RhoA gene expression and cancer metastasis. Nat. Cell Biol. 2010, 12, 457–467. [Google Scholar] [CrossRef]
- Shajahan-Haq, A.N.; Cook, K.L.; Schwartz-Roberts, J.L.; Eltayeb, A.E.; Demas, D.M.; Warri, A.M.; Facey, C.O.; Hilakivi-Clarke, L.A.; Clarke, R. MYC regulates the unfolded protein response and glucose and glutamine uptake in endocrine resistant breast cancer. Mol. Cancer 2014, 13, 239. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.K.; Wang, Y.Z.; Warden, C.; Chen, S.A. Cross-talk between ER and HER2 regulates c-MYC-mediated glutamine metabolism in aromatase inhibitor resistant breast cancer cells. J. Steroid Biochem. 2015, 149, 118–127. [Google Scholar] [CrossRef] [Green Version]
- Miller, T.W.; Balko, J.M.; Ghazoui, Z.; Dunbier, A.; Anderson, H.; Dowsett, M.; Gonzalez-Angulo, A.M.; Mills, G.B.; Miller, W.R.; Wu, H.Y.; et al. A Gene Expression Signature from Human Breast Cancer Cells with Acquired Hormone Independence Identifies MYC as a Mediator of Antiestrogen Resistance. Clin. Cancer Res. 2011, 17, 2024–2034. [Google Scholar] [CrossRef] [Green Version]
- Garg, H.; Suri, P.; Gupta, J.C.; Talwar, G.P.; Dubey, S. Survivin: A unique target for tumor therapy. Cancer Cell Int. 2016, 16, 49. [Google Scholar] [CrossRef] [Green Version]
- Yu, P.; Li, A.X.; Chen, X.S.; Tian, M.; Wang, H.Y.; Wang, X.L.; Zhang, Y.; Wang, K.S.; Cheng, Y. PKM2-c-Myc-Survivin Cascade Regulates the Cell Proliferation, Migration, and Tamoxifen Resistance in Breast Cancer. Front. Pharmacol. 2020, 11. [Google Scholar] [CrossRef]
- Mandlekar, S.; Kong, A.N.T. Mechanisms of tamoxifen-induced apoptosis. Apoptosis 2001, 6, 469–477. [Google Scholar] [CrossRef]
- Dong, Y.; Tu, R.F.; Liu, H.D.; Qing, G.L. Regulation of cancer cell metabolism: Oncogenic MYC in the driver’s seat. Signal Transduct. Target. 2020, 5, 124. [Google Scholar] [CrossRef] [PubMed]
- van Riggelen, J.; Yetil, A.; Felsher, D.W. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer 2010, 10, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Destefanis, F.; Manara, V.; Bellosta, P. Myc as a Regulator of Ribosome Biogenesis and Cell Competition: A Link to Cancer. Int. J. Mol. Sci. 2020, 21, 4037. [Google Scholar] [CrossRef] [PubMed]
- Bassler, J.; Hurt, E. Eukaryotic Ribosome Assembly. Annu. Rev. Biochem. 2019, 88, 281–306. [Google Scholar] [CrossRef] [PubMed]
- Truitt, M.L.; Ruggero, D. New frontiers in translational control of the cancer genome. Nat. Rev. Cancer 2017, 17, 332, Erratum in Nat. Rev. Cancer 2016, 16, 288. [Google Scholar] [CrossRef]
- Bastide, A.; David, A. The ribosome, (slow) beating heart of cancer (stem) cell. Oncogenesis 2018, 7, 34. [Google Scholar] [CrossRef] [Green Version]
- Zhai, W.; Comai, L. Repression of RNA polymerase I transcription by the tumor suppressor p53. Mol. Cell. Biol. 2000, 20, 5930–5938. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.C.; Liu, X.Y.; Teng, L.; Ji, Q.; Wu, Y.Y.; Li, J.M.; Gao, W.; Zhang, Y.Y.; La, T.; Tabatabaee, H.; et al. c-Myc inactivation of p53 through the pan-cancer lncRNA MILIP drives cancer pathogenesis. Nat. Commun. 2020, 11, 4980. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, J.W.; Yin, J.; Gan, Y.C.; Xu, S.L.; Gu, Y.; Huang, W.D. Alternative approaches to target Myc for cancer treatment. Signal Transduct. Target. 2021, 6, 117. [Google Scholar] [CrossRef]
- Schlosser, I.; Holzel, M.; Murnseer, M.; Burtscher, H.; Weidle, U.H.; Eick, D. A role for c-Myc in the regulation of ribosomal RNA processing. Nucleic Acids Res. 2003, 31, 6148–6156. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Li, Q.; Dang, C.V.; Lee, L.A. Induction of ribosomal genes and hepatocyte hypertrophy by adenovirus-mediated expression of c-Myc in vivo. Proc. Natl. Acad. Sci. USA 2000, 97, 11198–11202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coller, H.A.; Grandori, C.; Tamayo, P.; Colbert, T.; Lander, E.S.; Eisenman, R.N.; Golub, T.R. Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling, and adhesion. Proc. Natl. Acad. Sci. USA 2000, 97, 3260–3265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.H.; Sahoo, D.; Arvanitis, C.; Bradon, N.; Dill, D.L.; Felsher, D.W. Combined Analysis of Murine and Human Microarrays and ChIP Analysis Reveals Genes Associated with the Ability of MYC To Maintain Tumorigenesis. PLoS Genet. 2008, 4, e1000090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaiser, C.; Dobrikova, E.Y.; Bradrick, S.S.; Shveygert, M.; Herbert, J.T.; Gromeier, M. Activation of cap-independent translation by variant eukaryotic initiation factor 4G in vivo. RNA 2008, 14, 2170–2182. [Google Scholar] [CrossRef] [Green Version]
- Thoma, C.; Fraterman, S.; Gentzel, M.; Wilm, M.; Hentze, M.W. Translation initiation by the c-myc mRNA internal ribosome entry sequence and the poly(A) tail. RNA 2008, 14, 1579–1589. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.L.; Boone, D.; Hann, S.R. Nucleophosmin interacts directly with c-Myc and controls c-Myc-induced hyperproliferation and transformation. Proc. Natl. Acad. Sci. USA 2008, 105, 18794–18799. [Google Scholar] [CrossRef] [Green Version]
- Mitrea, D.M.; Cika, J.A.; Guy, C.S.; Ban, D.; Banerjee, P.R.; Stanley, C.B.; Nourse, A.; Deniz, A.A.; Kriwacki, R.W. Nucleophosmin integrates within the nucleolus via multi-modal interactions with proteins displaying R-rich linear motifs and rRNA. eLife 2016, 5, e13571. [Google Scholar] [CrossRef]
- Lindstrom, M.S. NPM1/B23: A Multifunctional Chaperone in Ribosome Biogenesis and Chromatin Remodeling. Biochem. Res. Int. 2011, 2011, 195209. [Google Scholar] [CrossRef] [Green Version]
- Kenneth, N.S.; Ramsbottom, B.A.; Gomez-Roman, N.; Marshall, L.; Cole, P.A.; White, R.J. TRRAP and GCN5 are used by c-Myc to activate RNA polymerase III transcription. Proc. Natl. Acad. Sci. USA 2007, 104, 14917–14922. [Google Scholar] [CrossRef] [Green Version]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, Metabolism, and Cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef] [Green Version]
- Morcelle, C.; Menoyo, S.; Moron-Duran, F.D.; Tauler, A.; Kozma, S.C.; Thomas, G.; Gentilella, A. Oncogenic MYC Induces the Impaired Ribosome Biogenesis Checkpoint and Stabilizes p53 Independent of Increased Ribosome Content. Cancer Res. 2019, 79, 4348–4359. [Google Scholar] [CrossRef] [Green Version]
- Xing, Y.; Draper, D.E. Stabilization of a ribosomal RNA tertiary structure by ribosomal protein L11. J. Mol. Biol. 1995, 249, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, P.; Woodson, S.A. Global stabilization of rRNA structure by ribosomal proteins S4, S17, and S20. J. Mol. Biol. 2009, 392, 666–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, Y.W.; Lamond, A.I.; Mann, M.; Andersen, J.S. Analysis of nucleolar protein dynamics reveals the nuclear degradation of ribosomal proteins. Curr. Biol. 2007, 17, 749–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, M.K.; Porras-Yakushi, T.R.; Reitsma, J.M.; Huber, F.M.; Sweredoski, M.J.; Hoelz, A.; Hesse, S.; Deshaies, R.J. A conserved quality-control pathway that mediates degradation of unassembled ribosomal proteins. eLife 2016, 5, e19105. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; O’Donnell, K.A.; Zeller, K.I.; Nguyen, T.; Osthus, R.C.; Li, F. The c-Myc target gene network. Semin. Cancer Biol. 2006, 16, 253–264. [Google Scholar] [CrossRef]
- Cheng, R.; Liu, Y.J.; Cui, J.W.; Yang, M.; Liu, X.L.; Li, P.; Wang, Z.; Zhu, L.Z.; Lu, S.Y.; Zou, L.; et al. Aspirin regulation of c-myc and cyclinD1 proteins to overcome tamoxifen resistance in estrogen receptor-positive breast cancer cells. Oncotarget 2017, 8, 30252–30264. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, E.V. The role of c-myc in regulation of translation initiation. Oncogene 2004, 23, 3217–3221. [Google Scholar] [CrossRef] [Green Version]
- Ramanathan, A.; Robb, G.B.; Chan, S.H. mRNA capping: Biological functions and applications. Nucleic Acids Res. 2016, 44, 7511–7526. [Google Scholar] [CrossRef]
- Ruggero, D.; Montanaro, L.; Ma, L.; Xu, W.; Londei, P.; Cardon-Cardo, C.; Pandolfi, P.P. The translation factor eIF-4E promotes tumor formation and cooperates with c-Myc in lymphomagenesis. Nat. Med. 2004, 10, 484–486. [Google Scholar] [CrossRef]
- Sonneveld, S.; Verhagen, B.M.P.; Tanenbaum, M.E. Heterogeneity in mRNA Translation. Trends Cell Biol. 2020, 30, 606–618. [Google Scholar] [CrossRef]
- Koromilas, A.E.; Lazaris-Karatzas, A.; Sonenberg, N. mRNAs containing extensive secondary structure in their 5’ non-coding region translate efficiently in cells overexpressing initiation factor eIF-4E. EMBO J. 1992, 11, 4153–4158. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Tsoi, H.; Mok, K.C.; Cheung, J.; Man, E.P.S.; Fujino, K.; Wong, A.H.; Lam, E.W.F.; Khoo, U.S. Phosphorylation independent eIF4E translational reprogramming of selective mRNAs determines tamoxifen resistance in breast cancer. Oncogene 2020, 39, 3206–3217. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.C.L.; Kanellos, G.; Vlahov, N.; Alexandrou, C.; Willis, A.E.; Knight, J.R.P.; Sansom, O.J. Translation initiation in cancer at a glance. J. Cell Sci. 2021, 134, jcs248476. [Google Scholar] [CrossRef] [PubMed]
- De Benedetti, A.; Graff, J.R. eIF-4E expression and its role in malignancies and metastases. Oncogene 2004, 23, 3189–3199. [Google Scholar] [CrossRef] [Green Version]
- Uttam, S.; Wong, C.; Price, T.J.; Khoutorsky, A. eIF4E-Dependent Translational Control: A Central Mechanism for Regulation of Pain Plasticity. Front. Genet. 2018, 9, 470. [Google Scholar] [CrossRef] [PubMed]
- Geter, P.A.; Ernlund, A.W.; Bakogianni, S.; Alard, A.; Arju, R.; Giashuddin, S.; Gadi, A.; Bromberg, J.; Schneider, R.J. Hyperactive mTOR and MNK1 phosphorylation of eIF4E confer tamoxifen resistance and estrogen independence through selective mRNA translation reprogramming. Gene Dev. 2017, 31, 2235–2249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.J.; Shin, H.L.; Kim, B.S.; Kim, H.J.; Ryoo, H.M. RUNX2-modifying enzymes: Therapeutic targets for bone diseases. Exp. Mol. Med. 2020, 52, 1178–1184. [Google Scholar] [CrossRef]
- Wu, H.; Whitfield, T.W.; Gordon, J.A.R.; Dobson, J.R.; Tai, P.W.L.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; Lian, J.B. Genomic occupancy of Runx2 with global expression profiling identifies a novel dimension to control of osteoblastogenesis. Genome Biol. 2014, 15, R52. [Google Scholar] [CrossRef] [Green Version]
- Chimge, N.O.; Frenkel, B. The RUNX family in breast cancer: Relationships with estrogen signaling. Oncogene 2013, 32, 2121–2130. [Google Scholar] [CrossRef] [Green Version]
- Won, H.S.; Lee, K.M.; Oh, J.E.; Nam, E.M.; Lee, K.E. Inhibition of beta-Catenin to Overcome Endocrine Resistance in Tamoxifen-Resistant Breast Cancer Cell Line. PLoS ONE 2016, 11, e0155983. [Google Scholar] [CrossRef]
- Colak, S.; Ten Dijke, P. Targeting TGF-beta Signaling in Cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Deng, K.; Huang, J.; Zeng, R.; Zuo, J. Progress in the Understanding of the Mechanism of Tamoxifen Resistance in Breast Cancer. Front. Pharmacol. 2020, 11, 592912. [Google Scholar] [CrossRef] [PubMed]
- Kalathil, D.; John, S.; Nair, A.S. FOXM1 and Cancer: Faulty Cellular Signaling Derails Homeostasis. Front. Oncol. 2021, 10, 3472. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Li, W.; Sheng, Y.; Li, L.P.; Huang, Y.; Zhang, Z.H.; Zhu, T.Y.; Peace, D.; Quigley, J.G.; Wu, W.S.; et al. The transcription factor Foxm1 is essential for the quiescence and maintenance of hematopoietic stem cells. Nat. Immunol. 2015, 16, 810–818. [Google Scholar] [CrossRef] [Green Version]
- Halasi, M.; Gartel, A.L. Suppression of FOXM1 Sensitizes Human Cancer Cells to Cell Death Induced by DNA-Damage. PLoS ONE 2012, 7, e31761. [Google Scholar] [CrossRef]
- Pelletier, J.; Thomas, G.; Volarevic, S. Ribosome biogenesis in cancer: New players and therapeutic avenues. Nat. Rev. Cancer 2018, 18, 51–63. [Google Scholar] [CrossRef]
- Barna, M.; Pusic, A.; Zollo, O.; Costa, M.; Kondrashov, N.; Rego, E.; Rao, P.H.; Ruggero, D. Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature 2008, 456, 971–975. [Google Scholar] [CrossRef]
- Wu, Q.; Gou, Y.W.; Wang, Q.M.; Jin, H.F.; Cui, L.N.; Zhang, Y.G.; He, L.J.; Wang, J.B.; Nie, Y.Z.; Shi, Y.Q.; et al. Downregulation of RPL6 by siRNA Inhibits Proliferation and Cell Cycle Progression of Human Gastric Cancer Cell Lines. PLoS ONE 2011, 6, e26401. [Google Scholar] [CrossRef] [Green Version]
- Tao, W.; Wang, Z.Y.; Zeng, L.Y.; Gao, Y.Z.; Yan, Y.X.; Zhang, Q. Down-Regulation of Ribosomal Protein RPS21 Inhibits Invasive Behavior of Osteosarcoma Cells Through the Inactivation of MAPK Pathway. Cancer Manag. Res. 2020, 12, 4949–4955. [Google Scholar] [CrossRef]
- Yang, S.X.; Cui, J.; Yang, Y.S.; Liu, Z.P.; Yan, H.Y.; Tang, C.H.; Wang, H.; Qin, H.F.; Li, X.Y.; Li, J.J.; et al. Over-expressed RPL34 promotes malignant proliferation of non-small cell lung cancer cells. Gene 2016, 576, 421–428. [Google Scholar] [CrossRef] [Green Version]
- Ebright, R.Y.; Lee, S.; Wittner, B.S.; Niederhoffer, K.L.; Nicholson, B.T.; Bardia, A.; Truesdell, S.; Wiley, D.F.; Wesley, B.; Li, S.; et al. Deregulation of ribosomal protein expression and translation promotes breast cancer metastasis. Science 2020, 367, 1468–1473. [Google Scholar] [CrossRef]
- Dolezal, J.M.; Dash, A.P.; Prochownik, E.V. Diagnostic and prognostic implications of ribosomal protein transcript expression patterns in human cancers. BMC Cancer 2018, 18, 275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, Y.K.; Li, X.; Chang, C.K.; He, F.X.Q.; Zhao, Y.S.; Wu, L.Y. Ribosomal protein L23 negatively regulates cellular apoptosis via the RPL23/Miz-1/c-Myc circuit in higher-risk myelodysplastic syndrome. Sci. Rep. 2017, 7, 2323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poortinga, G.; Quinn, L.M.; Hannan, R.D. Targeting RNA polymerase I to treat MYC-driven cancer. Oncogene 2015, 34, 403–412. [Google Scholar] [CrossRef]
- Choi, S.H.; Mahankali, M.; Lee, S.J.; Hull, M.; Petrassi, H.M.; Chatterjee, A.K.; Schultz, P.G.; Jones, K.A.; Shen, W.J. Targeted Disruption of Myc-Max Oncoprotein Complex by a Small Molecule. ACS Chem. Biol. 2017, 12, 2715–2719. [Google Scholar] [CrossRef] [PubMed]
- Castell, A.; Yan, Q.Z.; Fawkner, K.; Hydbring, P.; Zhang, F.; Verschut, V.; Franco, M.; Zakaria, S.M.; Bazzar, W.; Goodwin, J.; et al. A selective high affinity MYC-binding compound inhibits MYC: MAX interaction and MYC-dependent tumor cell proliferation. Sci. Rep. 2018, 8, 10064. [Google Scholar] [CrossRef] [Green Version]
- Jeong, K.C.; Ahn, K.O.; Yang, C.H. Small-molecule inhibitors of c-Myc transcriptional factor suppress proliferation and induce apoptosis of promyelocytic leukemia cell via cell cycle arrest. Mol. Biosyst. 2010, 6, 1503–1509. [Google Scholar] [CrossRef]
- Demma, M.J.; Mapelli, C.; Sun, A.; Bodea, S.; Ruprecht, B.; Javaid, S.; Wiswell, D.; Muise, E.; Chen, S.Y.; Zelina, J.; et al. Omomyc Reveals New Mechanisms To Inhibit the MYC Oncogene. Mol. Cell. Biol. 2019, 39, 22. [Google Scholar] [CrossRef] [Green Version]
- Soucek, L.; Whitfield, J.; Martins, C.P.; Finch, A.J.; Murphy, D.J.; Sodir, N.M.; Karnezis, A.N.; Swigart, L.B.; Nasi, S.; Evan, G.I. Modelling Myc inhibition as a cancer therapy. Nature 2008, 455, 679–683. [Google Scholar] [CrossRef] [Green Version]
- Drygin, D.; Lin, A.; Bliesath, J.; Ho, C.B.; O’Brien, S.E.; Proffitt, C.; Omori, M.; Haddach, M.; Schwaebe, M.K.; Siddiqui-Jain, A.; et al. Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 2011, 71, 1418–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khot, A.; Brajanovski, N.; Cameron, D.P.; Hein, N.; Maclachlan, K.H.; Sanij, E.; Lim, J.; Soong, J.; Link, E.; Blombery, P.; et al. First-in-Human RNA Polymerase I Transcription Inhibitor CX-5461 in Patients with Advanced Hematologic Cancers: Results of a Phase I Dose-Escalation Study. Cancer Discov. 2019, 9, 1036–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drygin, D.; Siddiqui-Jain, A.; O’Brien, S.; Schwaebe, M.; Lin, A.; Bliesath, J.; Ho, C.B.; Proffitt, C.; Trent, K.; Whitten, J.P.; et al. Anticancer activity of CX-3543: A direct inhibitor of rRNA biogenesis. Cancer Res. 2009, 69, 7653–7661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Y.X.; Xu, B.H.; Zhang, Y. CX-3543 Promotes Cell Apoptosis through Downregulation of CCAT1 in Colon Cancer Cells. Biomed. Res. Int. 2018, 2018, 9701957. [Google Scholar] [CrossRef]
- Pellegrino, S.; Meyer, M.; Zorbas, C.; Bouchta, S.A.; Saraf, K.; Pelly, S.C.; Yusupova, G.; Evidente, A.; Mathieu, V.; Kornienko, A.; et al. The Amaryllidaceae Alkaloid Haemanthamine Binds the Eukaryotic Ribosome to Repress Cancer Cell Growth. Structure 2018, 26, 416–425.e4. [Google Scholar] [CrossRef] [Green Version]
- Havelek, R.; Seifrtova, M.; Kralovec, K.; Bruckova, L.; Cahlikova, L.; Dalecka, M.; Vavrova, J.; Rezacova, M.; Opletal, L.; Bilkova, Z. The effect of Amaryllidaceae alkaloids haemanthamine and haemanthidine on cell cycle progression and apoptosis in p53-negative human leukemic Jurkat cells. Phytomedicine 2014, 21, 479–490. [Google Scholar] [CrossRef]
- Devlin, J.R.; Hannan, K.M.; Hein, N.; Cullinane, C.; Kusnadi, E.; Ng, P.Y.; George, A.J.; Shortt, J.; Bywater, M.J.; Poortinga, G.; et al. Combination Therapy Targeting Ribosome Biogenesis and mRNA Translation Synergistically Extends Survival in MYC-Driven Lymphoma. Cancer Discov. 2016, 6, 59–70. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.G.; Obinata, D.; Sandhu, S.; Selth, L.A.; Wong, S.Q.; Porter, L.H.; Lister, N.; Pook, D.; Pezaro, C.J.; Goode, D.L.; et al. Patient-derived Models of Abiraterone- and Enzalutamide-resistant Prostate Cancer Reveal Sensitivity to Ribosome-directed Therapy. Eur. Urol. 2018, 74, 562–572. [Google Scholar] [CrossRef]
- McNeil, C.M.; Sergio, C.M.; Anderson, L.R.; Inman, C.K.; Eggleton, S.A.; Murphy, N.C.; Millar, E.K.A.; Crea, P.; Kench, J.G.; Alles, M.C.; et al. c-Myc overexpression and endocrine resistance in breast cancer. J. Steroid. Biochem. 2006, 102, 147–155. [Google Scholar] [CrossRef]
- Steifensand, F.; Gallwas, J.; Bauerschmitz, G.; Grundker, C. Inhibition of Metabolism as a Therapeutic Option for Tamoxifen-Resistant Breast Cancer Cells. Cells 2021, 10, 2398. [Google Scholar] [CrossRef]
- Sanij, E.; Hannan, K.M.; Xuan, J.; Yan, S.; Ahern, J.E.; Trigos, A.S.; Brajanovski, N.; Son, J.; Chan, K.T.; Kondrashova, O.; et al. CX-5461 activates the DNA damage response and demonstrates therapeutic efficacy in high-grade serous ovarian cancer. Nat. Commun. 2020, 11, 2641. [Google Scholar] [CrossRef] [PubMed]
- Priedigkeit, N.; Ding, K.; Horne, W.; Kolls, J.K.; Du, T.; Lucas, P.C.; Blohmer, J.U.; Denkert, C.; Machleidt, A.; Ingold-Heppner, B.; et al. Acquired mutations and transcriptional remodeling in long-term estrogen-deprived locoregional breast cancer recurrences. Breast Cancer Res. 2021, 23, 1. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
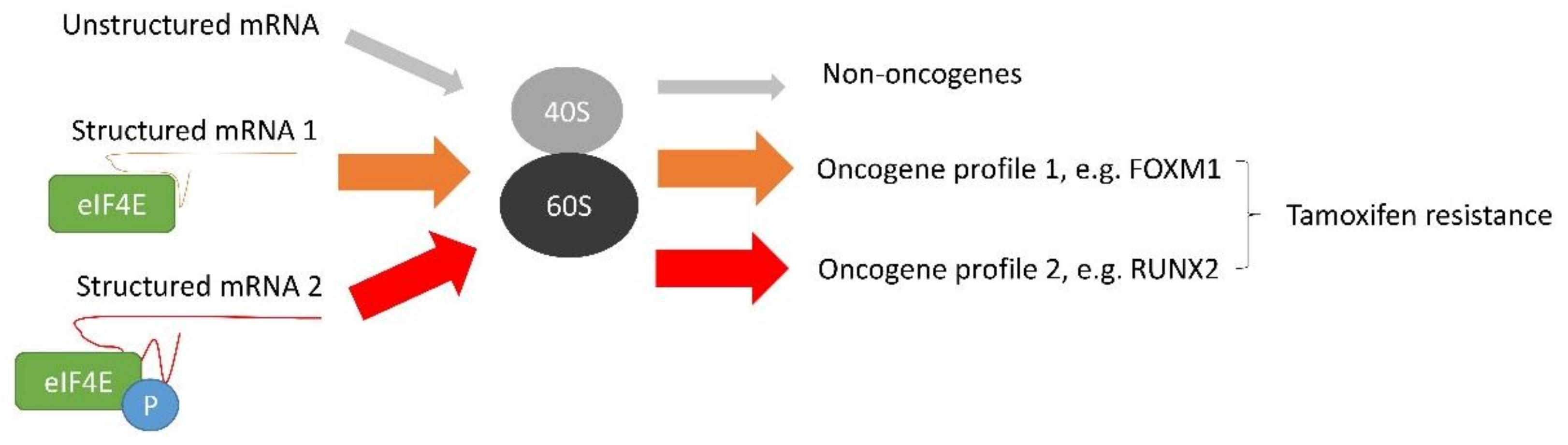{kind=link}
| Functional Roles | Candidate Proteins | References |
|---|---|---|
| Structural proteins of ribosomes | RPL3, RPL6, RPL23, RPL35, RPL44, RPS3 | [61] |
| RPS19, RPS17, RPS11, RPS24 | [60] | |
| RPL24, RPS11, RPS21, RPS25, RPL10a, RPS24, RPL6, RPL36a, RPS27, RPL3, RPS5 | [63] | |
| RPL5, RPL11 | [71] | |
| Factors to facilitate rRNA processing | Fibrillarin (FBL) | [60] |
| Nucleolin (NCL) | [61] | |
| Nucleophosmin (NPM1) | [66] | |
| rDNA transcription | TRRAP | [69] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsoi, H.; You, C.-P.; Leung, M.-H.; Man, E.P.S.; Khoo, U.-S. Targeting Ribosome Biogenesis to Combat Tamoxifen Resistance in ER+ve Breast Cancer. Cancers 2022, 14, 1251. https://doi.org/10.3390/cancers14051251
Tsoi H, You C-P, Leung M-H, Man EPS, Khoo U-S. Targeting Ribosome Biogenesis to Combat Tamoxifen Resistance in ER+ve Breast Cancer. Cancers. 2022; 14(5):1251. https://doi.org/10.3390/cancers14051251
Chicago/Turabian StyleTsoi, Ho, Chan-Ping You, Man-Hong Leung, Ellen P. S. Man, and Ui-Soon Khoo. 2022. "Targeting Ribosome Biogenesis to Combat Tamoxifen Resistance in ER+ve Breast Cancer" Cancers 14, no. 5: 1251. https://doi.org/10.3390/cancers14051251
APA StyleTsoi, H., You, C.-P., Leung, M.-H., Man, E. P. S., & Khoo, U.-S. (2022). Targeting Ribosome Biogenesis to Combat Tamoxifen Resistance in ER+ve Breast Cancer. Cancers, 14(5), 1251. https://doi.org/10.3390/cancers14051251