
Understanding Retinoblastoma Post-Translational Regulation for the Design of Targeted Cancer Therapies

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Rb1 Function and Downstream Signaling
2.1. E2F-Dependent Signaling
2.2. E2F-Independent Signaling
3. The Role of Rb1 in Cancer Development and Progression
3.1. Genetic and Epigenetic Alteration of the Rb1 Locus
3.2. Canonical Tumor Suppressor Role of Rb1
3.3. Non-Canonical Function of Rb1
4. Upstream Regulation and Post-Translational Modifications
4.1. Phosphorylation
4.2. Acetylation, Methylation and Sumoylation
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lele, K.P.; Penrose, L.S.; Stallard, H.B. Chromosome deletion in a case of Retinoblastoma. Ann. Hum. Genet. 1963, 27, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.G.; Ebbin, A.J.; Towner, J.W.; Spencer, W.H. Chromosomal anomalies in patients with retinoblastoma. Clin. Genet. 1977, 12, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Comings, D.E. A general theory of carcinogenesis. Proc. Natl. Acad. Sci. USA 1973, 70, 3324–3328. [Google Scholar] [CrossRef] [Green Version]
- Sparkes, R.S.; Murphree, A.L.; Lingua, R.W.; Sparkes, M.C.; Field, L.L.; Funderburk, S.J.; Benedict, W.F. Gene for hereditary retinoblastoma assigned to human chromosome 13 by linkage to esterase D. Science 1983, 219, 971–973. [Google Scholar] [CrossRef]
- Godbout, R.; Dryja, T.P.; Squire, J.; Gallie, B.L.; Phillips, R.A. Somatic inactivation of genes on chromosome 13 is a common event in retinoblastoma. Nature 1983, 304, 451–453. [Google Scholar] [CrossRef]
- Lee, W.H.; Bookstein, R.; Hong, F.; Young, L.J.; Shew, J.Y.; Lee, E.Y. Human retinoblastoma susceptibility gene: Cloning, identification, and sequence. Science 1987, 235, 1394–1399. [Google Scholar] [CrossRef] [Green Version]
- Claudio, P.P.; Tonini, T.; Giordano, A. The retinoblastoma family: Twins or distant cousins? Genome Biol. 2002, 3, reviews3012.1. [Google Scholar] [CrossRef]
- Henley, S.A.; Dick, F.A. The retinoblastoma family of proteins and their regulatory functions in the mammalian cell division cycle. Cell Div. 2012, 7, 10. [Google Scholar] [CrossRef] [Green Version]
- Xiao, B.; Spencer, J.; Clements, A.; Ali-Khan, N.; Mittnacht, S.; Broceno, C.; Burghammer, M.; Perrakis, A.; Marmorstein, R.; Gamblin, S.J. Crystal structure of the retinoblastoma tumor suppressor protein bound to E2F and the molecular basis of its regulation. Proc. Natl. Acad. Sci. USA 2003, 100, 2363–2368. [Google Scholar] [CrossRef] [Green Version]
- Hiebert, S.W.; Chellappan, S.P.; Horowitz, J.M.; Nevins, J.R. The interaction of RB with E2F coincides with an inhibition of the transcriptional activity of E2F. Genes Dev. 1992, 6, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, W.; Schneider, J.W.; Condorelli, G.; Kaushal, S.; Mahdavi, V.; Nadal-Ginard, B. Interaction of myogenic factors and the retinoblastoma protein mediates muscle cell commitment and differentiation. Cell 1993, 72, 309–324. [Google Scholar] [CrossRef]
- Chen, P.L.; Riley, D.J.; Chen-Kiang, S.; Lee, W.H. Retinoblastoma protein directly interacts with and activates the transcription factor NF-IL6. Proc. Natl. Acad. Sci. USA 1996, 93, 465–469. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.L.; Riley, D.J.; Chen, Y.; Lee, W.H. Retinoblastoma protein positively regulates terminal adipocyte differentiation through direct interaction with C/EBPs. Genes Dev. 1996, 10, 2794–2804. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, l1. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyson, N.J. RB1: A prototype tumor suppressor and an enigma. Genes Dev. 2016, 30, 1492–1502. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Chang, J.H.; Lee, H.S.; Cho, Y. Structural basis for the recognition of the E2F transactivation domain by the retinoblastoma tumor suppressor. Genes Dev. 2002, 16, 3199–3212. [Google Scholar] [CrossRef] [Green Version]
- Chan, H.M.; Smith, L.; La Thangue, N.B. Role of LXCXE motif-dependent interactions in the activity of the retinoblastoma protein. Oncogene 2001, 20, 6152–6163. [Google Scholar] [CrossRef] [Green Version]
- Dick, F.A.; Dyson, N. pRB contains an E2F1-specific binding domain that allows E2F1-induced apoptosis to be regulated separately from other E2F activities. Mol. Cell 2003, 12, 639–649. [Google Scholar] [CrossRef]
- Chau, B.N.; Pan, C.W.; Wang, J.Y. Separation of anti-proliferation and anti-apoptotic functions of retinoblastoma protein through targeted mutations of its A/B domain. PLoS ONE 2006, 1, e82. [Google Scholar] [CrossRef] [PubMed]
- Julian, L.M.; Palander, O.; Seifried, L.A.; Foster, J.E.; Dick, F.A. Characterization of an E2F1-specific binding domain in pRB and its implications for apoptotic regulation. Oncogene 2008, 27, 1572–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubin, S.M.; Gall, A.L.; Zheng, N.; Pavletich, N.P. Structure of the Rb C-terminal domain bound to E2F1-DP1: A mechanism for phosphorylation-induced E2F release. Cell 2005, 123, 1093–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, X.Q.; Chittenden, T.; Livingston, D.M.; Kaelin, W.G., Jr. Identification of a growth suppression domain within the retinoblastoma gene product. Genes Dev. 1992, 6, 953–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeGregori, J.; Kowalik, T.; Nevins, J.R. Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol. Cell Biol. 1995, 15, 4215–4224. [Google Scholar] [CrossRef] [Green Version]
- Hagemeier, C.; Cook, A.; Kouzarides, T. The retinoblastoma protein binds E2F residues required for activation in vivo and TBP binding in vitro. Nucleic Acids Res. 1993, 21, 4998–5004. [Google Scholar] [CrossRef] [Green Version]
- Weintraub, S.J.; Chow, K.N.; Luo, R.X.; Zhang, S.H.; He, S.; Dean, D.C. Mechanism of active transcriptional repression by the retinoblastoma protein. Nature 1995, 375, 812–815. [Google Scholar] [CrossRef]
- Magnaghi-Jaulin, L.; Groisman, R.; Naguibneva, I.; Robin, P.; Lorain, S.; Le Villain, J.P.; Troalen, F.; Trouche, D.; Harel-Bellan, A. Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature 1998, 391, 601–605. [Google Scholar] [CrossRef]
- Brehm, A.; Miska, E.A.; McCance, D.J.; Reid, J.L.; Bannister, A.J.; Kouzarides, T. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature 1998, 391, 597–601. [Google Scholar] [CrossRef]
- Luo, R.X.; Postigo, A.A.; Dean, D.C. Rb interacts with histone deacetylase to repress transcription. Cell 1998, 92, 463–473. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Jin, X.; Ma, J.; Ding, D.; Huang, Z.; Sheng, H.; Yan, Y.; Pan, Y.; Wei, T.; Wang, L.; et al. HDAC5 Loss Impairs RB Repression of Pro-Oncogenic Genes and Confers CDK4/6 Inhibitor Resistance in Cancer. Cancer Res. 2021, 81, 1486–1499. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.J.; Schneider, R.; Bauer, U.M.; Bannister, A.J.; Morrison, A.; O’Carroll, D.; Firestein, R.; Cleary, M.; Jenuwein, T.; Herrera, R.E.; et al. Rb targets histone H3 methylation and HP1 to promoters. Nature 2001, 412, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Ait-Si-Ali, S.; Guasconi, V.; Fritsch, L.; Yahi, H.; Sekhri, R.; Naguibneva, I.; Robin, P.; Cabon, F.; Polesskaya, A.; Harel-Bellan, A. A Suv39h-dependent mechanism for silencing S-phase genes in differentiating but not in cycling cells. EMBO J. 2004, 23, 605–615. [Google Scholar] [CrossRef] [Green Version]
- Dunaief, J.L.; Strober, B.E.; Guha, S.; Khavari, P.A.; Alin, K.; Luban, J.; Begemann, M.; Crabtree, G.R.; Goff, S.P. The retinoblastoma protein and BRG1 form a complex and cooperate to induce cell cycle arrest. Cell 1994, 79, 119–130. [Google Scholar] [CrossRef]
- Singh, P.; Coe, J.; Hong, W. A role for retinoblastoma protein in potentiating transcriptional activation by the glucocorticoid receptor. Nature 1995, 374, 562–565. [Google Scholar] [CrossRef]
- Schnitzler, G.; Sif, S.; Kingston, R.E. Human SWI/SNF interconverts a nucleosome between its base state and a stable remodeled state. Cell 1998, 94, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Cobrinik, D. Pocket proteins and cell cycle control. Oncogene 2005, 24, 2796–2809. [Google Scholar] [CrossRef] [Green Version]
- Robertson, K.D.; Ait-Si-Ali, S.; Yokochi, T.; Wade, P.A.; Jones, P.L.; Wolffe, A.P. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat. Genet. 2000, 25, 338–342. [Google Scholar] [CrossRef]
- Ishak, C.A.; Marshall, A.E.; Passos, D.T.; White, C.R.; Kim, S.J.; Cecchini, M.J.; Ferwati, S.; MacDonald, W.A.; Howlett, C.J.; Welch, I.D.; et al. An RB-EZH2 Complex Mediates Silencing of Repetitive DNA Sequences. Mol. Cell 2016, 64, 1074–1087. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Marmorstein, R. When viral oncoprotein meets tumor suppressor: A structural view. Genes Dev. 2006, 20, 2332–2337. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.O.; Russo, A.A.; Pavletich, N.P. Structure of the retinoblastoma tumour-suppressor pocket domain bound to a peptide from HPV E7. Nature 1998, 391, 859–865. [Google Scholar] [CrossRef] [PubMed]
- Dyson, N.; Howley, P.M.; Munger, K.; Harlow, E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 1989, 243, 934–937. [Google Scholar] [CrossRef] [PubMed]
- DeCaprio, J.A.; Ludlow, J.W.; Figge, J.; Shew, J.Y.; Huang, C.M.; Lee, W.H.; Marsilio, E.; Paucha, E.; Livingston, D.M. SV40 large tumor antigen forms a specific complex with the product of the retinoblastoma susceptibility gene. Cell 1988, 54, 275–283. [Google Scholar] [CrossRef]
- Whyte, P.; Buchkovich, K.J.; Horowitz, J.M.; Friend, S.H.; Raybuck, M.; Weinberg, R.A.; Harlow, E. Association between an oncogene and an anti-oncogene: The adenovirus E1A proteins bind to the retinoblastoma gene product. Nature 1988, 334, 124–129. [Google Scholar] [CrossRef]
- Singh, M.; Krajewski, M.; Mikolajka, A.; Holak, T.A. Molecular determinants for the complex formation between the retinoblastoma protein and LXCXE sequences. J. Biol. Chem. 2005, 280, 37868–37876. [Google Scholar] [CrossRef] [Green Version]
- Morrison, A.J.; Sardet, C.; Herrera, R.E. Retinoblastoma protein transcriptional repression through histone deacetylation of a single nucleosome. Mol. Cell Biol. 2002, 22, 856–865. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, R.; Magnaghi-Jaulin, L.; Robin, P.; Harel-Bellan, A.; Trouche, D. The three members of the pocket proteins family share the ability to repress E2F activity through recruitment of a histone deacetylase. Proc. Natl. Acad. Sci. USA 1998, 95, 10493–10498. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.Z.; Tsai, S.Y.; Leone, G. Emerging roles of E2Fs in cancer: An exit from cell cycle control. Nat. Rev. Cancer 2009, 9, 785–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyson, N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998, 12, 2245–2262. [Google Scholar] [CrossRef] [Green Version]
- Lees, J.A.; Saito, M.; Vidal, M.; Valentine, M.; Look, T.; Harlow, E.; Dyson, N.; Helin, K. The retinoblastoma protein binds to a family of E2F transcription factors. Mol. Cell Biol. 1993, 13, 7813–7825. [Google Scholar]
- Moberg, K.; Starz, M.A.; Lees, J.A. E2F-4 switches from p130 to p107 and pRB in response to cell cycle reentry. Mol. Cell Biol. 1996, 16, 1436–1449. [Google Scholar] [CrossRef] [Green Version]
- Humbert, P.O.; Rogers, C.; Ganiatsas, S.; Landsberg, R.L.; Trimarchi, J.M.; Dandapani, S.; Brugnara, C.; Erdman, S.; Schrenzel, M.; Bronson, R.T.; et al. E2F4 is essential for normal erythrocyte maturation and neonatal viability. Mol. Cell 2000, 6, 281–291. [Google Scholar] [CrossRef]
- Danielian, P.S.; Hess, R.A.; Lees, J.A. E2f4 and E2f5 are essential for the development of the male reproductive system. Cell Cycle 2016, 15, 250–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuitino, M.C.; Pecot, T.; Sun, D.; Kladney, R.; Okano-Uchida, T.; Shinde, N.; Saeed, R.; Perez-Castro, A.J.; Webb, A.; Liu, T.; et al. Two Distinct E2F Transcriptional Modules Drive Cell Cycles and Differentiation. Cell Rep. 2019, 27, 3547–3560. [Google Scholar] [CrossRef] [Green Version]
- Miccadei, S.; Provenzano, C.; Mojzisek, M.; Natali, P.G.; Civitareale, D. Retinoblastoma protein acts as Pax 8 transcriptional coactivator. Oncogene 2005, 24, 6993–7001. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.M.; Carty, S.A.; Piscopo, D.M.; Lee, J.S.; Wang, W.F.; Forrester, W.C.; Hinds, P.W. The retinoblastoma protein acts as a transcriptional coactivator required for osteogenic differentiation. Mol. Cell 2001, 8, 303–316. [Google Scholar] [CrossRef]
- Kadri, Z.; Shimizu, R.; Ohneda, O.; Maouche-Chretien, L.; Gisselbrecht, S.; Yamamoto, M.; Romeo, P.H.; Leboulch, P.; Chretien, S. Direct binding of pRb/E2F-2 to GATA-1 regulates maturation and terminal cell division during erythropoiesis. PLoS. Biol. 2009, 7, e1000123. [Google Scholar] [CrossRef] [PubMed]
- MacLellan, W.R.; Xiao, G.; Abdellatif, M.; Schneider, M.D. A novel Rb- and p300-binding protein inhibits transactivation by MyoD. Mol. Cell Biol. 2000, 20, 8903–8915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iavarone, A.; Garg, P.; Lasorella, A.; Hsu, J.; Israel, M.A. The helix-loop-helix protein Id-2 enhances cell proliferation and binds to the retinoblastoma protein. Genes Dev. 1994, 8, 1270–1284. [Google Scholar] [CrossRef] [Green Version]
- Takebayashi, T.; Higashi, H.; Sudo, H.; Ozawa, H.; Suzuki, E.; Shirado, O.; Katoh, H.; Hatakeyama, M. NF-kappa B-dependent induction of cyclin D1 by retinoblastoma protein (pRB) family proteins and tumor-derived pRB mutants. J. Biol. Chem. 2003, 278, 14897–14905. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Ding, D.; Yan, Y.; Li, H.; Wang, B.; Ma, L.; Ye, Z.; Ma, T.; Wu, Q.; Rodrigues, D.N.; et al. Phosphorylated RB Promotes Cancer Immunity by Inhibiting NF-kappaB Activation and PD-L1 Expression. Mol. Cell 2019, 73, 22–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benevolenskaya, E.V.; Murray, H.L.; Branton, P.; Young, R.A.; Kaelin, W.G., Jr. Binding of pRB to the PHD protein RBP2 promotes cellular differentiation. Mol. Cell 2005, 18, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Isaac, C.E.; Francis, S.M.; Martens, A.L.; Julian, L.M.; Seifried, L.A.; Erdmann, N.; Binne, U.K.; Harrington, L.; Sicinski, P.; Berube, N.G.; et al. The retinoblastoma protein regulates pericentric heterochromatin. Mol. Cell Biol. 2006, 26, 3659–3671. [Google Scholar] [CrossRef] [Green Version]
- Gonzalo, S.; Garcia-Cao, M.; Fraga, M.F.; Schotta, G.; Peters, A.H.; Cotter, S.E.; Eguia, R.; Dean, D.C.; Esteller, M.; Jenuwein, T.; et al. Role of the RB1 family in stabilizing histone methylation at constitutive heterochromatin. Nat. Cell Biol. 2005, 7, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, P. The function of APC/CCdh1 in cell cycle and beyond. Cell Div. 2009, 4, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakayama, K.I.; Nakayama, K. Regulation of the cell cycle by SCF-type ubiquitin ligases. Semin. Cell Dev. Biol. 2005, 16, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Ji, P.; Jiang, H.; Rekhtman, K.; Bloom, J.; Ichetovkin, M.; Pagano, M.; Zhu, L. An Rb-Skp2-p27 pathway mediates acute cell cycle inhibition by Rb and is retained in a partial-penetrance Rb mutant. Mol. Cell 2004, 16, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Binne, U.K.; Classon, M.K.; Dick, F.A.; Wei, W.; Rape, M.; Kaelin, W.G.; Naar, A.M., Jr.; Dyson, N.J. Retinoblastoma protein and anaphase-promoting complex physically interact and functionally cooperate during cell-cycle exit. Nat. Cell Biol. 2007, 9, 225–232. [Google Scholar] [CrossRef]
- Ramanujan, A.; Tiwari, S. APC/C and retinoblastoma interaction: Cross-talk of retinoblastoma protein with the ubiquitin proteasome pathway. Biosci. Rep. 2016, 36, e00377. [Google Scholar] [CrossRef] [Green Version]
- Wiggs, J.; Nordenskjold, M.; Yandell, D.; Rapaport, J.; Grondin, V.; Janson, M.; Werelius, B.; Petersen, R.; Craft, A.; Riedel, K.; et al. Prediction of the risk of hereditary retinoblastoma, using DNA polymorphisms within the retinoblastoma gene. N. Engl. J. Med. 1988, 318, 151–157. [Google Scholar] [CrossRef]
- Friend, S.H.; Horowitz, J.M.; Gerber, M.R.; Wang, X.F.; Bogenmann, E.; Li, F.P.; Weinberg, R.A. Deletions of a DNA sequence in retinoblastomas and mesenchymal tumors: Organization of the sequence and its encoded protein. Proc. Natl. Acad. Sci. USA 1987, 84, 9059–9063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berge, E.O.; Knappskog, S.; Geisler, S.; Staalesen, V.; Pacal, M.; Borresen-Dale, A.L.; Puntervoll, P.; Lillehaug, J.R.; Lonning, P.E. Identification and characterization of retinoblastoma gene mutations disturbing apoptosis in human breast cancers. Mol. Cancer 2010, 9, 173. [Google Scholar] [CrossRef] [Green Version]
- Condorelli, R.; Spring, L.; O’Shaughnessy, J.; Lacroix, L.; Bailleux, C.; Scott, V.; Dubois, J.; Nagy, R.J.; Lanman, R.B.; Iafrate, A.J.; et al. Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Ann. Oncol. 2018, 29, 640–645. [Google Scholar] [CrossRef] [PubMed]
- McCartney, A.; Migliaccio, I.; Bonechi, M.; Biagioni, C.; Romagnoli, D.; De, L.F.; Galardi, F.; Risi, E.; De, S.I.; Benelli, M.; et al. Mechanisms of Resistance to CDK4/6 Inhibitors: Potential Implications and Biomarkers for Clinical Practice. Front. Oncol. 2019, 9, 666. [Google Scholar] [CrossRef]
- Gansauge, S.; Gansauge, F.; Ramadani, M.; Stobbe, H.; Rau, B.; Harada, N.; Beger, H.G. Overexpression of cyclin D1 in human pancreatic carcinoma is associated with poor prognosis. Cancer Res. 1997, 57, 1634–1637. [Google Scholar]
- Wang, L.; Shao, Z.M. Cyclin e expression and prognosis in breast cancer patients: A meta-analysis of published studies. Cancer Investig. 2006, 24, 581–587. [Google Scholar] [CrossRef]
- Tsihlias, J.; Kapusta, L.; Slingerland, J. The prognostic significance of altered cyclin-dependent kinase inhibitors in human cancer. Annu. Rev. Med. 1999, 50, 401–423. [Google Scholar] [CrossRef]
- Straume, O.; Sviland, L.; Akslen, L.A. Loss of nuclear p16 protein expression correlates with increased tumor cell proliferation (Ki-67) and poor prognosis in patients with vertical growth phase melanoma. Clin. Cancer Res. 2000, 6, 1845–1853. [Google Scholar] [PubMed]
- Stirzaker, C.; Millar, D.S.; Paul, C.L.; Warnecke, P.M.; Harrison, J.; Vincent, P.C.; Frommer, M.; Clark, S.J. Extensive DNA methylation spanning the Rb promoter in retinoblastoma tumors. Cancer Res. 1997, 57, 2229–2237. [Google Scholar]
- Ohtani-Fujita, N.; Fujita, T.; Aoike, A.; Osifchin, N.E.; Robbins, P.D.; Sakai, T. CpG methylation inactivates the promoter activity of the human retinoblastoma tumor-suppressor gene. Oncogene 1993, 8, 1063–1067. [Google Scholar]
- Gonzalez-Gomez, P.; Bello, M.J.; Arjona, D.; Lomas, J.; Alonso, M.E.; De Campos, J.M.; Vaquero, J.; Isla, A.; Gutierrez, M.; Rey, J.A. Promoter hypermethylation of multiple genes in astrocytic gliomas. Int. J. Oncol. 2003, 22, 601–608. [Google Scholar] [PubMed]
- Price, E.A.; Kolkiewicz, K.; Patel, R.; Hashim, S.; Karaa, E.; Scheimberg, I.; Sagoo, M.S.; Reddy, M.A.; Onadim, Z. Detection and reporting of RB1 promoter hypermethylation in diagnostic screening. Ophthalmic Genet. 2018, 39, 526–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Gomez, P.; Bello, M.J.; Alonso, M.E.; Arjona, D.; Lomas, J.; De Campos, J.M.; Isla, A.; Rey, J.A. CpG island methylation status and mutation analysis of the RB1 gene essential promoter region and protein-binding pocket domain in nervous system tumours. Br. J. Cancer 2003, 88, 109–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witkiewicz, A.K.; Knudsen, K.E.; Dicker, A.P.; Knudsen, E.S. The meaning of p16(ink4a) expression in tumors: Functional significance, clinical associations and future developments. Cell Cycle 2011, 10, 2497–2503. [Google Scholar] [CrossRef] [Green Version]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [Green Version]
- Cam, H.; Dynlacht, B.D. Emerging roles for E2F: Beyond the G1/S transition and DNA replication. Cancer Cell 2003, 3, 311–316. [Google Scholar] [CrossRef] [Green Version]
- Fry, D.W.; Harvey, P.J.; Keller, P.R.; Elliott, W.L.; Meade, M.; Trachet, E.; Albassam, M.; Zheng, X.; Leopold, W.R.; Pryer, N.K.; et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol. Cancer Ther. 2004, 3, 1427–1438. [Google Scholar]
- Musgrove, E.A.; Caldon, C.E.; Barraclough, J.; Stone, A.; Sutherland, R.L. Cyclin D as a therapeutic target in cancer. Nat. Rev. Cancer 2011, 11, 558–572. [Google Scholar] [CrossRef]
- Sherr, C.J.; Beach, D.; Shapiro, G.I. Targeting CDK4 and CDK6: From Discovery to Therapy. Cancer Discov. 2016, 6, 353–367. [Google Scholar] [CrossRef] [Green Version]
- Patnaik, A.; Rosen, L.S.; Tolaney, S.M.; Tolcher, A.W.; Goldman, J.W.; Gandhi, L.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Hilton, J.F.; et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov. 2016, 6, 740–753. [Google Scholar] [CrossRef] [Green Version]
- DeMichele, A.; Clark, A.S.; Tan, K.S.; Heitjan, D.F.; Gramlich, K.; Gallagher, M.; Lal, P.; Feldman, M.; Zhang, P.; Colameco, C.; et al. CDK 4/6 inhibitor palbociclib (PD0332991) in Rb+ advanced breast cancer: Phase II activity, safety, and predictive biomarker assessment. Clin. Cancer Res. 2015, 21, 995–1001. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.S.; Dering, J.; Conklin, D.; Kalous, O.; Cohen, D.J.; Desai, A.J.; Ginther, C.; Atefi, M.; Chen, I.; Fowst, C.; et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009, 11, R77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knudsen, E.S.; Witkiewicz, A.K. The Strange Case of CDK4/6 Inhibitors: Mechanisms, Resistance, and Combination Strategies. Trends Cancer 2017, 3, 39–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brantley, M.A., Jr.; Harbour, J.W. Inactivation of retinoblastoma protein in uveal melanoma by phosphorylation of sites in the COOH-terminal region. Cancer Res. 2000, 60, 4320–4323. [Google Scholar]
- Hinds, P.W.; Mittnacht, S.; Dulic, V.; Arnold, A.; Reed, S.I.; Weinberg, R.A. Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell 1992, 70, 993–1006. [Google Scholar] [CrossRef]
- Lundberg, A.S.; Weinberg, R.A. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol. Cell Biol. 1998, 18, 753–761. [Google Scholar] [CrossRef] [Green Version]
- Alsina, M.; Landolfi, S.; Aura, C.; Caci, K.; Jimenez, J.; Prudkin, L.; Castro, S.; Moreno, D.; Navalpotro, B.; Tabernero, J.; et al. Cyclin E amplification/overexpression is associated with poor prognosis in gastric cancer. Ann. Oncol. 2015, 26, 438–439. [Google Scholar] [CrossRef]
- Velez-Cruz, R.; Manickavinayaham, S.; Biswas, A.K.; Clary, R.W.; Premkumar, T.; Cole, F.; Johnson, D.G. RB localizes to DNA double-strand breaks and promotes DNA end resection and homologous recombination through the recruitment of BRG1. Genes Dev. 2016, 30, 2500–2512. [Google Scholar] [CrossRef] [Green Version]
- Hays, E.; Nettleton, E.; Carter, C.; Morales, M.; Vo, L.; Passo, M.; Velez-Cruz, R. The SWI/SNF ATPase BRG1 stimulates DNA end resection and homologous recombination by reducing nucleosome density at DNA double strand breaks and by promoting the recruitment of the CtIP nuclease. Cell Cycle 2020, 19, 3096–3114. [Google Scholar] [CrossRef]
- Jiang, Y.; Yam, J.C.; Tham, C.C.; Pang, C.P.; Chu, W.K. RB Regulates DNA Double Strand Break Repair Pathway Choice by Mediating CtIP Dependent End Resection. Int. J. Mol. Sci. 2020, 21, 9176. [Google Scholar] [CrossRef]
- Chen, L.; Nievera, C.J.; Lee, A.Y.; Wu, X. Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J. Biol. Chem. 2008, 283, 7713–7720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, R.; Zoumpoulidou, G.; Luczynski, M.T.; Rieger, S.; Moquet, J.; Spanswick, V.J.; Hartley, J.A.; Rothkamm, K.; Huang, P.H.; Mittnacht, S. Direct involvement of retinoblastoma family proteins in DNA repair by non-homologous end-joining. Cell Rep. 2015, 10, 2006–2018. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, H.; Fox, S.R.; Gunawardena, R.W.; Knudsen, E.S. Loss of RB compromises specific heterochromatin modifications and modulates HP1alpha dynamics. J. Cell Physiol. 2007, 211, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Coschi, C.H.; Martens, A.L.; Ritchie, K.; Francis, S.M.; Chakrabarti, S.; Berube, N.G.; Dick, F.A. Mitotic chromosome condensation mediated by the retinoblastoma protein is tumor-suppressive. Genes Dev. 2010, 24, 1351–1363. [Google Scholar] [CrossRef] [Green Version]
- Coschi, C.H.; Ishak, C.A.; Gallo, D.; Marshall, A.; Talluri, S.; Wang, J.; Cecchini, M.J.; Martens, A.L.; Percy, V.; Welch, I.; et al. Haploinsufficiency of an RB-E2F1-Condensin II complex leads to aberrant replication and aneuploidy. Cancer Discov. 2014, 4, 840–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Nath, N.; Minden, A.; Chellappan, S. Regulation of Rb and E2F by signal transduction cascades: Divergent effects of JNK1 and p38 kinases. EMBO J. 1999, 18, 1559–1570. [Google Scholar] [CrossRef] [Green Version]
- Manning, A.L.; Longworth, M.S.; Dyson, N.J. Loss of pRB causes centromere dysfunction and chromosomal instability. Genes Dev. 2010, 24, 1364–1376. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.; Furge, K.; Koeman, J.; Dykema, K.; Su, Y.; Cutler, M.L.; Werts, A.; Haak, P.; Vande Woude, G.F. Chromosome instability, chromosome transcriptome, and clonal evolution of tumor cell populations. Proc. Natl. Acad. Sci. USA 2007, 104, 8995–9000. [Google Scholar] [CrossRef] [Green Version]
- Ruscetti, M.; Leibold, J.; Bott, M.J.; Fennell, M.; Kulick, A.; Salgado, N.R.; Chen, C.C.; Ho, Y.J.; Sanchez-Rivera, F.J.; Feucht, J.; et al. NK cell-mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science 2018, 362, 1416–1422. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabra, S.; Huang, W.; Liu, H.; Aref, A.R.; et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer Discov. 2018, 8, 216–233. [Google Scholar] [CrossRef] [Green Version]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Markey, M.P.; Bergseid, J.; Bosco, E.E.; Stengel, K.; Xu, H.; Mayhew, C.N.; Schwemberger, S.J.; Braden, W.A.; Jiang, Y.; Babcock, G.F.; et al. Loss of the retinoblastoma tumor suppressor: Differential action on transcriptional programs related to cell cycle control and immune function. Oncogene 2007, 26, 6307–6318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velez-Cruz, R.; Johnson, D.G. The Retinoblastoma (RB) Tumor Suppressor: Pushing Back against Genome Instability on Multiple Fronts. Int. J. Mol. Sci. 2017, 18, 1776. [Google Scholar] [CrossRef] [PubMed]
- Dick, F.A.; Goodrich, D.W.; Sage, J.; Dyson, N.J. Non-canonical functions of the RB protein in cancer. Nat. Rev. Cancer 2018, 18, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Selitsky, S.R.; Chai, S.; Armistead, P.M.; Vincent, B.G.; Serody, J.S. Alternative tumour-specific antigens. Nat. Rev. Cancer 2019, 19, 465–478. [Google Scholar] [CrossRef]
- Upadhyay, S.; Sharma, N.; Gupta, K.B.; Dhiman, M. Role of immune system in tumor progression and carcinogenesis. J. Cell Biochem. 2018, 119, 5028–5042. [Google Scholar] [CrossRef]
- Mardis, E.R. Neoantigens and genome instability: Impact on immunogenomic phenotypes and immunotherapy response. Genome Med. 2019, 11, 71. [Google Scholar] [CrossRef]
- Chae, Y.K.; Viveiros, P.; Lopes, G.; Sukhadia, B.; Sheikh, M.M.; Saravia, D.; Florou, V.; Sokol, E.S.; Frampton, G.M.; Chalmers, Z.R.; et al. Clinical and Immunological Implications of Frameshift Mutations in Lung Cancer. J. Thorac. Oncol. 2019, 14, 1807–1817. [Google Scholar] [CrossRef]
- Turajlic, S.; Litchfield, K.; Xu, H.; Rosenthal, R.; McGranahan, N.; Reading, J.L.; Wong, Y.N.S.; Rowan, A.; Kanu, N.; Al, B.M.; et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: A pan-cancer analysis. Lancet Oncol. 2017, 18, 1009–1021. [Google Scholar] [CrossRef] [Green Version]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellmann, M.D.; Ciuleanu, T.E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef] [PubMed]
- Bugide, S.; Janostiak, R.; Wajapeyee, N. Epigenetic Mechanisms Dictating Eradication of Cancer by Natural Killer Cells. Trends Cancer 2018, 4, 553–566. [Google Scholar] [CrossRef]
- Lentine, B.; Antonucci, L.; Hunce, R.; Edwards, J.; Marallano, V.; Krucher, N.A. Dephosphorylation of threonine-821 of the retinoblastoma tumor suppressor protein (Rb) is required for apoptosis induced by UV and Cdk inhibition. Cell Cycle 2012, 11, 3324–3330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gubern, A.; Joaquin, M.; Marques, M.; Maseres, P.; Garcia-Garcia, J.; Amat, R.; Gonzalez-Nunez, D.; Oliva, B.; Real, F.X.; de Nadal, E.; et al. The N-Terminal Phosphorylation of RB by p38 Bypasses Its Inactivation by CDKs and Prevents Proliferation in Cancer Cells. Mol. Cell 2016, 64, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Hassler, M.; Singh, S.; Yue, W.W.; Luczynski, M.; Lakbir, R.; Sanchez-Sanchez, F.; Bader, T.; Pearl, L.H.; Mittnacht, S. Crystal structure of the retinoblastoma protein N domain provides insight into tumor suppression, ligand interaction, and holoprotein architecture. Mol. Cell 2007, 28, 371–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dix, M.M.; Simon, G.M.; Wang, C.; Okerberg, E.; Patricelli, M.P.; Cravatt, B.F. Functional interplay between caspase cleavage and phosphorylation sculpts the apoptotic proteome. Cell 2012, 150, 426–440. [Google Scholar] [CrossRef] [Green Version]
- Burke, J.R.; Deshong, A.J.; Pelton, J.G.; Rubin, S.M. Phosphorylation-induced conformational changes in the retinoblastoma protein inhibit E2F transactivation domain binding. J. Biol. Chem. 2010, 285, 16286–16293. [Google Scholar] [CrossRef] [Green Version]
- Delston, R.B.; Matatall, K.A.; Sun, Y.; Onken, M.D.; Harbour, J.W. p38 phosphorylates Rb on Ser567 by a novel, cell cycle-independent mechanism that triggers Rb-Hdm2 interaction and apoptosis. Oncogene 2011, 30, 588–599. [Google Scholar] [CrossRef] [Green Version]
- Rousset-Roman, A.; Rebolloso-Gomez, Y.; Olivares-Illana, V. Expression and purification of the recombinant full-length retinoblastoma protein and characterisation of its interaction with the oncoprotein HDM2. Protein Expr. Purif. 2019, 162, 62–66. [Google Scholar] [CrossRef]
- Knudsen, E.S.; Wang, J.Y. Dual mechanisms for the inhibition of E2F binding to RB by cyclin-dependent kinase-mediated RB phosphorylation. Mol. Cell Biol. 1997, 17, 5771–5783. [Google Scholar] [CrossRef] [Green Version]
- Zarkowska, T.; Sally, U.; Harlow, E.; Mittnacht, S. Monoclonal antibodies specific for underphosphorylated retinoblastoma protein identify a cell cycle regulated phosphorylation site targeted by CDKs. Oncogene 1997, 14, 249–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, Y.; Kitagawa, M.; Taya, Y. Phosphorylation of pRB at Ser612 by Chk1/2 leads to a complex between pRB and E2F-1 after DNA damage. EMBO J. 2007, 26, 2083–2093. [Google Scholar] [CrossRef] [Green Version]
- Ledl, A.; Schmidt, D.; Muller, S. Viral oncoproteins E1A and E7 and cellular LxCxE proteins repress SUMO modification of the retinoblastoma tumor suppressor. Oncogene 2005, 24, 3810–3818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Kuehn, M.R. SENP1-modulated sumoylation regulates retinoblastoma protein (RB) and Lamin A/C interaction and stabilization. Oncogene 2016, 35, 6429–6438. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.Y.; Wang, D.H.; Campbell, M.; Huerta, S.B.; Shevchenko, B.; Izumiya, C.; Izumiya, Y. PRMT4-mediated arginine methylation negatively regulates retinoblastoma tumor suppressor protein and promotes E2F-1 dissociation. Mol. Cell Biol. 2015, 35, 238–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitagawa, M.; Higashi, H.; Jung, H.K.; Suzuki-Takahashi, I.; Ikeda, M.; Tamai, K.; Kato, J.; Segawa, K.; Yoshida, E.; Nishimura, S.; et al. The consensus motif for phosphorylation by cyclin D1-Cdk4 is different from that for phosphorylation by cyclin A/E-Cdk2. EMBO J. 1996, 15, 7060–7069. [Google Scholar] [CrossRef]
- Nair, J.S.; Ho, A.L.; Tse, A.N.; Coward, J.; Cheema, H.; Ambrosini, G.; Keen, N.; Schwartz, G.K. Aurora B kinase regulates the postmitotic endoreduplication checkpoint via phosphorylation of the retinoblastoma protein at serine 780. Mol. Biol. Cell 2009, 20, 2218–2228. [Google Scholar] [CrossRef] [Green Version]
- Mishra, S.; Melino, G.; Murphy, L.J. Transglutaminase 2 kinase activity facilitates protein kinase A-induced phosphorylation of retinoblastoma protein. J. Biol. Chem. 2007, 282, 18108–18115. [Google Scholar] [CrossRef] [Green Version]
- Nagano, K.; Itagaki, C.; Izumi, T.; Nunomura, K.; Soda, Y.; Tani, K.; Takahashi, N.; Takenawa, T.; Isobe, T. Rb plays a role in survival of Abl-dependent human tumor cells as a downstream effector of Abl tyrosine kinase. Oncogene 2006, 25, 493–502. [Google Scholar] [CrossRef] [Green Version]
- Carr, S.M.; Munro, S.; Kessler, B.; Oppermann, U.; La Thangue, N.B. Interplay between lysine methylation and Cdk phosphorylation in growth control by the retinoblastoma protein. EMBO J. 2011, 30, 317–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carr, S.M.; Munro, S.; Sagum, C.A.; Fedorov, O.; Bedford, M.T.; La Thangue, N.B. Tudor-domain protein PHF20L1 reads lysine methylated retinoblastoma tumour suppressor protein. Cell Death Differ. 2017, 24, 2139–2149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youn, C.K.; Cho, H.J.; Kim, S.H.; Kim, H.B.; Kim, M.H.; Chang, I.Y.; Lee, J.S.; Chung, M.H.; Hahm, K.S.; You, H.J. Bcl-2 expression suppresses mismatch repair activity through inhibition of E2F transcriptional activity. Nat. Cell Biol. 2005, 7, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; MacDonald, J.I.; Dick, F.A. Phosphorylation of the RB C-terminus regulates condensin II release from chromatin. J. Biol. Chem. 2021, 296, 100108. [Google Scholar] [CrossRef]
- Mertins, P.; Qiao, J.W.; Patel, J.; Udeshi, N.D.; Clauser, K.R.; Mani, D.R.; Burgess, M.W.; Gillette, M.A.; Jaffe, J.D.; Carr, S.A. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat. Methods 2013, 10, 634–637. [Google Scholar] [CrossRef]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Markham, D.; Munro, S.; Soloway, J.; O’Connor, D.P.; La Thangue, N.B. DNA-damage-responsive acetylation of pRb regulates binding to E2F-1. EMBO Rep. 2006, 7, 192–198. [Google Scholar] [CrossRef]
- Nguyen, D.X.; Baglia, L.A.; Huang, S.M.; Baker, C.M.; McCance, D.J. Acetylation regulates the differentiation-specific functions of the retinoblastoma protein. EMBO J. 2004, 23, 1609–1618. [Google Scholar] [CrossRef] [Green Version]
- Alcolea, M.P.; Casado, P.; Rodriguez-Prados, J.C.; Vanhaesebroeck, B.; Cutillas, P.R. Phosphoproteomic analysis of leukemia cells under basal and drug-treated conditions identifies markers of kinase pathway activation and mechanisms of resistance. Mol. Cell Proteomics 2012, 11, 453–466. [Google Scholar] [CrossRef] [Green Version]
- Kettenbach, A.N.; Schweppe, D.K.; Faherty, B.K.; Pechenick, D.; Pletnev, A.A.; Gerber, S.A. Quantitative phosphoproteomics identifies substrates and functional modules of Aurora and Polo-like kinase activities in mitotic cells. Sci. Signal. 2011, 4, rs5. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.T.; Tsai, C.F.; Hong, T.C.; Tsou, C.C.; Lin, P.Y.; Pan, S.H.; Hong, T.M.; Yang, P.C.; Sung, T.Y.; Hsu, W.L.; et al. An informatics-assisted label-free quantitation strategy that depicts phosphoproteomic profiles in lung cancer cell invasion. J. Proteome Res. 2010, 9, 5582–5597. [Google Scholar] [CrossRef] [PubMed]
- Stokes, M.P.; Farnsworth, C.L.; Moritz, A.; Silva, J.C.; Jia, X.; Lee, K.A.; Guo, A.; Polakiewicz, R.D.; Comb, M.J. PTMScan direct: Identification and quantification of peptides from critical signaling proteins by immunoaffinity enrichment coupled with LC-MS/MS. Mol. Cell Proteomics 2012, 11, 187–201. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.Q.; Yang, Q.K.; Lu, B.W.; Yi, W.; Cantin, G.; Chen, Y.L.; Fearns, C.; Yates, J.R., III; Lee, J.D. CDC25B mediates rapamycin-induced oncogenic responses in cancer cells. Cancer Res. 2009, 69, 2663–2668. [Google Scholar] [PubMed] [Green Version]
- Malec, V.; Coulson, J.M.; Urbe, S.; Clague, M.J. Combined Analyses of the VHL and Hypoxia Signaling Axes in an Isogenic Pairing of Renal Clear Cell Carcinoma Cells. J. Proteome Res. 2015, 14, 5263–5272. [Google Scholar] [CrossRef] [PubMed]
- Bennetzen, M.V.; Larsen, D.H.; Bunkenborg, J.; Bartek, J.; Lukas, J.; Andersen, J.S. Site-specific phosphorylation dynamics of the nuclear proteome during the DNA damage response. Mol. Cell Proteomics 2010, 9, 1314–1323. [Google Scholar] [PubMed] [Green Version]
- Stuart, S.A.; Houel, S.; Lee, T.; Wang, N.; Old, W.M.; Ahn, N.G. A Phosphoproteomic Comparison of B-RAFV600E and MKK1/2 Inhibitors in Melanoma Cells. Mol. Cell Proteomics 2015, 14, 1599–1615. [Google Scholar] [CrossRef] [Green Version]
- Dephoure, N.; Zhou, C.; Villen, J.; Beausoleil, S.A.; Bakalarski, C.E.; Elledge, S.J.; Gygi, S.P. A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. USA 2008, 105, 10762–10767. [Google Scholar] [CrossRef] [Green Version]
- Sharma, K.; D’Souza, R.C.; Tyanova, S.; Schaab, C.; Wisniewski, J.R.; Cox, J.; Mann, M. Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 2014, 8, 1583–1594. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Di, P.S.; Preisinger, C.; Peng, M.; Polat, A.N.; Heck, A.J.; Mohammed, S. Toward a comprehensive characterization of a human cancer cell phosphoproteome. J. Proteome Res. 2013, 12, 260–271. [Google Scholar] [CrossRef]
- Weinberg, R.A. The retinoblastoma protein and cell cycle control. Cell 1995, 81, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Connell-Crowley, L.; Harper, J.W.; Goodrich, D.W. Cyclin D1/Cdk4 regulates retinoblastoma protein-mediated cell cycle arrest by site-specific phosphorylation. Mol. Biol. Cell 1997, 8, 287–301. [Google Scholar] [CrossRef] [Green Version]
- Narasimha, A.M.; Kaulich, M.; Shapiro, G.S.; Choi, Y.J.; Sicinski, P.; Dowdy, S.F. Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. Elife 2014, 3, e02872. [Google Scholar] [CrossRef]
- Sanidas, I.; Morris, R.; Fella, K.A.; Rumde, P.H.; Boukhali, M.; Tai, E.C.; Ting, D.T.; Lawrence, M.S.; Haas, W.; Dyson, N.J. A Code of Mono-phosphorylation Modulates the Function of RB. Mol. Cell 2019, 73, 985–1000. [Google Scholar]
- Buchkovich, K.; Duffy, L.A.; Harlow, E. The retinoblastoma protein is phosphorylated during specific phases of the cell cycle. Cell 1989, 58, 1097–1105. [Google Scholar] [CrossRef]
- Ezhevsky, S.A.; Nagahara, H.; Vocero-Akbani, A.M.; Gius, D.R.; Wei, M.C.; Dowdy, S.F. Hypo-phosphorylation of the retinoblastoma protein (pRb) by cyclin D:Cdk4/6 complexes results in active pRb. Proc. Natl. Acad. Sci. USA 1997, 94, 10699–10704. [Google Scholar] [CrossRef] [Green Version]
- Zarkowska, T.; Mittnacht, S. Differential phosphorylation of the retinoblastoma protein by G1/S cyclin-dependent kinases. J. Biol. Chem. 1997, 272, 12738–12746. [Google Scholar] [CrossRef] [Green Version]
- Ren, S.; Rollins, B.J. Cyclin C/cdk3 promotes Rb-dependent G0 exit. Cell 2004, 117, 239–251. [Google Scholar] [CrossRef] [Green Version]
- Burke, J.R.; Hura, G.L.; Rubin, S.M. Structures of inactive retinoblastoma protein reveal multiple mechanisms for cell cycle control. Genes Dev. 2012, 26, 1156–1166. [Google Scholar] [CrossRef] [Green Version]
- Olsen, J.V.; Vermeulen, M.; Santamaria, A.; Kumar, C.; Miller, M.L.; Jensen, L.J.; Gnad, F.; Cox, J.; Jensen, T.S.; Nigg, E.A.; et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 2010, 3, ra3. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; Ertel, A.; McFalls, J.; Valsecchi, M.E.; Schwartz, G.; Knudsen, E.S. RB-pathway disruption is associated with improved response to neoadjuvant chemotherapy in breast cancer. Clin. Cancer Res. 2012, 18, 5110–5122. [Google Scholar] [CrossRef] [Green Version]
- Zagorski, W.A.; Knudsen, E.S.; Reed, M.F. Retinoblastoma deficiency increases chemosensitivity in lung cancer. Cancer Res. 2007, 67, 8264–8273. [Google Scholar] [CrossRef] [Green Version]
- Coussy, F.; El-Botty, R.; Chateau-Joubert, S.; Dahmani, A.; Montaudon, E.; Leboucher, S.; Morisset, L.; Painsec, P.; Sourd, L.; Huguet, L.; et al. BRCAness, SLFN11, and RB1 loss predict response to topoisomerase I inhibitors in triple-negative breast cancers. Sci. Transl. Med. 2020, 12, eaax2625. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Limon, A.; Joaquin, M.; Caballero, M.; Posas, F.; de Nadal, E. The p38 Pathway: From Biology to Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 1913. [Google Scholar] [CrossRef] [Green Version]
- Faust, D.; Schmitt, C.; Oesch, F.; Oesch-Bartlomowicz, B.; Schreck, I.; Weiss, C.; Dietrich, C. Differential p38-dependent signalling in response to cellular stress and mitogenic stimulation in fibroblasts. Cell Commun. Signal. 2012, 10, 6. [Google Scholar] [CrossRef] [Green Version]
- Joaquin, M.; de Nadal, E.; Posas, F. An RB insensitive to CDK regulation. Mol. Cell. Oncol. 2016, 4, e1268242. [Google Scholar] [CrossRef] [Green Version]
- Yu-Lee, L.Y.; Yu, G.; Lee, Y.C.; Lin, S.C.; Pan, J.; Pan, T.; Yu, K.J.; Liu, B.; Creighton, C.J.; Rodriguez-Canales, J.; et al. Osteoblast-Secreted Factors Mediate Dormancy of Metastatic Prostate Cancer in the Bone via Activation of the TGFbetaRIII-p38MAPK-pS249/T252RB Pathway. Cancer Res. 2018, 78, 2911–2924. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Ferraris, J.D.; Izumi, Y.; Dmitrieva, N.; Ramkissoon, K.; Wang, G.; Gucek, M.; Burg, M.B. Global discovery of high-NaCl-induced changes of protein phosphorylation. Am. J. Physiol Cell Physiol. 2014, 307, C442–C454. [Google Scholar] [CrossRef] [Green Version]
- Carnevale, J.; Palander, O.; Seifried, L.A.; Dick, F.A. DNA damage signals through differentially modified E2F1 molecules to induce apoptosis. Mol. Cell Biol. 2012, 32, 900–912. [Google Scholar] [CrossRef] [Green Version]
- Tomas-Loba, A.; Manieri, E.; Gonzalez-Teran, B.; Mora, A.; Leiva-Vega, L.; Santamans, A.M.; Romero-Becerra, R.; Rodriguez, E.; Pintor-Chocano, A.; Feixas, F.; et al. p38gamma is essential for cell cycle progression and liver tumorigenesis. Nature 2019, 568, 557–560. [Google Scholar] [CrossRef] [Green Version]
- Gong, X.; Du, J.; Parsons, S.H.; Merzoug, F.F.; Webster, Y.; Iversen, P.W.; Chio, L.C.; Van Horn, R.D.; Lin, X.; Blosser, W.; et al. Aurora A Kinase Inhibition Is Synthetic Lethal with Loss of the RB1 Tumor Suppressor Gene. Cancer Discov. 2019, 9, 248–263. [Google Scholar] [CrossRef] [Green Version]
- Oser, M.G.; Fonseca, R.; Chakraborty, A.A.; Brough, R.; Spektor, A.; Jennings, R.B.; Flaifel, A.; Novak, J.S.; Gulati, A.; Buss, E.; et al. Cells Lacking the RB1 Tumor Suppressor Gene Are Hyperdependent on Aurora B Kinase for Survival. Cancer Discov. 2019, 9, 230–247. [Google Scholar] [CrossRef] [Green Version]
- Lyu, J.; Yang, E.J.; Zhang, B.; Wu, C.; Pardeshi, L.; Shi, C.; Mou, P.K.; Liu, Y.; Tan, K.; Shim, J.S. Synthetic lethality of RB1 and aurora A is driven by stathmin-mediated disruption of microtubule dynamics. Nat. Commun. 2020, 11, 5105. [Google Scholar] [CrossRef]
- Alberts, A.S.; Thorburn, A.M.; Shenolikar, S.; Mumby, M.C.; Feramisco, J.R. Regulation of cell cycle progression and nuclear affinity of the retinoblastoma protein by protein phosphatases. Proc. Natl. Acad. Sci. USA 1993, 90, 388–392. [Google Scholar] [CrossRef] [Green Version]
- Ludlow, J.W.; Glendening, C.L.; Livingston, D.M.; DeCarprio, J.A. Specific enzymatic dephosphorylation of the retinoblastoma protein. Mol. Cell Biol. 1993, 13, 367–372. [Google Scholar]
- Dohadwala, M.; da Cruz e Silva, E.F.; Hall, F.L.; Williams, R.T.; Carbonaro-Hall, D.A.; Nairn, A.C.; Greengard, P.; Berndt, N. Phosphorylation and inactivation of protein phosphatase 1 by cyclin-dependent kinases. Proc. Natl. Acad. Sci. USA 1994, 91, 6408–6412. [Google Scholar] [CrossRef] [Green Version]
- Berndt, N.; Dohadwala, M.; Liu, C.W. Constitutively active protein phosphatase 1alpha causes Rb-dependent G1 arrest in human cancer cells. Curr. Biol. 1997, 7, 375–386. [Google Scholar] [CrossRef] [Green Version]
- Tamrakar, S.; Ludlow, J.W. The carboxyl-terminal region of the retinoblastoma protein binds non-competitively to protein phosphatase type 1alpha and inhibits catalytic activity. J. Biol. Chem. 2000, 275, 27784–27789. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, M.; Villa-Moruzzi, E. Binding of phosphatase-1 delta to the retinoblastoma protein pRb involves domains that include substrate recognition residues and a pRB binding motif. Biochem. Biophys. Res. Commun. 2001, 280, 1–3. [Google Scholar] [CrossRef]
- Tamrakar, S.; Mittnacht, S.; Ludlow, J.W. Binding of select forms of pRB to protein phosphatase type 1 independent of catalytic activity. Oncogene 1999, 18, 7803–7809. [Google Scholar]
- Rubin, E.; Mittnacht, S.; Villa-Moruzzi, E.; Ludlow, J.W. Site-specific and temporally-regulated retinoblastoma protein dephosphorylation by protein phosphatase type 1. Oncogene 2001, 20, 3776–3785. [Google Scholar] [CrossRef] [Green Version]
- Kiss, A.; Lontay, B.; Becsi, B.; Markasz, L.; Olah, E.; Gergely, P.; Erdodi, F. Myosin phosphatase interacts with and dephosphorylates the retinoblastoma protein in THP-1 leukemic cells: Its inhibition is involved in the attenuation of daunorubicin-induced cell death by calyculin-A. Cell Signal. 2008, 20, 2059–2070. [Google Scholar] [CrossRef]
- Udho, E.; Tedesco, V.C.; Zygmunt, A.; Krucher, N.A. PNUTS (phosphatase nuclear targeting subunit) inhibits retinoblastoma-directed PP1 activity. Biochem. Biophys. Res. Commun. 2002, 297, 463–467. [Google Scholar] [CrossRef]
- Verdugo-Sivianes, E.M.; Carnero, A. Role of the Holoenzyme PP1-SPN in the Dephosphorylation of the RB Family of Tumor Suppressors During Cell Cycle. Cancers 2021, 13, 2226. [Google Scholar] [CrossRef]
- Garriga, J.; Jayaraman, A.L.; Limon, A.; Jayadeva, G.; Sotillo, E.; Truongcao, M.; Patsialou, A.; Wadzinski, B.E.; Grana, X. A dynamic equilibrium between CDKs and PP2A modulates phosphorylation of pRB, p107 and p130. Cell Cycle 2004, 3, 1320–1330. [Google Scholar] [CrossRef]
- Avni, D.; Yang, H.; Martelli, F.; Hofmann, F.; ElShamy, W.M.; Ganesan, S.; Scully, R.; Livingston, D.M. Active localization of the retinoblastoma protein in chromatin and its response to S phase DNA damage. Mol. Cell 2003, 12, 735–746. [Google Scholar] [CrossRef]
- Cicchillitti, L.; Fasanaro, P.; Biglioli, P.; Capogrossi, M.C.; Martelli, F. Oxidative stress induces protein phosphatase 2A-dependent dephosphorylation of the pocket proteins pRb, p107, and p130. J. Biol. Chem. 2003, 278, 19509–19517. [Google Scholar] [CrossRef] [Green Version]
- Voorhoeve, P.M.; Watson, R.J.; Farlie, P.G.; Bernards, R.; Lam, E.W. Rapid dephosphorylation of p107 following UV irradiation. Oncogene 1999, 18, 679–688. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [Green Version]
- Chan, H.M.; Krstic-Demonacos, M.; Smith, L.; Demonacos, C.; La Thangue, N.B. Acetylation control of the retinoblastoma tumour-suppressor protein. Nat. Cell Biol. 2001, 3, 667–674. [Google Scholar] [CrossRef]
- Lin, W.C.; Lin, F.T.; Nevins, J.R. Selective induction of E2F1 in response to DNA damage, mediated by ATM-dependent phosphorylation. Genes Dev. 2001, 15, 1833–1844. [Google Scholar]
- Stevens, C.; Smith, L.; La Thangue, N.B. Chk2 activates E2F-1 in response to DNA damage. Nat. Cell Biol. 2003, 5, 401–409. [Google Scholar] [CrossRef]
- Carr, S.M.; Munro, S.; Zalmas, L.P.; Fedorov, O.; Johansson, C.; Krojer, T.; Sagum, C.A.; Bedford, M.T.; Oppermann, U.; La Thangue, N.B. Lysine methylation-dependent binding of 53BP1 to the pRb tumor suppressor. Proc. Natl. Acad. Sci. USA 2014, 111, 11341–11346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trere, D.; Brighenti, E.; Donati, G.; Ceccarelli, C.; Santini, D.; Taffurelli, M.; Montanaro, L.; Derenzini, M. High prevalence of retinoblastoma protein loss in triple-negative breast cancers and its association with a good prognosis in patients treated with adjuvant chemotherapy. Ann. Oncol. 2009, 20, 1818–1823. [Google Scholar] [CrossRef] [PubMed]
- Robinson, T.J.; Liu, J.C.; Vizeacoumar, F.; Sun, T.; Maclean, N.; Egan, S.E.; Schimmer, A.D.; Datti, A.; Zacksenhaus, E. RB1 status in triple negative breast cancer cells dictates response to radiation treatment and selective therapeutic drugs. PLoS ONE 2013, 8, e78641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
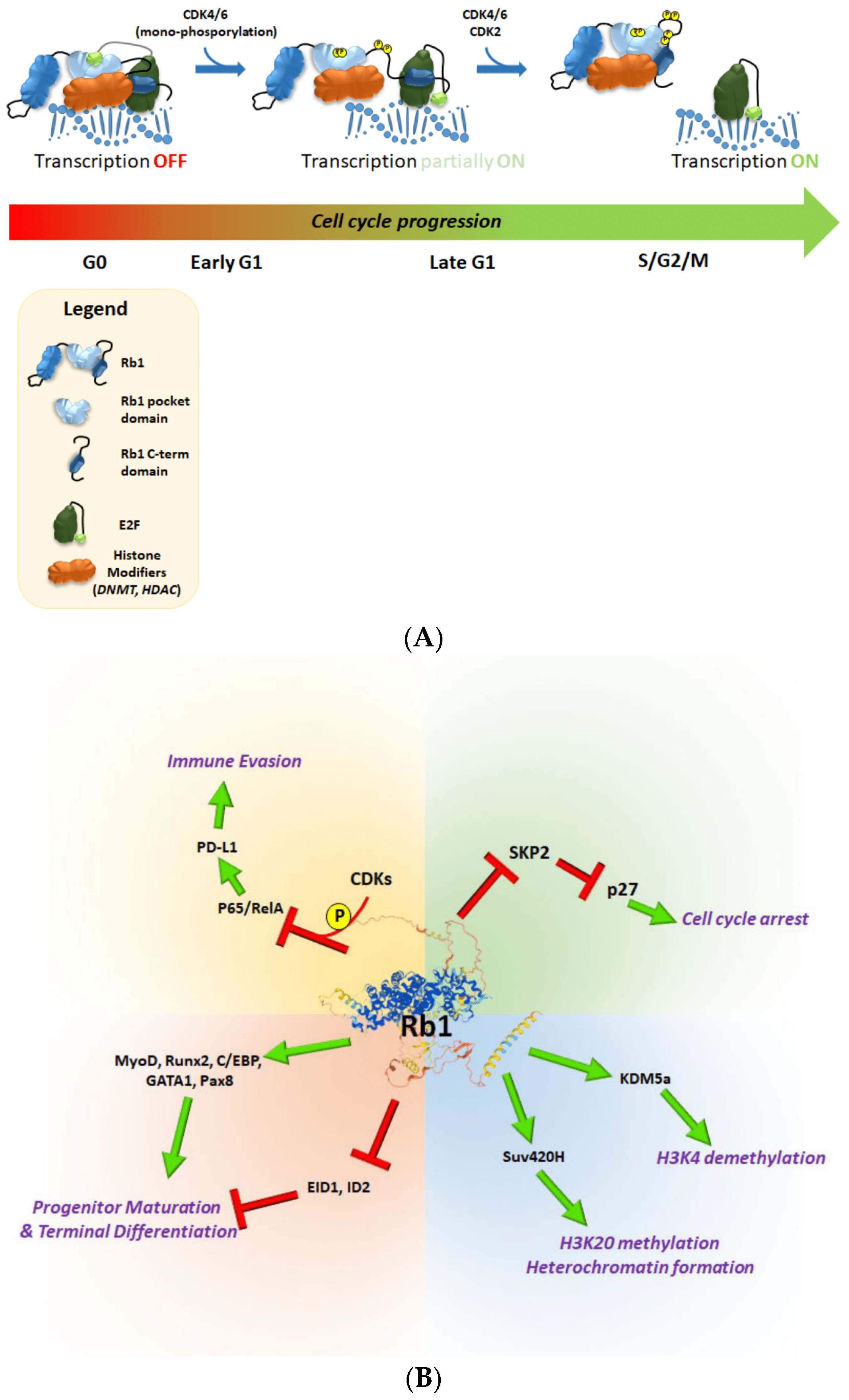
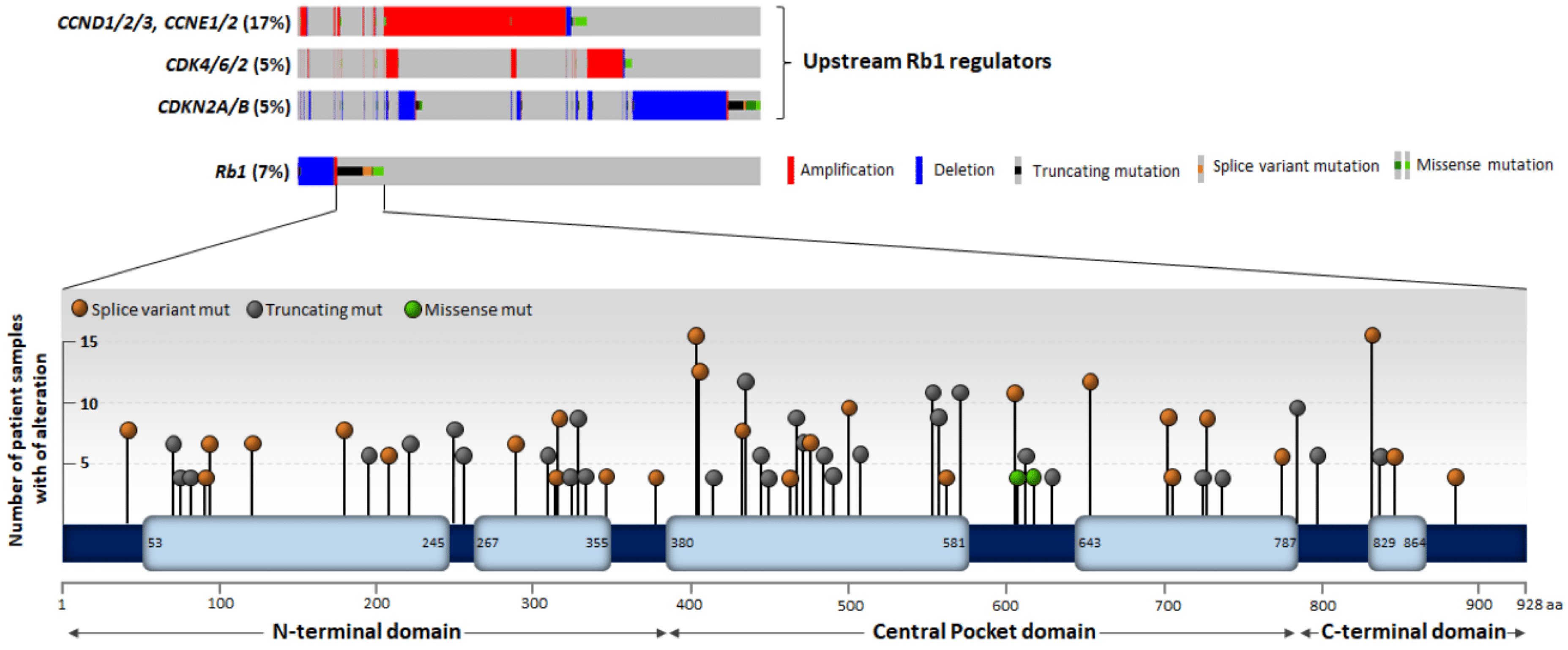


{kind=link}
{kind=link}
{kind=link}
| Site | Modification | Modifier | Molecular Function | Outcome | Reference |
|---|---|---|---|---|---|
| T5 | Phosphorylation | N.A. | N.A. | Apoptosis inhibition | [124] |
| S230 | Phosphorylation | N.A. | N.A. | Apoptosis inhibition | [124] |
| S249 | Phosphorylation | p38α | Increase affinity to E2F1 | Cell cycle inhibition | [125] |
| CDK1/2 | Reduce affinity to HDAC5 and EID1 | Transcription regulation | [31] [126] | ||
| T252 | Phosphorylation | p38α | Increase affinity to E2F1 | Cell cycle inhibition | [125] |
| CDK1/2 | Reduce affinity to HDAC5 and EID1 | Transcription regulation | [31] [126] | ||
| S347 | Phosphorylation | N/A | Promote caspase cleavage | Increased Rb proteolysis during apoptosis | [127] |
| T356 | Phosphorylation | CDK2 | Reduce affinity to E2F1 | Cell cycle entry | [124,128] |
| T373 | Phosphorylation | CDKs | Reduce affinity to E2F1 | Cell cycle entry | [128] |
| S567 | Phosphorylation | p38α | Increase affinity to HDM2 | Rb degradation and apoptosis | [129,130] |
| S608 | Phosphorylation | CDK2 | Reduce affinity to E2F1 | Cell cycle entry | [128,131,132] |
| S612 | Phosphorylation | CDK2 | Reduce affinity to E2F1 | Cell cycle entry | [128] |
| Chk1/ Chk2 | Increase affinity to E2F1 | Cell survival upon DNA damage | [133] | ||
| K720 | Sumoylation | N/A | Reduce affinity to E2F1 | Cell cycle entry | [134,135] |
| R775 | Methylation | PRMT4 | Reduce affinity to E2F1 | Cell cycle entry | [136] |
| S780 | Phosphorylation | CDK4 | Reduce affinity to E2F1 | Cell cycle entry | [137] |
| Aurora B | Increase affinity to E2F1 | Prevents endoreduplication | [138] | ||
| TG2 | Reduce affinity to E2F1 | Cell cycle entry | [139] | ||
| R787 | Methylation | PRMT4 | Reduce affinity to E2F1 | Cell cycle entry | [136] |
| S788 | Phosphorylation | CDKs | Reduce affinity to E2F1 | Cell cycle entry | [23] |
| S795 | Phosphorylation | CDK4 | Reduce affinity to E2F1 | Cell cycle entry | [23] |
| R798 | Methylation | PRMT4 | Reduce affinity to E2F1 | Cell cycle entry | [136] |
| Y805 | Phosphorylation | Abl tyrosine kinase | N.A. | Necessary for survival of Abl-dependent tumor cells | [140] |
| S807 | Phosphorylation | CDKs | Reduce affinity to E2F1 | Cell cycle entry | [131] |
| K810 | Methylation | Set7/9 Smyd2 | Inhibits Cdk-directed phosphorylation | Cell cycle arrest | [141,142] |
| S811 | Phosphorylation | CDKs | Reduce affinity to E2F1 | Cell cycle entry | [131,143] |
| T821 | Phosphorylation | CDK2 | Reduce affinity to E2F1 | Cell cycle entry | [23] |
| Reduce affinity to HDAC5 | [31] | ||||
| T826 | Phosphorylation | CDKs | Reduce affinity to E2F1 | Cell cycle entry | [23] |
| S838 | Phosphorylation | p38α | Disrupts condensin II interaction with chromatin | Chromatin decondensation | [144,145] |
| T841 | Phosphorylation | p38α | Disrupts condensin II interaction with chromatin | Chromatin decondensation | [144,146] |
| K873 | Acetylation | N/A | Increase affinity to MDM2 Reduce affinity to E2F1 | Cell cycle exit and cell differentiation | [147,148] |
| K874 | Acetylation | N/A | Increase affinity to MDM2 | Cell cycle exit and cell differentiation | [147,148] |
| S882 | Phosphorylation | N.A. | Promote caspase cleavage | Increased Rb proteolysis | [127,147] |
| Rb modification without assigned function (identified through HTP proteomics) | |||||
| S350 | Phosphorylation | [145] | |||
| T353 | Phosphorylation | [149,150,151] | |||
| S624 | Phosphorylation | [145] | |||
| T774 | Phosphorylation | [152] | |||
| T778 | Phosphorylation | [145,152] | |||
| Y790 | Phosphorylation | [152,153] | |||
| Y813 | Phosphorylation | [153] | |||
| S816 | Phosphorylation | [154,155,156] | |||
| T823 | Phosphorylation | [146,156,157] | |||
| 855 | Phosphorylation | [145,158,159] | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janostiak, R.; Torres-Sanchez, A.; Posas, F.; de Nadal, E. Understanding Retinoblastoma Post-Translational Regulation for the Design of Targeted Cancer Therapies. Cancers 2022, 14, 1265. https://doi.org/10.3390/cancers14051265
Janostiak R, Torres-Sanchez A, Posas F, de Nadal E. Understanding Retinoblastoma Post-Translational Regulation for the Design of Targeted Cancer Therapies. Cancers. 2022; 14(5):1265. https://doi.org/10.3390/cancers14051265
Chicago/Turabian StyleJanostiak, Radoslav, Ariadna Torres-Sanchez, Francesc Posas, and Eulàlia de Nadal. 2022. "Understanding Retinoblastoma Post-Translational Regulation for the Design of Targeted Cancer Therapies" Cancers 14, no. 5: 1265. https://doi.org/10.3390/cancers14051265
APA StyleJanostiak, R., Torres-Sanchez, A., Posas, F., & de Nadal, E. (2022). Understanding Retinoblastoma Post-Translational Regulation for the Design of Targeted Cancer Therapies. Cancers, 14(5), 1265. https://doi.org/10.3390/cancers14051265