
Targeting the DNA Damage Response Pathway as a Novel Therapeutic Strategy in Colorectal Cancer

 , , , ,
, , , , 
Abstract
:Simple Summary
Abstract
1. Introduction
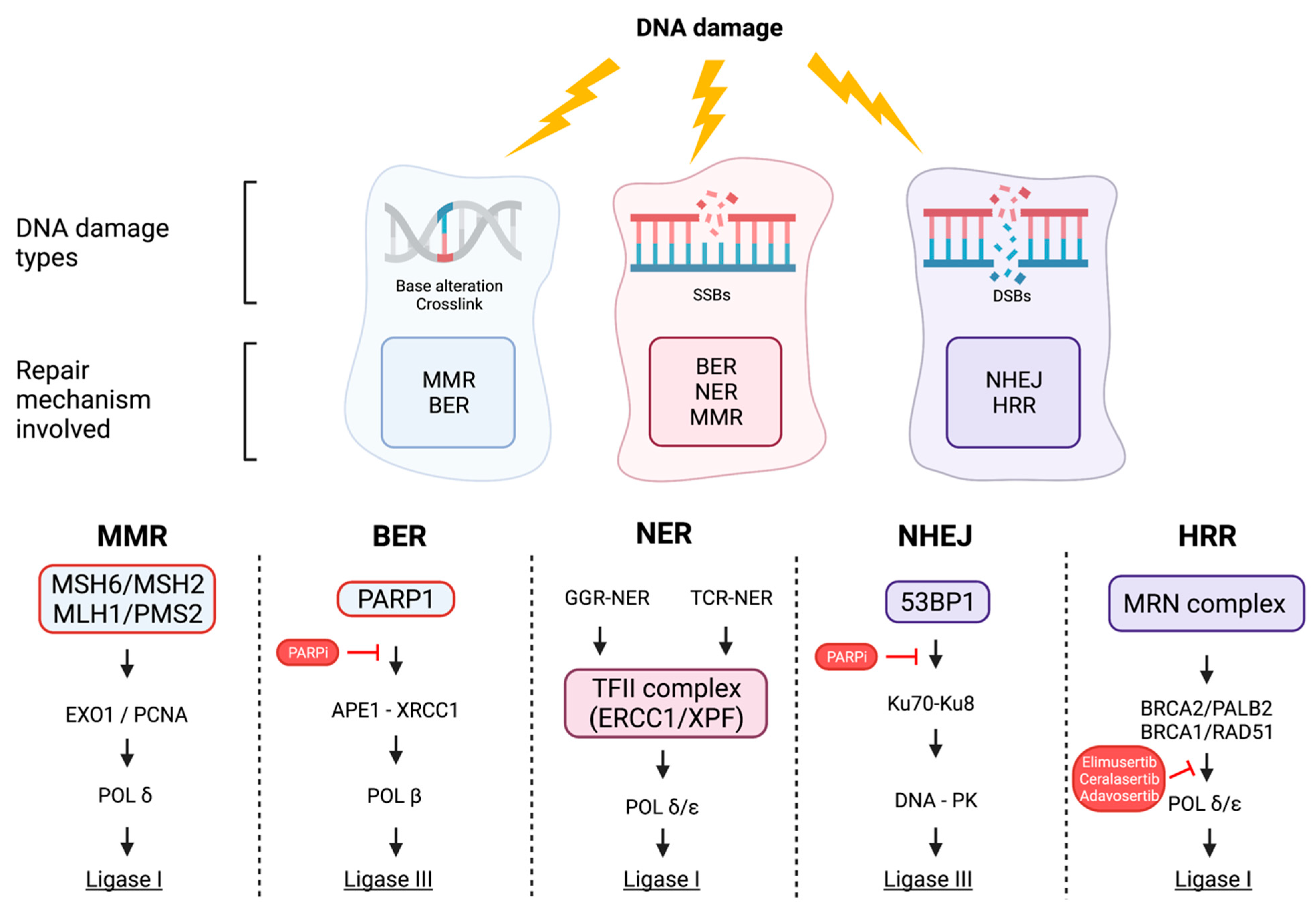
2. The DNA Damage Response (DDR) Pathway
3. Biomarkers of DDR: How to Select Patients
4. Preclinical Data in Colorectal Cancer
5. Clinical Data and Ongoing Trials in CRC
5.1. PARP Inhibitors
5.2. Not Only PARPi: Other Inhibitors of the DDR System
6. Hereditary Implications
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Prz. Gastroenterol. 2019, 14, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Mattiuzzi, C.; Sanchis-Gomar, F.; Lippi, G. Concise update on colorectal cancer epidemiology. Ann. Transl. Med. 2019, 7, 609. [Google Scholar] [CrossRef] [PubMed]
- Benson, A.B.; Venook, A.P.; Al-Hawary, M.M.; Arain, M.A.; Chen, Y.J.; Ciombor, K.K.; Cohen, S.; Cooper, H.S.; Deming, D.; Farkas, L.; et al. Colon Cancer, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2021, 19, 329–359. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Cervantes, A.; Adam, R.; Sobrero, A.; Van Krieken, J.H.; Aderka, D.; Aranda Aguilar, E.; Bardelli, A.; Benson, A.; Bodoky, G.; et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann. Oncol. 2016, 27, 1386–1422. [Google Scholar] [CrossRef]
- Antoniotti, C.; Moretto, R.; Rossini, D.; Masi, G.; Falcone, A.; Cremolini, C. Treatments after first progression in metastatic colorectal cancer. A literature review and evidence-based algorithm. Cancer Treat. Rev. 2021, 92, 102135. [Google Scholar] [CrossRef] [PubMed]
- Cremolini, C.; Antoniotti, C.; Moretto, R.; Masi, G.; Falcone, A. First-line therapy for mCRC—The influence of primary tumour location on the therapeutic algorithm. Nat. Rev. Clin. Oncol. 2017, 14, 113. [Google Scholar] [CrossRef] [Green Version]
- Van Cutsem, E.; Cervantes, A.; Nordlinger, B.; Arnold, D.; Group, E.G.W. Metastatic colorectal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2014, 25 (Suppl. 3), iii1–iii9. [Google Scholar] [CrossRef] [PubMed]
- Macedo, L.T.; da Costa Lima, A.B.; Sasse, A.D. Addition of bevacizumab to first-line chemotherapy in advanced colorectal cancer: A systematic review and meta-analysis, with emphasis on chemotherapy subgroups. BMC Cancer 2012, 12, 89. [Google Scholar] [CrossRef] [Green Version]
- Mao, C.; Liao, R.Y.; Qiu, L.X.; Wang, X.W.; Ding, H.; Chen, Q. BRAF V600E mutation and resistance to anti-EGFR monoclonal antibodies in patients with metastatic colorectal cancer: A meta-analysis. Mol. Biol. Rep. 2011, 38, 2219–2223. [Google Scholar] [CrossRef]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef] [Green Version]
- Sartore-Bianchi, A.; Trusolino, L.; Martino, C.; Bencardino, K.; Lonardi, S.; Bergamo, F.; Zagonel, V.; Leone, F.; Depetris, I.; Martinelli, E.; et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): A proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 738–746. [Google Scholar] [CrossRef]
- Strickler, J.H.; Ng, K.; Cercek, A.; Fountzilas, C.; Sanchez, F.A.; Hubbard, J.M.; Wu, C.; Siena, S.; Tabernero, J.; Cutsem, E.V.; et al. MOUNTAINEER:open-label, phase II study of tucatinib combined with trastuzumab for HER2-positive metastatic colorectal cancer (SGNTUC-017, trial in progress). J. Clin. Oncol. 2021, 39, TPS153. [Google Scholar] [CrossRef]
- Siena, S.; Di Bartolomeo, M.; Raghav, K.; Masuishi, T.; Loupakis, F.; Kawakami, H.; Yamaguchi, K.; Nishina, T.; Fakih, M.; Elez, E.; et al. Trastuzumab deruxtecan (DS-8201) in patients with HER2-expressing metastatic colorectal cancer (DESTINY-CRC01): A multicentre, open-label, phase 2 trial. Lancet Oncol. 2021, 22, 779–789. [Google Scholar] [CrossRef]
- Yoshino, T.; Bartolomeo, M.D.; Raghav, K.P.S.; Masuishi, T.; Kawakami, H.; Yamaguchi, K.; Nishina, T.; Wainberg, Z.A.; Elez, E.; Rodriguez, J.; et al. Trastuzumab deruxtecan (T-DXd; DS-8201) in patients (pts) with HER2-expressing metastatic colorectal cancer (mCRC): Final results from a phase 2, multicenter, open-label study (DESTINY-CRC01). J. Clin. Oncol. 2022, 40, 119. [Google Scholar] [CrossRef]
- Andre, T.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Lenz, H.-J.; Cutsem, E.V.; Limon, M.L.; Wong, K.Y.M.; Hendlisz, A.; Aglietta, M.; García-Alfonso, P.; Neyns, B.; Luppi, G.; Cardin, D.B.; et al. First-Line Nivolumab Plus Low-Dose Ipilimumab for Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: The Phase II CheckMate 142 Study. J. Clin. Oncol. 2022, 40, 161–170. [Google Scholar] [CrossRef]
- Siena, S.; Demetri, G.; Doebele, R.; Chae, Y.; Conkling, P.; Garrido-Laguna, I.; Garrido, P.; Rolfo, C.; Sigal, D.; Eng, S.; et al. Entrectinib in NTRK-fusion positive gastrointestinal cancers: Integrated analysis of patients enrolled in three trials (STARTRK-2, STARTRK-1, and ALKA-372-001). Ann. Oncol. 2019, 30, iv134. [Google Scholar] [CrossRef]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Jiang, M.; Jia, K.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Wang, H.; Zhao, S.; He, Y.; Zhou, C. Alterations of DNA damage response pathway: Biomarker and therapeutic strategy for cancer immunotherapy. Acta Pharm. Sin. B 2021, 11, 2983–2994. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Perol, D.; Gonzalez-Martin, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Maenpaa, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- Collins, T.S.; Hurwitz, H.I. Targeting vascular endothelial growth factor and angiogenesis for the treatment of colorectal cancer. Semin. Oncol. 2005, 32, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, D.C.; Chalmers, A.J.; El-Khamisy, S.F. Topoisomerase I inhibition in colorectal cancer: Biomarkers and therapeutic targets. Br. J. Cancer 2012, 106, 18–24. [Google Scholar] [CrossRef] [Green Version]
- Oh, M.; McBride, A.; Yun, S.; Bhattacharjee, S.; Slack, M.; Martin, J.R.; Jeter, J.; Abraham, I. BRCA1 and BRCA2 Gene Mutations and Colorectal Cancer Risk: Systematic Review and Meta-analysis. J. Natl. Cancer Inst. 2018, 110, 1178–1189. [Google Scholar] [CrossRef] [Green Version]
- AlDubayan, S.H.; Giannakis, M.; Moore, N.D.; Han, G.C.; Reardon, B.; Hamada, T.; Mu, X.J.; Nishihara, R.; Qian, Z.; Liu, L.; et al. Inherited DNA-Repair Defects in Colorectal Cancer. Am. J. Hum. Genet. 2018, 102, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Heeke, A.L.; Pishvaian, M.J.; Lynce, F.; Xiu, J.; Brody, J.R.; Chen, W.J.; Baker, T.M.; Marshall, J.L.; Isaacs, C. Prevalence of Homologous Recombination-Related Gene Mutations Across Multiple Cancer Types. JCO Precis. Oncol. 2018, 2018, 1–13. [Google Scholar] [CrossRef]
- Moretto, R.; Elliott, A.; Zhang, J.; Arai, H.; Germani, M.M.; Conca, V.; Xiu, J.; Stafford, P.; Oberley, M.; Abraham, J.; et al. Homologous Recombination Deficiency Alterations in Colorectal Cancer: Clinical, Molecular, and Prognostic Implications. J. Natl. Cancer Inst. 2021, 114, 271–279. [Google Scholar] [CrossRef]
- Arai, H.; Elliott, A.; Xiu, J.; Wang, J.; Battaglin, F.; Kawanishi, N.; Soni, S.; Zhang, W.; Millstein, J.; Sohal, D.; et al. The Landscape of Alterations in DNA Damage Response Pathways in Colorectal Cancer. Clin. Cancer Res. 2021, 27, 3234–3242. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, K.; Kocher, F.; Puccini, A.; Seeber, A. Targeting BRCA and DNA Damage Repair Genes in GI Cancers: Pathophysiology and Clinical Perspectives. Front. Oncol. 2021, 11, 662055. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Jia, K.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Wang, H.; Zhao, S.; He, Y.; Zhou, C. Alterations of DNA damage repair in cancer: From mechanisms to applications. Ann. Transl. Med. 2020, 8, 1685. [Google Scholar] [CrossRef]
- Gerson, S.L. MGMT: Its role in cancer aetiology and cancer therapeutics. Nat. Rev. Cancer 2004, 4, 296–307. [Google Scholar] [CrossRef]
- Kaina, B.; Christmann, M.; Naumann, S.; Roos, W.P. MGMT: Key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair 2007, 6, 1079–1099. [Google Scholar] [CrossRef]
- Kumar, M.; Shukla, V.K.; Misra, P.K.; Raman, M.J. Dysregulated Expression and Subcellular Localization of Base Excision Repair (BER) Pathway Enzymes in Gallbladder Cancer. Int. J. Mol. Cell. Med. 2018, 7, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Pecina-Slaus, N.; Kafka, A.; Salamon, I.; Bukovac, A. Mismatch Repair Pathway, Genome Stability and Cancer. Front. Mol. Biosci. 2020, 7, 122. [Google Scholar] [CrossRef] [PubMed]
- Trenner, A.; Sartori, A.A. Harnessing DNA Double-Strand Break Repair for Cancer Treatment. Front. Oncol. 2019, 9, 1388. [Google Scholar] [CrossRef]
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef] [Green Version]
- Daley, J.M.; Sung, P. 53BP1, BRCA1, and the choice between recombination and end joining at DNA double-strand breaks. Mol. Cell Biol. 2014, 34, 1380–1388. [Google Scholar] [CrossRef] [Green Version]
- Difilippantonio, M.J.; Zhu, J.; Chen, H.T.; Meffre, E.; Nussenzweig, M.C.; Max, E.E.; Ried, T.; Nussenzweig, A. DNA repair protein Ku80 suppresses chromosomal aberrations and malignant transformation. Nature 2000, 404, 510–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef] [PubMed]
- Pilie, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef] [PubMed]
- Yurgelun, M.B.; Kulke, M.H.; Fuchs, C.S.; Allen, B.A.; Uno, H.; Hornick, J.L.; Ukaegbu, C.I.; Brais, L.K.; McNamara, P.G.; Mayer, R.J.; et al. Cancer Susceptibility Gene Mutations in Individuals with Colorectal Cancer. J. Clin. Oncol. 2017, 35, 1086–1095. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.M.; Lonigro, R.J.; Vats, P.; Cobain, E.; Everett, J.; Cao, X.; Rabban, E.; Kumar-Sinha, C.; Raymond, V.; et al. Integrative clinical genomics of metastatic cancer. Nature 2017, 548, 297–303. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Sishc, B.J.; Davis, A.J. The Role of the Core Non-Homologous End Joining Factors in Carcinogenesis and Cancer. Cancers 2017, 9, 81. [Google Scholar] [CrossRef] [Green Version]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across the Cancer Genome Atlas. Cell Rep. 2018, 23, 239–254.e6. [Google Scholar] [CrossRef] [Green Version]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef]
- NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines® Breast Cancer Version 8.2021. 13 September 2021. Available online: https://www.nccn.org/professionals/physician_gls/pdf/breast.pdf (accessed on 10 February 2022).
- NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines® Ovarian Cancer Version 3.2021. 9 September 2021. Available online: https://www.nccn.org/professionals/physician_gls/pdf/ovarian.pdf (accessed on 10 February 2022).
- FDA Approves Olaparib for HRR Gene-Mutated Metastatic Castration-Resistant Prostate Cancer. 19 May 2020. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-olaparib-hrr-gene-mutated-metastatic-castration-resistant-prostate-cancer (accessed on 10 February 2022).
- FDA Grants Accelerated Approval to Rucaparib for BRCA-Mutated Metastatic Castration-Resistant Prostate Cancer. 15 May 2020. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-rucaparib-brca-mutated-metastatic-castration-resistant-prostate (accessed on 10 February 2022).
- Golan, T.; Hammel, P.; Reni, M.; Cutsem, E.V.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Overall survival from the phase 3 POLO trial: Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. J. Clin. Oncol. 2021, 39, 378. [Google Scholar] [CrossRef]
- FDA Approves Olaparib for gBRCAm. Metastatic Pancreatic Adenocarcinoma. 27 December 2019. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-olaparib-gbrcam-metastatic-pancreatic-adenocarcinoma (accessed on 10 February 2022).
- Cullinane, C.M.; Creavin, B.; O’Connell, E.P.; Kelly, L.; O’Sullivan, M.J.; Corrigan, M.A.; Redmond, H.P. Risk of colorectal cancer associated with BRCA1 and/or BRCA2 mutation carriers: Systematic review and meta-analysis. Br. J. Surg. 2020, 107, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Mauri, G.; Sartore-Bianchi, A.; Russo, A.G.; Marsoni, S.; Bardelli, A.; Siena, S. Early-onset colorectal cancer in young individuals. Mol. Oncol. 2019, 13, 109–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phelan, C.M.; Iqbal, J.; Lynch, H.T.; Lubinski, J.; Gronwald, J.; Moller, P.; Ghadirian, P.; Foulkes, W.D.; Armel, S.; Eisen, A.; et al. Incidence of colorectal cancer in BRCA1 and BRCA2 mutation carriers: Results from a follow-up study. Br. J. Cancer 2014, 110, 530–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grinshpun, A.; Halpern, N.; Granit, R.Z.; Hubert, A.; Hamburger, T.; Laitman, Y.; Shacham-Shmueli, E.; Peerless, Y.; Friedman, E.; Peretz, T. Phenotypic characteristics of colorectal cancer in BRCA1/2 mutation carriers. Eur. J. Hum. Genet. 2018, 26, 382–386. [Google Scholar] [CrossRef]
- Sayed, I.M.; Chakraborty, A.; Abd El-Hafeez, A.A.; Sharma, A.; Sahan, A.Z.; Huang, W.J.M.; Sahoo, D.; Ghosh, P.; Hazra, T.K.; Das, S. The DNA Glycosylase NEIL2 Suppresses Fusobacterium-Infection-Induced Inflammation and DNA Damage in Colonic Epithelial Cells. Cells 2020, 9, 1980. [Google Scholar] [CrossRef]
- Mauri, G.; Arena, S.; Siena, S.; Bardelli, A.; Sartore-Bianchi, A. The DNA damage response pathway as a land of therapeutic opportunities for colorectal cancer. Ann. Oncol. 2020, 31, 1135–1147. [Google Scholar] [CrossRef]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef] [Green Version]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Hoppe, M.M.; Sundar, R.; Tan, D.S.P.; Jeyasekharan, A.D. Biomarkers for Homologous Recombination Deficiency in Cancer. J. Natl. Cancer Inst. 2018, 110, 704–713. [Google Scholar] [CrossRef] [Green Version]
- Watkins, J.A.; Irshad, S.; Grigoriadis, A.; Tutt, A.N. Genomic scars as biomarkers of homologous recombination deficiency and drug response in breast and ovarian cancers. Breast Cancer Res. 2014, 16, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Telli, M.L.; Timms, K.M.; Reid, J.; Hennessy, B.; Mills, G.B.; Jensen, K.C.; Szallasi, Z.; Barry, W.T.; Winer, E.P.; Tung, N.M.; et al. Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin. Cancer Res. 2016, 22, 3764–3773. [Google Scholar] [CrossRef] [Green Version]
- Ford, L.; Wolford, J.E.; Brown, S.M.; Randall, L.M. A profile on the FoundationFocus CDxBRCA tests. Expert Rev. Mol. Diagn. 2020, 20, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, E.; Amillano, K.; Cortés, A.; Juan, M.J.; Márquez, A.; Ruiz, M.; Cortes, J. Abstract CT165: A two-stage Simon Design phase II study for NOn-BRCA metastatic BReast cancer (MBC)patients with homologous recombination deficiency treated with OLAparib single agent. (NOBROLA study). Cancer Res. 2018, 78, CT165. [Google Scholar] [CrossRef] [Green Version]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Funingana, I.G.; Reinius, M.A.V.; Petrillo, A.; Ang, J.E.; Brenton, J.D. Can integrative biomarker approaches improve prediction of platinum and PARP inhibitor response in ovarian cancer? Semin. Cancer Biol. 2021, 77, 67–82. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 2017, 23, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Kubota, E.; Williamson, C.T.; Ye, R.; Elegbede, A.; Peterson, L.; Lees-Miller, S.P.; Bebb, D.G. Low ATM protein expression and depletion of p53 correlates with olaparib sensitivity in gastric cancer cell lines. Cell Cycle 2014, 13, 2129–2137. [Google Scholar] [CrossRef] [Green Version]
- Bang, Y.J.; Im, S.A.; Lee, K.W.; Cho, J.Y.; Song, E.K.; Lee, K.H.; Kim, Y.H.; Park, J.O.; Chun, H.G.; Zang, D.Y.; et al. Randomized, Double-Blind Phase II Trial with Prospective Classification by ATM Protein Level to Evaluate the Efficacy and Tolerability of Olaparib Plus Paclitaxel in Patients with Recurrent or Metastatic Gastric Cancer. J. Clin. Oncol. 2015, 33, 3858–3865. [Google Scholar] [CrossRef]
- Gilardini Montani, M.S.; Prodosmo, A.; Stagni, V.; Merli, D.; Monteonofrio, L.; Gatti, V.; Gentileschi, M.P.; Barila, D.; Soddu, S. ATM-depletion in breast cancer cells confers sensitivity to PARP inhibition. J. Exp. Clin. Cancer Res. 2013, 32, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; Peng, Y.; Wei, L.; Zhang, W.; Yang, L.; Lan, L.; Kapoor, P.; Ju, Z.; Mo, Q.; Shih, I.-M.; et al. ARID1A Deficiency Impairs the DNA Damage Checkpoint and Sensitizes Cells to PARP Inhibitors. Cancer Discov. 2015, 5, 752–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Pak, H.; Hammond-Martel, I.; Ghram, M.; Rodrigue, A.; Daou, S.; Barbour, H.; Corbeil, L.; Hebert, J.; Drobetsky, E.; et al. Tumor suppressor and deubiquitinase BAP1 promotes DNA double-strand break repair. Proc. Natl. Acad. Sci. USA 2014, 111, 285–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrotta, R.; Okonska, A.; Ronner, M.; Weder, W.; Stahel, R.; Penengo, L.; Felley-Bosco, E. A Novel BRCA1-Associated Protein-1 Isoform Affects Response of Mesothelioma Cells to Drugs Impairing BRCA1-Mediated DNA Repair. J. Thorac. Oncol. 2017, 12, 1309–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajrami, I.; Frankum, J.R.; Konde, A.; Miller, R.E.; Rehman, F.L.; Brough, R.; Campbell, J.; Sims, D.; Rafiq, R.; Hooper, S.; et al. Genome-wide profiling of genetic synthetic lethality identifies CDK12 as a novel determinant of PARP1/2 inhibitor sensitivity. Cancer Res. 2014, 74, 287–297. [Google Scholar] [CrossRef] [Green Version]
- Goodall, J.; Mateo, J.; Yuan, W.; Mossop, H.; Porta, N.; Miranda, S.; Perez-Lopez, R.; Dolling, D.; Robinson, D.R.; Sandhu, S.; et al. Circulating Cell-Free DNA to Guide Prostate Cancer Treatment with PARP Inhibition. Cancer Discov. 2017, 7, 1006–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, G.; Chun-Jen Lin, C.; Mo, W.; Dai, H.; Park, Y.Y.; Kim, S.M.; Peng, Y.; Mo, Q.; Siwko, S.; Hu, R.; et al. Genome-wide transcriptome profiling of homologous recombination DNA repair. Nat. Commun. 2014, 5, 3361. [Google Scholar] [CrossRef] [Green Version]
- Arena, S.; Corti, G.; Durinikova, E.; Montone, M.; Reilly, N.M.; Russo, M.; Lorenzato, A.; Arcella, P.; Lazzari, L.; Rospo, G.; et al. A Subset of Colorectal Cancers with Cross-Sensitivity to Olaparib and Oxaliplatin. Clin. Cancer Res. 2020, 26, 1372–1384. [Google Scholar] [CrossRef]
- Allison Stewart, C.; Tong, P.; Cardnell, R.J.; Sen, T.; Li, L.; Gay, C.M.; Masrorpour, F.; Fan, Y.; Bara, R.O.; Feng, Y.; et al. Dynamic variations in epithelial-to-mesenchymal transition (EMT), ATM, and SLFN11 govern response to PARP inhibitors and cisplatin in small cell lung cancer. Oncotarget 2017, 8, 28575–28587. [Google Scholar] [CrossRef] [Green Version]
- Byers, L.A.; Wang, J.; Nilsson, M.B.; Fujimoto, J.; Saintigny, P.; Yordy, J.; Giri, U.; Peyton, M.; Fan, Y.H.; Diao, L.; et al. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Discov. 2012, 2, 798–811. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Kwintkiewicz, J.; Liu, Y.; Tech, K.; Frady, L.N.; Su, Y.T.; Bautista, W.; Moon, S.I.; MacDonald, J.; Ewend, M.G.; et al. Chemosensitivity of IDH1-Mutated Gliomas Due to an Impairment in PARP1-Mediated DNA Repair. Cancer Res. 2017, 77, 1709–1718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAndrew, E.N.; Lepage, C.C.; McManus, K.J. The synthetic lethal killing of RAD54B-deficient colorectal cancer cells by PARP1 inhibition is enhanced with SOD1 inhibition. Oncotarget 2016, 7, 87417–87430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Jette, N.; Moussienko, D.; Bebb, D.G.; Lees-Miller, S.P. ATM-Deficient Colorectal Cancer Cells Are Sensitive to the PARP Inhibitor Olaparib. Transl. Oncol. 2017, 10, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Ozden, O.; Bishehsari, F.; Bauer, J.; Park, S.H.; Jana, A.; Baik, S.H.; Sporn, J.C.; Staudacher, J.J.; Yazici, C.; Krett, N.; et al. Expression of an Oncogenic BARD1 Splice Variant Impairs Homologous Recombination and Predicts Response to PARP-1 Inhibitor Therapy in Colon Cancer. Sci. Rep. 2016, 6, 26273. [Google Scholar] [CrossRef]
- Smeby, J.; Kryeziu, K.; Berg, K.C.G.; Eilertsen, I.A.; Eide, P.W.; Johannessen, B.; Guren, M.G.; Nesbakken, A.; Bruun, J.; Lothe, R.A.; et al. Molecular correlates of sensitivity to PARP inhibition beyond homologous recombination deficiency in pre-clinical models of colorectal cancer point to wild-type TP53 activity. EBioMedicine 2020, 59, 102923. [Google Scholar] [CrossRef]
- Xu, K.; Chen, Z.; Cui, Y.; Qin, C.; He, Y.; Song, X. Combined olaparib and oxaliplatin inhibits tumor proliferation and induces G2/M arrest and gamma-H2AX foci formation in colorectal cancer. OncoTargets Ther. 2015, 8, 3047–3054. [Google Scholar] [CrossRef] [Green Version]
- Genther Williams, S.M.; Kuznicki, A.M.; Andrade, P.; Dolinski, B.M.; Elbi, C.; O’Hagan, R.C.; Toniatti, C. Treatment with the PARP inhibitor, niraparib, sensitizes colorectal cancer cell lines to irinotecan regardless of MSI/MSS status. Cancer Cell Int. 2015, 15, 14. [Google Scholar] [CrossRef] [Green Version]
- Tahara, M.; Inoue, T.; Sato, F.; Miyakura, Y.; Horie, H.; Yasuda, Y.; Fujii, H.; Kotake, K.; Sugano, K. The use of Olaparib (AZD2281) potentiates SN-38 cytotoxicity in colon cancer cells by indirect inhibition of Rad51-mediated repair of DNA double-strand breaks. Mol. Cancer Ther. 2014, 13, 1170–1180. [Google Scholar] [CrossRef] [Green Version]
- Augustine, T.; Maitra, R.; Zhang, J.; Nayak, J.; Goel, S. Sensitization of colorectal cancer to irinotecan therapy by PARP inhibitor rucaparib. Investig. New Drugs 2019, 37, 948–960. [Google Scholar] [CrossRef]
- Greene, J.; Nguyen, A.; Bagby, S.M.; Jones, G.N.; Tai, W.M.; Quackenbush, K.S.; Schreiber, A.; Messersmith, W.A.; Devaraj, K.M.; Blatchford, P.; et al. The novel ATM inhibitor (AZ31) enhances antitumor activity in patient derived xenografts that are resistant to irinotecan monotherapy. Oncotarget 2017, 8, 110904–110913. [Google Scholar] [CrossRef] [Green Version]
- Berlin, J.; Ramanathan, R.K.; Strickler, J.H.; Subramaniam, D.S.; Marshall, J.; Kang, Y.K.; Hetman, R.; Dudley, M.W.; Zeng, J.; Nickner, C.; et al. A phase 1 dose-escalation study of veliparib with bimonthly FOLFIRI in patients with advanced solid tumours. Br. J. Cancer 2018, 118, 938–946. [Google Scholar] [CrossRef]
- Samol, J.; Ranson, M.; Scott, E.; Macpherson, E.; Carmichael, J.; Thomas, A.; Cassidy, J. Safety and tolerability of the poly(ADP-ribose) polymerase (PARP) inhibitor, olaparib (AZD2281) in combination with topotecan for the treatment of patients with advanced solid tumors: A phase I study. Investig. New Drugs 2012, 30, 1493–1500. [Google Scholar] [CrossRef] [PubMed]
- Kummar, S.; Chen, A.; Ji, J.; Zhang, Y.; Reid, J.M.; Ames, M.; Jia, L.; Weil, M.; Speranza, G.; Murgo, A.J.; et al. Phase I study of PARP inhibitor ABT-888 in combination with topotecan in adults with refractory solid tumors and lymphomas. Cancer Res. 2011, 71, 5626–5634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czito, B.G.; Deming, D.A.; Jameson, G.S.; Mulcahy, M.F.; Vaghefi, H.; Dudley, M.W.; Holen, K.D.; DeLuca, A.; Mittapalli, R.K.; Munasinghe, W.; et al. Safety and tolerability of veliparib combined with capecitabine plus radiotherapy in patients with locally advanced rectal cancer: A phase 1b study. Lancet Gastroenterol. Hepatol. 2017, 2, 418–426. [Google Scholar] [CrossRef]
- Leijen, S.; van Geel, R.M.; Pavlick, A.C.; Tibes, R.; Rosen, L.; Razak, A.R.; Lam, R.; Demuth, T.; Rose, S.; Lee, M.A.; et al. Phase I Study Evaluating WEE1 Inhibitor AZD1775 As Monotherapy and in Combination with Gemcitabine, Cisplatin, or Carboplatin in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2016, 34, 4371–4380. [Google Scholar] [CrossRef] [PubMed]
- Leichman, L.; Groshen, S.; O’Neil, B.H.; Messersmith, W.; Berlin, J.; Chan, E.; Leichman, C.G.; Cohen, S.J.; Cohen, D.; Lenz, H.J.; et al. Phase II Study of Olaparib (AZD-2281) After Standard Systemic Therapies for Disseminated Colorectal Cancer. Oncologist 2016, 21, 172–177. [Google Scholar] [CrossRef] [Green Version]
- Gorbunova, V.; Beck, J.T.; Hofheinz, R.D.; Garcia-Alfonso, P.; Nechaeva, M.; Cubillo Gracian, A.; Mangel, L.; Elez Fernandez, E.; Deming, D.A.; Ramanathan, R.K.; et al. A phase 2 randomised study of veliparib plus FOLFIRI+/-bevacizumab versus placebo plus FOLFIRI+/-bevacizumab in metastatic colorectal cancer. Br. J. Cancer 2019, 120, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Pishvaian, M.J.; Slack, R.S.; Jiang, W.; He, A.R.; Hwang, J.J.; Hankin, A.; Dorsch-Vogel, K.; Kukadiya, D.; Weiner, L.M.; Marshall, J.L.; et al. A phase 2 study of the PARP inhibitor veliparib plus temozolomide in patients with heavily pretreated metastatic colorectal cancer. Cancer 2018, 124, 2337–2346. [Google Scholar] [CrossRef]
- A Study to Evaluate Rucaparib in Patients with Solid Tumors and with Deleterious Mutations in HRR Genes (LODESTAR). Available online: https://clinicaltrials.gov/ct2/show/NCT04171700 (accessed on 10 February 2022).
- Davidson, D.; Wang, Y.; Aloyz, R.; Panasci, L. The PARP inhibitor ABT-888 synergizes irinotecan treatment of colon cancer cell lines. Investig. New Drugs 2013, 31, 461–468. [Google Scholar] [CrossRef]
- Goldstein, M.; Kastan, M.B. The DNA damage response: Implications for tumor responses to radiation and chemotherapy. Annu. Rev. Med. 2015, 66, 129–143. [Google Scholar] [CrossRef] [Green Version]
- Reilly, N.M.; Novara, L.; Di Nicolantonio, F.; Bardelli, A. Exploiting DNA repair defects in colorectal cancer. Mol. Oncol. 2019, 13, 681–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakkenist, C.J.; Lee, J.J.; Schmitz, J.C. ATM Is Required for the Repair of Oxaliplatin-Induced DNA Damage in Colorectal Cancer. Clin. Colorectal Cancer 2018, 17, 255–257. [Google Scholar] [CrossRef] [PubMed]
- Sundar, R.; Miranda, S.; Rodrigues, D.N.; Chenard-Poirier, M.; Dolling, D.; Clarke, M.; Figueiredo, I.; Bertan, C.; Yuan, W.; Ferreira, A.; et al. Ataxia Telangiectasia Mutated Protein Loss and Benefit From Oxaliplatin-based Chemotherapy in Colorectal Cancer. Clin. Colorectal Cancer 2018, 17, 280–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Ioannes, P.; Malu, S.; Cortes, P.; Aggarwal, A.K. Structural basis of DNA ligase IV-Artemis interaction in nonhomologous end-joining. Cell Rep. 2012, 2, 1505–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Pinch, B.J.; Olson, C.M.; Donovan, K.A.; Nowak, R.P.; Mills, C.E.; Scott, D.A.; Doctor, Z.M.; Eleuteri, N.A.; Chung, M.; et al. Development and Characterization of a Wee1 Kinase Degrader. Cell Chem Biol. 2020, 27, 57–65.e9. [Google Scholar] [CrossRef]
- Ha, D.H.; Min, A.; Kim, S.; Jang, H.; Kim, S.H.; Kim, H.J.; Ryu, H.S.; Ku, J.L.; Lee, K.H.; Im, S.A. Antitumor effect of a WEE1 inhibitor and potentiation of olaparib sensitivity by DNA damage response modulation in triple-negative breast cancer. Sci. Rep. 2020, 10, 9930. [Google Scholar] [CrossRef]
- Phase I/Ib Trial of ATR Inhibitor BAY 1895344 in Combination with FOLFIRI in GI Malignancies with a Focus on Metastatic Colorectal and Gastric/Gastroesophageal Cancers. Available online: https://clinicaltrials.gov/ct2/show/NCT04535401 (accessed on 10 February 2022).
- Kim, S.T.; Smith, S.A.; Mortimer, P.; Loembe, A.B.; Cho, H.; Kim, K.M.; Smith, C.; Willis, S.; Irurzun-Arana, I.; Berges, A.; et al. Phase I Study of Ceralasertib (AZD6738), a Novel DNA Damage Repair Agent, in Combination with Weekly Paclitaxel in Refractory Cancer. Clin. Cancer Res. 2021, 27, 4700–4709. [Google Scholar] [CrossRef]
- Kim, R.; Kwon, M.; An, M.; Kim, S.T.; Smith, S.A.; Loembe, A.B.; Mortimer, P.G.S.; Armenia, J.; Lukashchuk, N.; Shah, N.; et al. Phase II study of ceralasertib (AZD6738) in combination with durvalumab in patients with advanced/metastatic melanoma who have failed prior anti-PD-1 therapy. Ann. Oncol. 2021, 33, 193–203. [Google Scholar] [CrossRef]
- Yap, T.A.; Krebs, M.G.; Postel-Vinay, S.; El-Khouiery, A.; Soria, J.C.; Lopez, J.; Berges, A.; Cheung, S.A.; Irurzun-Arana, I.; Goldwin, A.; et al. Ceralasertib (AZD6738), an Oral ATR Kinase Inhibitor, in Combination with Carboplatin in Patients with Advanced Solid Tumors: A Phase I Study. Clin. Cancer Res. 2021. [Google Scholar] [CrossRef]
- Phase 1/1B Study of DS-8201a in Combination with ATR Inhibition (AZD6738) in Advanced Solid Tumors with HER2 Expression (DASH Trial). Available online: https://clinicaltrials.gov/ct2/show/NCT04704661 (accessed on 10 February 2022).
- A Phase Ib Study Combining Irinotecan with AZD1775, a Selective Wee 1 Inhibitor, in RAS (KRAS or NRAS) or BRAF Mutated Metastatic Colorectal Cancer Patients Who Have Progressed on First Line Therapy. Available online: https://clinicaltrials.gov/ct2/show/NCT02906059 (accessed on 10 February 2022).
- Seligmann, J.F.; Fisher, D.J.; Brown, L.C.; Adams, R.A.; Graham, J.; Quirke, P.; Richman, S.D.; Butler, R.; Domingo, E.; Blake, A.; et al. Inhibition of WEE1 Is Effective in TP53- and RAS-Mutant Metastatic Colorectal Cancer: A Randomized Trial (FOCUS4-C) Comparing Adavosertib (AZD1775) with Active Monitoring. J. Clin. Oncol. 2021, 39, 3705–3715. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Incorvaia, L.; Capoluongo, E.; Tagliaferri, P.; Galvano, A.; Del Re, M.; Malapelle, U.; Chiari, R.; Conte, P.; Danesi, R.; et al. The challenge of the Molecular Tumor Board empowerment in clinical oncology practice: A Position Paper on behalf of the AIOM-SIAPEC/IAP-SIBioC-SIC-SIF-SIGU-SIRM Italian Scientific Societies. Crit. Rev. Oncol. Hematol. 2022, 169, 103567. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, C.; Sud, A.; Houlston, R.S. Cancer genetics, precision prevention and a call to action. Nat. Genet. 2018, 50, 1212–1218. [Google Scholar] [CrossRef]
- Lesueur, F.; Easton, D.F.; Renault, A.L.; Tavtigian, S.V.; Bernstein, J.L.; Kote-Jarai, Z.; Eeles, R.A.; Plaseska-Karanfia, D.; Feliubadalo, L.; Arun, B.; et al. First international workshop of the ATM and cancer risk group (4–5 December 2019). Fam. Cancer 2021, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Toss, A.; Tenedini, E.; Piombino, C.; Venturelli, M.; Marchi, I.; Gasparini, E.; Barbieri, E.; Razzaboni, E.; Domati, F.; Caggia, F.; et al. Clinicopathologic Profile of Breast Cancer in Germline ATM and CHEK2 Mutation Carriers. Genes 2021, 12, 616. [Google Scholar] [CrossRef] [PubMed]
- Hannan, Z.; Yu, S.; Domchek, S.; Mamtani, R.; Reiss, K.A. Clinical Characteristics of Patients with Pancreatic Cancer and Pathogenic ATM Alterations. JNCI Cancer Spectr. 2021, 5, pkaa121. [Google Scholar] [CrossRef]
- Lorans, M.; Dow, E.; Macrae, F.A.; Winship, I.M.; Buchanan, D.D. Update on Hereditary Colorectal Cancer: Improving the Clinical Utility of Multigene Panel Testing. Clin. Colorectal Cancer 2018, 17, e293–e305. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
| Authors/Year | Phase | Patient Population | Drugs | Results | Ref. |
|---|---|---|---|---|---|
| Leichman et al., 2016 | 2 | CRC, 33 patients (20 MSS; 13 MSI-H) | Olaparib (AZD-2281) | No complete or partial responses were reported; ORR 0% | [101] |
| Gorbunova et al., 2019 | 2 | mCRC, 130 patients | Veliparib + FOLFIRI ± bevacizumab vs. Placebo + FOLFIRI ± bevacizumab | mPFS 12 vs. 11 months mOS 25 vs. 27 months mDOR 11 vs. 9 months ORR 57% | [102] |
| Pishvaian et al., 2018 | 2 | mCRC, 75 patients | Veliparib + temozolomide | DCR 24% mPFS 1.8 months mOS 6.6 months | [103] |
| Czito et al., 2017 | 1b | Locally advanced RC, 32 patients | Veliparib + capecitabine + RT | 29% of patients achieved CR | [99] |
| Samol et al., 2012 | 1 | mCRC, 8 patients | Olaparib + topotecan | ORR 0% | [97] |
| Kummar et al., 2011 | 1 | CRC, 5 patients | Veliparib + topotecan | ORR 0% | [98] |
| Berlin et al., 2018 | 1 | CRC, 10 patients | Veliparib + FOLFIRI | ORR 20% | [96] |
| Clinical Trial | Phase | Patient Population | Mutations | Treatment Arm(s) |
|---|---|---|---|---|
| NCT02484404 | 1–2 | Advanced solid tumors | Not required | MEDI4736 (anti PD1) + olaparib and/or cediranib |
| NCT04171700 | 2 | Solid tumors | Deleterious mutation (germline or somatic) in BRCA1, BRCA2, PALB2, RAD51C, RAD51D, BARD1, BRIP1, FANCA, NBN, RAD51, or RAD51B | Rucaparib |
| NCT04166435 | 2 | mCRC | MGMT promoter hypermethylation | Temozolomide + olaparib |
| NCT03983993 | 2 | mCRC | Not required (RAS wildtype) | Niraparib + panitumumab |
| NCT04456699 | 3 | mCRC | Not required | Olaparib OR olaparib + bevacizumab Vs. bevacizumab + 5-FU |
| NCT04511039 | 1 | CRC or gastroesophageal cancer | Not required | Trifluridine and tipiracil hydrochloride + talazoparib * |
| NCT03337087 | 1–2 | Advanced pancreatic, colorectal, gastroesophageal, or biliary cancer | Only for pancreatic cancer: BRCA1 or BRCA2 or PALB2 mutation, or HRD (non-BRCA, non-PALB) | Rucaparib + liposomal irinotecan + fluorouracil + leucovorin calcium |
| NCT03842228 | 1 | Advanced solid tumors | Germline or somatic mutations in DDR genes (ARID1A, ATM, ATRX, BARD1, BRCA1, BRCA2, BRIP1, CDK12, CHEK1, CHEK2, FANCA, FANCL, MRE11A, MSH2, PALB2, PARP1, POLD1, PP2R2A, RAD51B, RAD51C, RAD51D, RAD54L, or XRCC2), actionable mutations in the PTEN gene, or hotspot mutations in the PIK3CA gene (E542, E545, or H1047) | Olaparib + MEDI4736 (durvalumab) + copanlisib hydrocloride |
| NCT04123366 | 2 | Advanced solid tumors | Known or suspected deleterious mutations in ≥1 of the specified 15 genes involved in HRR | Olaparib + pembrolizumab |
| NCT04497116 | 1–2 | Advanced solid tumors | ATR inhibitor-sensitizing mutations | RP-3500 (oral ATR inhibitor) ± talazoparib |
| NCT03127215 | 2 | Advanced solid tumors | Defective DNA repair via HRR | Trabectedin/olaparib vs. physician’s choice |
| NCT04276376 | 2 | Advanced solid tumors | In CRC cohort: ATM, BARD1, BRCA1, BRCA2, BRIP1, CDK12, CHEK2, PALB2, RAD51C, RAD51D, FANCA, NBN, RAD51, RAD54L | Rucaparib + atezolizumab |
| NCT03851614 | 2 | mCRC or Pancreatic adenocarcinoma or Leyomiosarcoma | Not required (CRC patients must have MMR proficiency disease) | Olaparib + durvalumab OR cediranib + durvalumab * |
| NCT04693468 | 1 | Metastatic solid tumors | Defect in DDR, MET, ALK, or ROS1 genes | Talazoparib + palbociclib OR talazoparib + axitinib OR talazoparib + crizotinib |
| NCT03772561 | 1 | Advanced solid tumors | Not required | AZD5363 + olaparib + durvalumab |
| NCT04672460 | 1 | Advanced solid tumors | Solid tumors with known or likely pathogenic germline or somatic variants in BRCA1 or BRCA2 that would benefit from PARPi therapy | Talazoparib capsule vs. talazoparib soft gel capsule |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Catalano, F.; Borea, R.; Puglisi, S.; Boutros, A.; Gandini, A.; Cremante, M.; Martelli, V.; Sciallero, S.; Puccini, A. Targeting the DNA Damage Response Pathway as a Novel Therapeutic Strategy in Colorectal Cancer. Cancers 2022, 14, 1388. https://doi.org/10.3390/cancers14061388
Catalano F, Borea R, Puglisi S, Boutros A, Gandini A, Cremante M, Martelli V, Sciallero S, Puccini A. Targeting the DNA Damage Response Pathway as a Novel Therapeutic Strategy in Colorectal Cancer. Cancers. 2022; 14(6):1388. https://doi.org/10.3390/cancers14061388
Chicago/Turabian StyleCatalano, Fabio, Roberto Borea, Silvia Puglisi, Andrea Boutros, Annalice Gandini, Malvina Cremante, Valentino Martelli, Stefania Sciallero, and Alberto Puccini. 2022. "Targeting the DNA Damage Response Pathway as a Novel Therapeutic Strategy in Colorectal Cancer" Cancers 14, no. 6: 1388. https://doi.org/10.3390/cancers14061388
APA StyleCatalano, F., Borea, R., Puglisi, S., Boutros, A., Gandini, A., Cremante, M., Martelli, V., Sciallero, S., & Puccini, A. (2022). Targeting the DNA Damage Response Pathway as a Novel Therapeutic Strategy in Colorectal Cancer. Cancers, 14(6), 1388. https://doi.org/10.3390/cancers14061388